Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Activation of Notch1 signaling alleviates dysfunction of bone marrow-derived mesenchymal stem cells induced by cigarette smoke extract
Authors Cheng Y, Gu W ![]() , Zhang G, Li X, Guo XJ
, Zhang G, Li X, Guo XJ
Received 12 July 2017
Accepted for publication 30 September 2017
Published 27 October 2017 Volume 2017:12 Pages 3133—3147
DOI https://doi.org/10.2147/COPD.S146201
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chunxue Bai
Yi Cheng,* Wen Gu,* Guorui Zhang, Xiaoming Li, Xuejun Guo
Department of Respiratory Medicine, Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
*These authors contributed equally to this work
Abstract: Bone marrow-derived mesenchymal stem cells (BM-MSCs) are considered attractive therapeutic agents for the treatment of COPD. However, little is known about the impact of Notch on the proliferation, migration, and survival of MSCs in a cigarette smoke (CS) microenvironment. Here, we used CS extract to mimic the CS microenvironment in vitro, with the intention to investigate the effect of Notch in regulating proliferation, migration, and survival of BM-MSCs. Rat bone marrow mesenchymal stem cells were infected with lentivirus vector containing the intracellular domain of Notch1 (N1ICD) and challenged with CS extract. Cell proliferation was detected by Ki67 staining and expression of cell cycle-related proteins. A transwell assay was used to measure cell migration and the expression of apoptotic proteins was examined. The proliferation of BM-MSCs overexpressing N1ICD significantly increased. Consistently, levels of cyclin D1, p-Rb, and E2F-1 increased in N1ICD overexpressing cells. N1ICD overexpression also increased cell migration compared with the control group. N1ICD overexpression equilibrated the expression of Bax and Bcl-2, and blocked caspase-3 cleavage, contributing to the inhibition of apoptosis. Moreover, blockade of the PI3K/Akt pathway suppressed the aforementioned cytoprotective effects of N1ICD. In conclusion, activation of Notch signaling improved proliferation, migration, and survival of BM-MSCs in a CS microenvironment partly through the PI3K/Akt pathway.
Keywords: mesenchymal stem cells, chronic obstructive pulmonary disease, cigarette smoke extract, Notch
Introduction
Mesenchymal stem cells (MSCs) are pluripotent stem cells with a fibroblast-like morphology and the capacity to differentiate into chondrocytes, osteoblasts, adipocytes, and muscle cells under different microenvironmental conditions, culture media, and supplements.1,2 MSCs are suitable for cell replacement. They do not possess the plasticity of embryonic stem cells, and they offer practical advantages in their ease of separation, propagation, and low immunogenicity. Their use also is not burdened by ethical issues often raised by the use of embryonic stem cells.3 In addition to their regenerative properties, MSCs have therapeutic potential for many diseases because of their anti-inflammatory, immunomodulatory, and anti-apoptotic properties.4
COPD is characterized by persistent inflammation and progressive alveolar destruction. It is becoming a major global health problem. Cigarette smoke (CS) is considered the chief causative agent in the development of COPD.5 In addition to the respiratory system, CS affects the function of numerous organs and systems.6 MSCs are known to possess potent therapeutic capacities in animal models of CS-induced or elastase-induced COPD/emphysema.7,8 However, little is known about the effect of CS on bone marrow (BM)-derived stem cells. Previous data indicate that BM is suppressed in patients with COPD, supported by the indirect finding that their circulating BM-derived progenitor cells were reduced.9,10 Furthermore, a direct effect of CS on the BM is supported by experimental studies showing that CS can adversely affect the performance of BM-derived stem cell recruitment to, and differentiation in, the uterus.11 Despite the observed beneficial effects of cell therapy, improving the function of MSCs in a CS microenvironment is particularly important to improve the therapeutic potential of BM-MSC transplantation. Unfortunately, no potential remedies for CS-induced BM dysfunction have been identified.
Notch proteins belong to a family of transmembrane receptor proteins (Notch1–4) that are the pivotal components of the Notch signaling pathway. Tumor necrosis factor-converting enzyme and the γ-secretase/presenilin complex mediate activation of Notch signaling sequentially.12 In the canonical Notch signaling pathway, activation of Notch receptors releases a signal in the form of the intracellular domain of Notch (NICD).13 In the absence of NICD, the DNA-binding protein CSL associates with ubiquitous co-repressor proteins to repress the transcription of target genes. When NICD enters the nucleus, its binding to CSL may trigger an allosteric change that facilitates displacement of transcriptional repressors with consequential activation of downstream effector proteins such as Hes and Hey protein,14 which are indispensable for the regulation of stem cell proliferation, differentiation, migration, and apoptosis.15 We hypothesized that the biological function of BM-MSCs would be improved by Notch signaling activation in the CS microenvironment. CS extract (CSE) contains all of the compounds inhaled by smokers and is frequently added to cells in culture to monitor specific cellular responses.16 To ascertain the particular biological effects of CS on MSCs, we used CSE to mimic the CS microenvironment in vitro. In the present study, we examined the beneficial effects of Notch1 on MSC survival, proliferation, and migration in the CS microenvironment, which partly depend on the PI3K/Akt signaling pathway.
Materials and methods
Ethics statement
The present study was conducted in accordance with the Declaration of Helsinki and the guidelines of the Ethics Committee of Xinhua Hospital affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China. The Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China, approved all experimental protocols.
Cell culture
The rat alveolar epithelial cell line, RLE-6TN, which was characterized and found to be similar to alveolar type 2 cells, was purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM: Nutrient mixture F-12 (DMEM/F-12; Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS, Gibco), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Gibco), in a humidified incubator with an atmosphere of 5% CO2 and 95% air at 37°C.
Isolation, culture, and identification of BM-MSCs
Briefly, BM samples from male Sprague Dawley (SD) rats were diluted with PBS and centrifuged at 1,200 rpm for 10 min. The buffy coat containing mononuclear cells was washed twice with PBS and plated in MSC culture growth medium, which consisted of DMEM/F-12 supplemented with 10% (v/v) FBS and 1% (v/v) penicillin–streptomycin. Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. After 24 h, non-adherent cells were discarded. Fresh medium was added and replaced every 3 days.
MSCs were analyzed by flow cytometry (BD Biosciences, San Diego, CA, USA) using anti-CD29, anti-CD90, anti-CD80, anti-CD86, (eBioscience, San Diego, CA, USA), CD34, CD45, CD73, and CD105 antibodies (BD Biosciences) at passage 3. Antibody isotypes were used for negative controls. Cells were incubated in the dark at 4°C with the antibodies for 30 min, then washed 3 times with 10 volumes of PBS and centrifuged at 300× g for 5 min. Next, the cells were resuspended in PBS supplemented with 1% FBS and measured by fluorescence-activated cell sorting (FACS). The acquired data from 4 independent experiments were analyzed using FCS Express V4 (BD Biosciences).
To determine tri-lineage differentiation capacity, MSCs were cultured in adipogenic, osteogenic, or chondrogenic medium (Cyagen Biosciences, Guangzhou, China) according to the manufacturer’s protocol. To demonstrate adipogenic differentiation, the cells were stained with 0.3% Oil Red O (O0625; Sigma, St Louis, MO, USA) in isopropanol for 30 min and rinsed with PBS. For osteogenic differentiation, the cells were stained with 2% Alizarin Red (A5533; Sigma) for 5 min and rinsed with PBS. For chondrogenic differentiation, cells were stained with 0.1% Alcian Blue (Cyagen Biosciences) for 30 min and rinsed with PBS. Stained cells were imaged under a Phase-contrast microscope (Leica, Wetzlar, Germany).
CSE preparation
CSE was prepared as previously described.17,18 Briefly, 400 mL of CS (containing 12 mg of tar and 2.5 mg of nicotine per cigarette; Da Qianmen, Shanghai, China) was drawn into a 50 mL plastic syringe through a 3-way stopcock and mixed with 20 mL of DMEM/F-12 by vigorous shaking. One cigarette was used per 20 mL CSE, and CSE was prepared no more than 30 min before being used in experiments. The CSE solution was filtered through an aseptic 0.22 μm filter and considered a 100% extract.
N1ICD overexpressing lentivirus and transfection
Rat Notch-1 overexpressing lentivirus and lentivirus control were obtained from Genechem (Shanghai, China). Rat Notch-1 complementary DNA (cDNA) (NM_001105721) was generated by polymerase chain reaction (PCR) using the forward primer: 5′-GAGGATCCCCGGGTACCGGTCGCCACCATGCGCAAGCGCAGGCGGCAGCATG-3′ and reverse primer: 5′-CACCCACATTCCAGAGGCATTTAAGGGTATGGACTACAAGGA-3′. Rat BM-MSCs were transfected with N1ICD overexpressing lentivirus (Lenti-N1ICD-MSCs) and empty vector lentivirus carrying enhanced green fluorescence protein (Lenti-V-MSCs), according to the manufacturer’s instructions. Cells were used for the experiments 72 h post-transfection.
Reagent treatment
To investigate the role of the PI3K/Akt pathway in N1ICD-mediated MSC survival, proliferation, and migration, LY294002 (20 μM; Cell Signaling Technology, Danvers, MA, USA) was added to the MSC medium for 2 h before CSE treatment to block PI3K/Akt pathway activation.
Cell viability assay
MSC viability was assessed by using a cell counting kit (CCK-8; Dojindo, Kumamoto, Japan). Test cells (5×103) were plated in 96-well plates. After treatment with CSE for different intervals (6, 12, 24, and 48 h), the medium was removed, and 100 μL of fresh medium containing 10 μL water soluble tetrazolium (WST-8) reagent was added to each well. The cells were incubated with WST-8 reagent at 37°C for 2 h. The cell viability in each well was determined by measuring the absorbance at 450 nm using a microplate reader. Data are expressed as the optical density value.
Cell proliferation
Cell proliferative capacities were determined by immunostaining. After treatment, the cells were fixed with 4% paraformaldehyde for 2 h at 37°C and permeabilized with 0.5% (v/v) triton. Then, the cells were incubated with a Ki67 antibody (BD Biosciences, 1:200) overnight at 4°C, followed by phycoerythrin (PE)-labeled goat anti-mouse secondary antibody (Jackson Immuno Research Laboratories, West Grove, PA, USA, 1:100) for 1 h at 37°C. The cells were washed 3 times with PBS, then stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) for 5 min and immediately visualized under a fluorescence microscope (Olympus, Tokyo, Japan). The total number of double-positive cells in 5 randomly chosen fields was counted at 100× magnification.
Cell migration
Chemotaxis experiments were performed in polycarbonate transwell inserts (8-μm pores; Corning Costar Corp., Corning, NY, USA). RLE-6TN cells (1×106) were added in the lower chamber. MSCs (1×105) in 100 μL of culture medium were seeded into the upper chamber in serum-free medium and cultured at 37°C for 24 h. The non-migrating cells in the upper chamber were carefully removed with a cotton swab. Migrating cells were fixed in 4% paraformaldehyde, stained with 1% crystal violet (Beyotime Institute of Biotechnology, Shanghai, China), and photographed. The number of migrated cells was calculated as the ratio of the experimental samples to the control samples ×100, by counting at least 5 randomly separated fields.
Western blot analysis
Following the appropriate treatments, cultured cells were resuspended in lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Nonidet P (NP)-40, 1 mM EDTA, 1 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride) (Beyotime Institute of Biotechnology) containing protease and phosphatase inhibitors. The suspension was incubated for 30 min at 4°C and lysed by ultrasound pyrolysis for 30 s. The lysate was centrifuged at 14,000 rpm for 20 min. The protein concentration of each sample was determined with a bicinchoninic acid protein assay (Beyotime Institute of Biotechnology). An equal amount of each protein sample was electrophoresed on a 6%–12% gradient sodium dodecyl sulfate-polyacrylamide gel and transferred to a polyvinylidenedifluoride membrane. The membranes were blocked with 5% milk in Tris-buffered saline and 1% Tween at 25°C for 1 h and then incubated with primary antibodies overnight at 4°C. The antibodies used were: β-actin and Flag (Sigma); C-X-C chemokine receptor type 4 (CXCR4; Abgent, San Diego, CA, USA); cleaved caspase-3, Bcl-2, and Bax (Cell Signaling Technology); and p-21, p-Rb, cyclin D1, E2F-1, t-AKT, and p-AKT (Abcam, Cambridge, UK). The blots were washed 3 times with Tris-buffered saline containing 1% Tween and incubated with a horseradish peroxidase-conjugated secondary antibody (Beyotime Institute of Biotechnology) for 1 h at 25°C. Membranes were washed with TBST 3 times for 10 min and the expression signals were detected with an enhanced chemiluminescence reagent (Millipore, Billerica, MA, USA). The relative quantities of proteins were determined by scanning densitometry (ChemiDoc XRS + Systems; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and analyzed using Image Lab 5.0 Software (Bio-Rad Laboratories, Inc.). β-actin was used as an internal loading control.
Real-time PCR (RT-PCR)
Total cellular RNA from MSCs was extracted with TRIzol reagent (Takara Biotechnology, Dalian, China), and used for reverse transcription to cDNA. cDNA was synthesized by using Prime Script RT Master Mix (Takara Biotechnology), according to the manufacturer’s instructions. mRNA levels of Hes-1, Hey1, HeyL, Sdf-1α, Cxcr4, G-Csf, and Csf3r were measured by quantitative PCR using SYBR Green Ex Taq™ (Takara) detected by an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The PCR protocol consisted of 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, and 60°C for 34 s, with a final dissociation of 95°C for 30 s. The relative transcript levels were calculated by using the 2−ΔΔCT method, in which ΔCT is the difference between the threshold cycle for the gene of interest and that for glyceraldehyde-3-phosphate dehydrogenase. The results were normalized to untreated controls. Primer sequences are listed in Table 1.
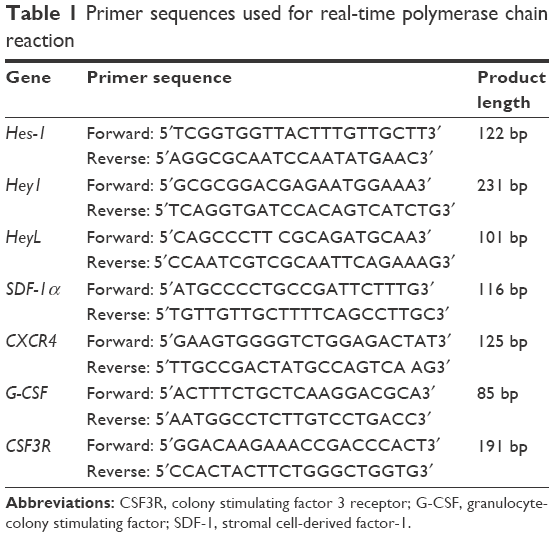 | Table 1 Primer sequences used for real-time polymerase chain reaction |
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) assay
MSCs were seeded onto cover slips and then treated with 10% CSE for 24 h to measure apoptosis. Subsequent to incubation, cover slips were processed for the TUNEL assay according to the manufacturer’s protocol. Briefly, cells were fixed for 1 h in 4% (w/v) paraformaldehyde at 25°C. After specific labeling was performed, the cells were exposed to DAPI in the dark for 5 min to stain nuclei. Then, the immunostained MSCs were observed by fluorescence microscopy. The degree of apoptosis was calculated as the number of double-positive cells per 500 MSC nuclei in 5 randomly chosen fields.
Immunocytochemistry (ICC)
Since caspase-3 activation represents a critical step in the apoptotic process, cleaved caspase-3 was detected in cells by ICC staining with an anti-cleaved-caspase-3 monoclonal antibody (Cell Signaling Technology) according to the manufacturer’s protocol. Apoptotic cells were evaluated manually as the number of anti-cleaved-caspase-3 positive cells per 500 counted MSCs.
Statistical analysis
Data are expressed as the mean ± standard deviation. After comparison of the homogeneity of variance, 1-way analysis of variance was used to determine whether there were any significant differences among the means of the different groups. All statistical analyses were performed using the software SPSS Base 24.0 for Windows (SPSS Inc., Chicago, IL, USA). Probability values P<0.05 were considered significant.
Results
MSC morphology and characterization
MSCs cultured in the medium demonstrated a spindle-like or fibroblast-like shape with directionality and regularity. After several days of induction toward the adipogenic lineage, considerable morphological changes with lipid vacuole accumulation were observed. Following 14 days of osteogenic differentiation, positive staining for Alizarin Red S confirmed the differentiation of MSCs into osteocytes. After 4 weeks of chondrogenic differentiation, characteristic Alcian Blue staining confirmed the differentiation of MSCs into chondrocytes (Figure S1). MSCs were characterized by surface marker expression via FACS analysis. The results presented in Figure 1 indicate that MSCs expressed CD29, CD73, CD90, and CD105, but these cells were negative for CD34, CD45, CD80, and CD86, consistent with a previous report.19
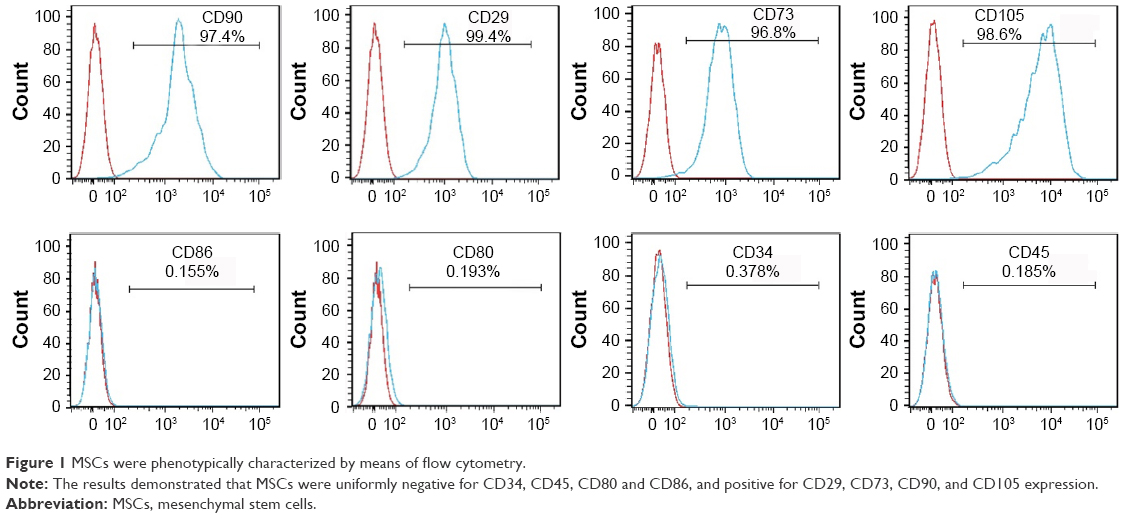 | Figure 1 MSCs were phenotypically characterized by means of flow cytometry. |
Effects of CSE and N1ICD overexpression on MSC viability, Notch signaling pathway activity, and the PI3K/Akt pathway
First, we assessed whether CSE and Notch overexpression had toxic effects on MSCs. We used a single concentration of CSE over an extended period (6–48 h). As shown in Figure 2A, over the entire time course, 10% CSE had little influence on cell viability up to 24 h when compared with untreated cells. However, when MSCs were exposed to 10% CSE for 48 h, their viability decreased significantly. When exposed to 10% CSE for 48 h, the viability of N1ICD overexpressing cells did not significantly decrease compared with that of CSE-exposed MSCs. These results indicated that 10% CSE had little influence on cell viability when exposure was <24 h and N1ICD overexpression can improve the response of MSCs to CSE cytotoxic effects.
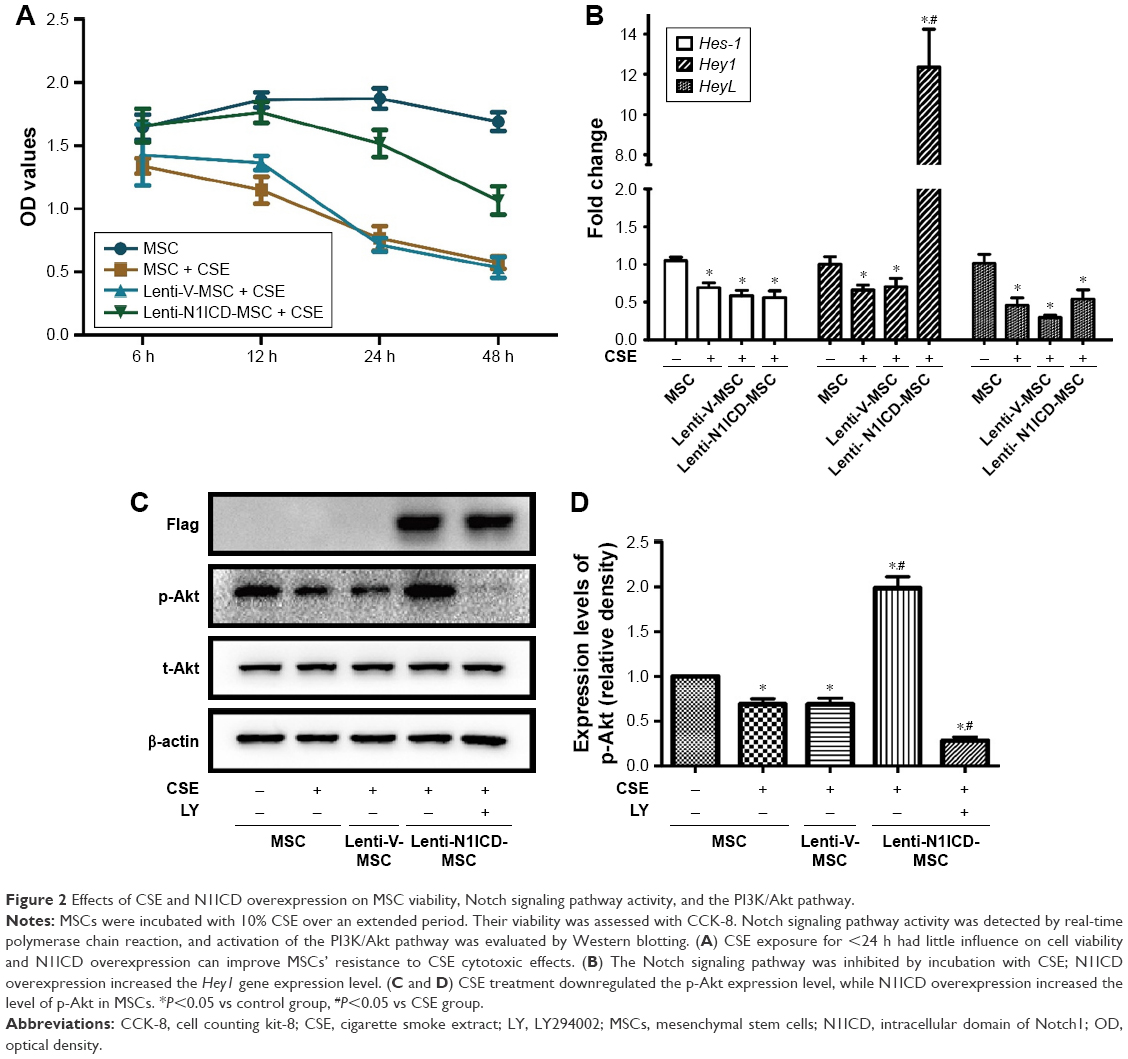 | Figure 2 Effects of CSE and N1ICD overexpression on MSC viability, Notch signaling pathway activity, and the PI3K/Akt pathway. |
To determine the effect of CSE and N1ICD overexpression on Notch signaling pathway activity, we examined the mRNA expression level of Notch1 downstream target genes. N1ICD overexpression increased the Hey1 mRNA gene expression level (P<0.05), which was inhibited by incubation with CSE (Figure 2B). CSE treatment decreased the expression levels of Hes-1 and HeyL compared with the control group (P<0.05); however, there was no difference between the N1ICD and CSE groups (P>0.05). These data suggest that the Notch1 signaling pathway was inhibited by exposure to CSE, while N1ICD overexpression can activate the endogenous Notch1 signaling pathway and Hey1, a target gene of Notch1, in MSCs.
The PI3K/Akt pathway exerts classical growth and survival effects on MSCs.20 Thus, we examined the effects of Notch on the PI3K/Akt pathway in MSCs under CSE exposure. As shown in Figure 2C and D, CSE treatment down-regulated the level of phosphorylated Akt (p-Akt), while N1ICD overexpression increased p-Akt in MSCs (all P<0.05). In the presence of PI3K/Akt inhibitors, p-Akt expression was strongly inhibited, indicating that Notch1 was the upstream activator of the PI3K/Akt pathway in MSCs.
PI3K/Akt is required for N1ICD-mediated proliferation
To establish the role of N1ICD in MSC growth, cells were cultured in the medium containing 10% CSE for 24 h. As shown in Figure 3A and B, the number of Ki67-positive cells was higher in the N1ICD group than that in the CSE group. However, pretreatment with LY294002 reversed this trend, suggesting that N1ICD overexpression caused an increase in the number of dividing cells under CSE exposure. Furthermore, we examined the expression level of cell cycle-related proteins and determined that the expression levels of cyclin D1, p-Rb, and E2F-1 (Figure 3C and D) increased, while that of p21 decreased in N1ICD overexpressing MSCs when compared with that in the CSE group. PI3K/Akt blockade reversed these expression levels. The release of p-Rb activates the transcription factor E2F-1, which allows the expression of genes that are necessary for DNA replication and mitosis.21 These data suggested that N1ICD increased the proliferative capacity of MSCs through the PI3K/Akt pathway, which probably promoted cell proliferation via a cyclin D1-Rb-E2F-1-dependent pathway.
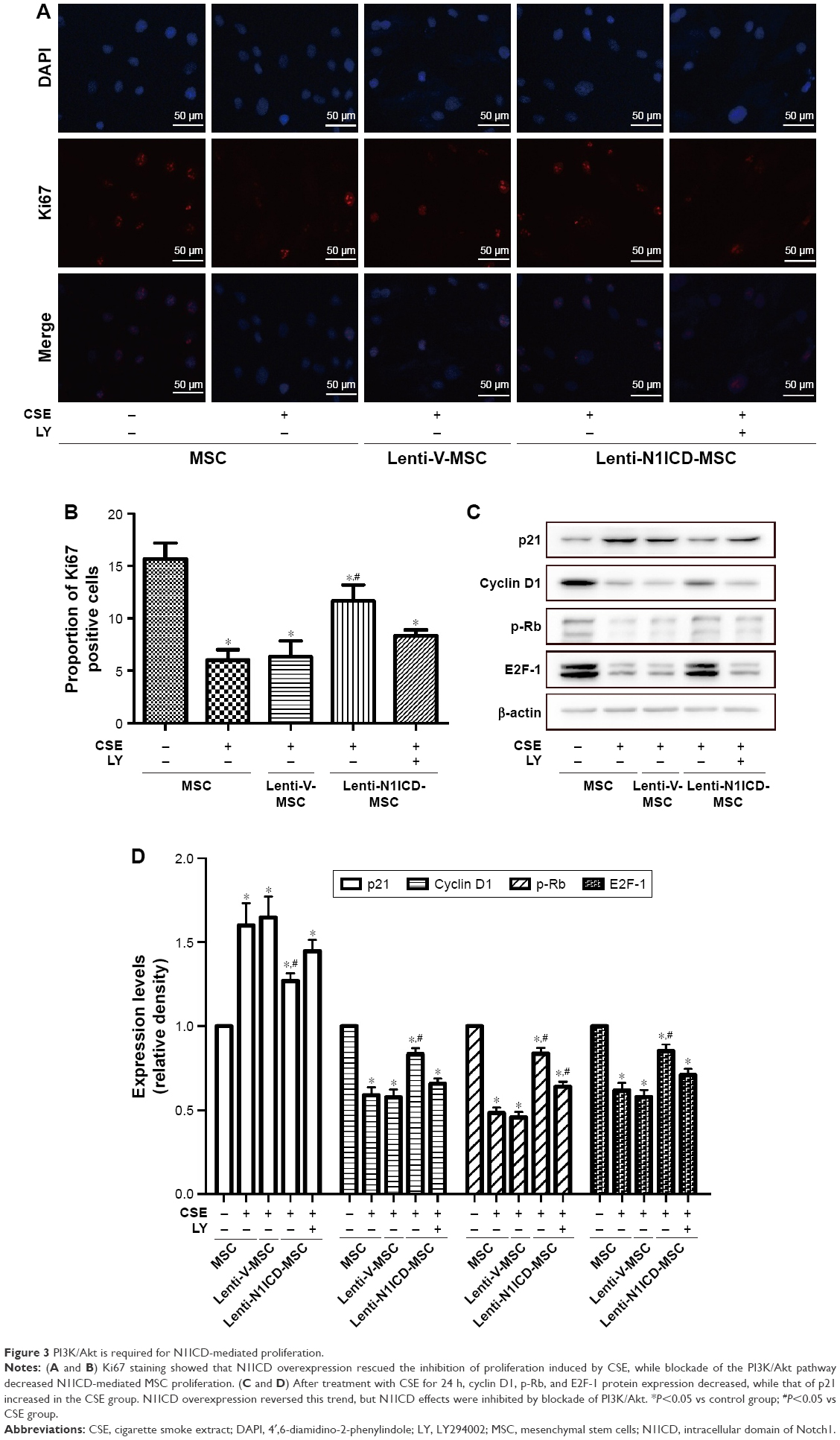 | Figure 3 PI3K/Akt is required for N1ICD-mediated proliferation. |
N1ICD improves CXCR4 expression and the subsequent migration capacity of MSCs
The stromal cell-derived factor 1 (SDF-1)/CXCR4 biological axis plays an important role in the mobilization and recruitment of MSCs to injured lung tissue after transplantation in a rat COPD model.22 In the present study, CSE treatment did not significantly alter CXCR4 protein expression in MSCs. There was no substantial difference between the control and CSE groups (P>0.05). In contrast, N1ICD overexpression markedly increased the CXCR4 protein level compared with that in the CSE group (P<0.05). However, pretreatment with a PI3K/Akt inhibitor partially decreased CXCR4 protein expression levels (Figure 4A and B), suggesting that the up-regulation of CXCR4 in N1ICD overexpressing MSCs involved the PI3K/Akt pathway.
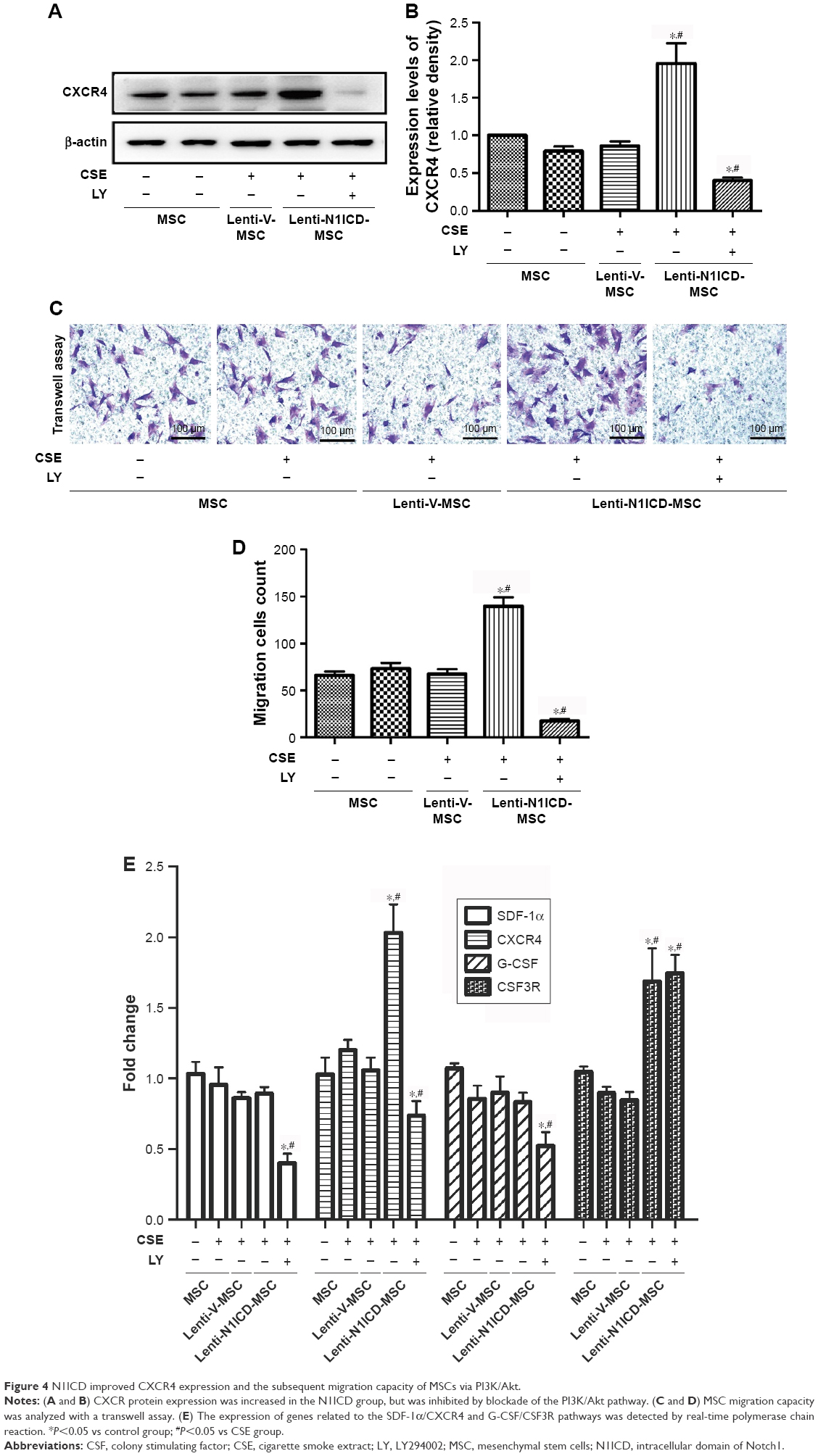 | Figure 4 N1ICD improved CXCR4 expression and the subsequent migration capacity of MSCs via PI3K/Akt. |
To determine whether altered CXCR4 expression in MSCs results in improved migration capacity, we performed a transwell assay to explore the chemoattractive response of MSCs. As shown in Figure 4C and D, no significant difference was observed between the control and CSE groups, and N1ICD overexpression in MSCs increased cell migration, indicating that MSCs with higher CXCR4 expression may have an increased migratory capacity. Interestingly, PI3K/Akt inhibition resulted in fewer cells located in the lower chamber when compared with the CSE group, suggesting that, in addition to Notch1, other signaling pathways may also be involved in the PI3K/Akt-dependent chemoattractive response. Furthermore, the gene expression of colony stimulating factor 3 receptor (CSF3R) was also increased in N1ICD overexpressing MSCs when compared with that in the CSE group, which was not reversed by LY294002 pretreatment. In contrast, the expression of SDF-1 and granulocyte-colony stimulating factor (G-CSF) were similar between the CSE and N1ICD groups, but all significantly decreased when cells were pretreated with LY294002 (Figure 4E).
Role of Notch and Akt in CSE-induced MSC apoptosis
CSE causes apoptotic cell death by inducing oxidative stress in vitro, which may contribute to the development of pulmonary emphysema.23 Under our experimental conditions, CSE was applied to induce MSC apoptosis. We assessed whether N1ICD overexpression influenced apoptosis-related proteins in MSCs. First, the TUNEL assay was performed to demonstrate the presence of DNA fragmentation (Figure 5A and B). The analysis showed N1ICD overexpression decreased in TUNEL + cells when cells were exposed to CSE. In addition, because caspase-3 activation represents a critical step in the apoptotic process, we evaluated cleaved caspase-3 staining by ICC (Figure 5C and D). The results showed that cleaved caspase-3 increased in the CSE group but decreased when MSCs were transfected with N1ICD overexpressing lentivirus. Furthermore, CSE led to an increase in the pro-apoptotic Bax/anti-apoptotic Bcl-2 ratio. In contrast, the Bax/Bcl-2 ratio decreased in N1ICD overexpressing cells (Figure 5E and F). These observations suggested that N1ICD overexpression could reverse CSE-mediated MSC apoptosis by modulating the balance between the expression of pro-apoptotic and anti-apoptotic proteins belonging to the Bcl-2 family and by decreasing the level of cleaved caspase-3. LY294002 pretreatment to block Akt inhibited the protective effects of N1ICD on CSE-treated MSCs, as evidenced by an increase in the number of apoptotic cells, higher cleaved caspase-3 levels, and a higher ratio of Bax/Bcl-2. These results indicated that Akt was partly responsible for the anti-apoptotic role of N1ICD in CSE-mediated MSC death.
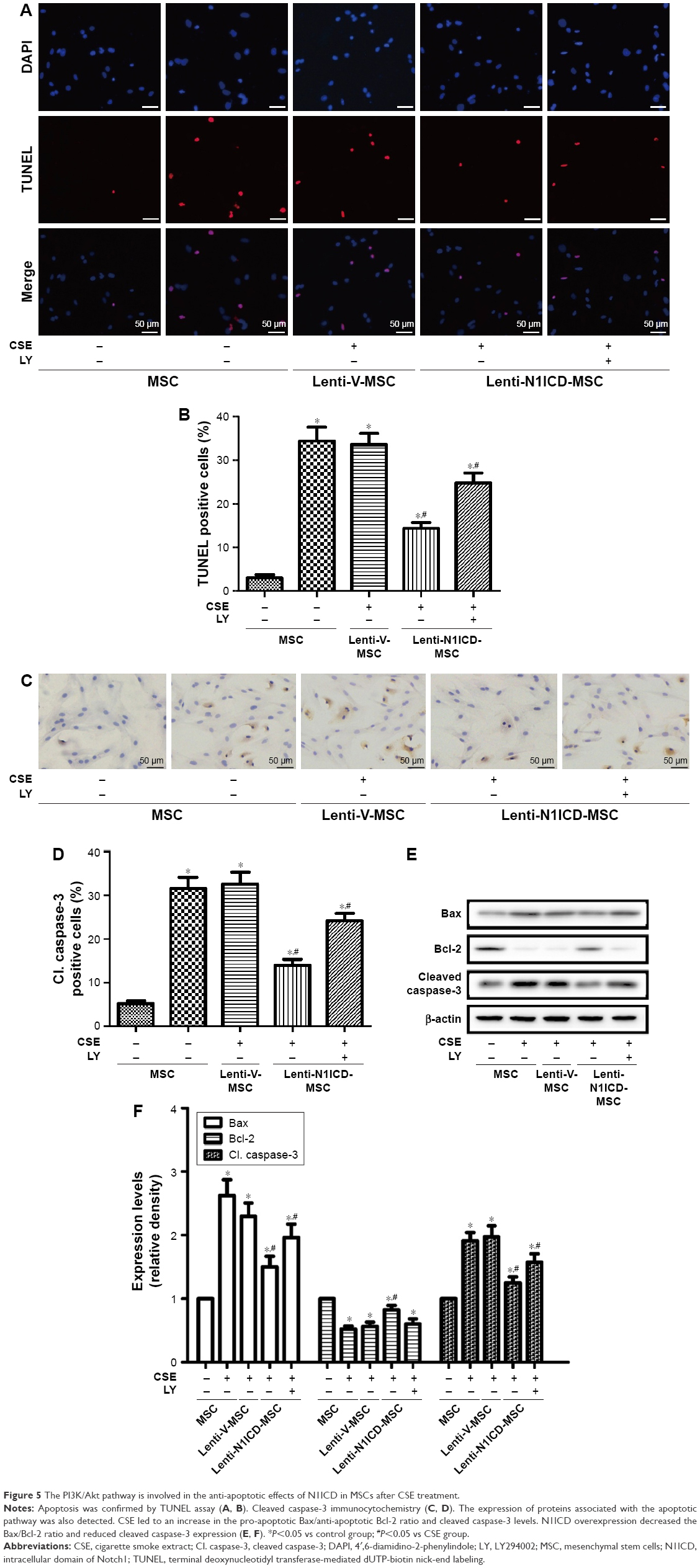 | Figure 5 The PI3K/Akt pathway is involved in the anti-apoptotic effects of N1ICD in MSCs after CSE treatment. |
Discussion
Stem cells have recently been proposed as a promising strategy for the treatment of lung diseases. MSCs have been widely administered in experimental models of COPD. The CS microenvironment inhibits the therapeutic potential of MSCs against COPD, which is a critical issue, hence the need to improve the properties of MSCs for transplantation in COPD. Herein, we investigated the effects of Notch on MSC proliferation, migration, and apoptosis in a CS microenvironment in vitro.
The present study reveals several novel findings indicating that Notch1 signaling may represent a critical cell-signaling pathway for maintaining key quality-related properties of MSCs. Our results are consistent with previous studies. Vujovic et al first investigated the role of Notch signaling in human MSCs by using γ-secretase inhibitors. They found that inhibiting γ-secretase reduced both the proliferation and differentiation capacity of hMSCs.24 Na et al treated human umbilical cord-derived MSCs with γ-secretase inhibitor I (GSI-I) and found that GSI-I treatment led to apoptosis, inhibited osteogenic differentiation, and reduced immunomodulatory properties of MSCs.25 However, the reagent inhibits both Notch signaling and the proteasome activities of MSCs. To investigate how Notch signaling and its interaction with other potential signaling pathway(s) regulate these critical properties of MSCs, we treated rat BM-MSCs by overexpressing N1ICD and confirmed that N1ICD overexpression improves MSC proliferation. The underlying molecular mechanism may involve the regulation of the cell cycle by Akt through inhibition of p21 protein, which is associated with cell cycle arrest. The phosphorylation of p21 by Akt inhibits its interaction with proliferating cell nuclear antigen and promotes the assembly of the cyclin D1/cyclin-dependent kinase 4 complex.26 Activation of the cyclin D1-P-Rb pathway facilitates the release of the transcription factor E2F-1 in MSCs, resulting in the expression of genes required for DNA synthesis. Moreover, PI3K inhibition reversed the effects of N1ICD on cell cycle protein expression and MSC growth.
Promoting BM stem cell mobilization is a common method to augment the cellular yield of peripheral blood apheresis for clinical stem cell transplantation.27 Selected mobilizing agents have been tested for in vivo beneficial effects following experimental injury, with modulation of the SDF-1α/CXCR4 axis being the most common strategy. Activation of Notch signaling up-regulated CXCR4 expression in BM-derived dendritic cells (DCs), and transduction with a CXCR4-expressing lentivirus relieved developmental arrest of recombination signal binding protein Jκ (RBP-Jκ)-deficient DCs.28 Down-regulation of CXCR4 expression in BM-MSCs has been observed in global hemizygous deletion of Notch1 (Notch1+/-) in mice.29 In the present study, after CSE treatment, CXCR4 protein expression was not substantially altered in MSCs, while N1ICD overexpression improved CXCR4 expression and the subsequent migratory response via the PI3k/Akt pathway as evidenced by transwell assay. Thus, our results provide a feasible means to improve the migration ability of MSCs during in vitro expansion. Furthermore, up-regulation of CSF3R expression in N1ICD overexpressing cells suggests that Notch1 may mediate the mobilization and migration of BM cells through CSF3R and CXCR4 signaling pathways. Indeed, G-CSF/CSF3R and SDF-1α/CXCR4 signaling pathways contribute to the recruitment of BM-derived cells to damaged tissues.29–31 Taken together, these findings suggest a critical role for Notch signaling in BM-derived MSC mobilization and migration.
Although the improved proliferative capacity and migration response of MSCs could contribute to higher transplantation efficiency in clinical applications, the hostile environment of injured lung tissue, including inflammation and lung tissue structural damage, may cause excessive cell death, leading to an urgent need to enhance the cells’ resistance to apoptosis. CS-induced in vivo apoptosis can be simulated in vitro by CSE treatment of isolated MSCs. The Notch signaling pathway plays important roles in regulating stem cell self-renewal and apoptosis. Notch1 signaling activation confers chemoresistance to rapamycin in lung cancer cells by inhibiting apoptosis via the PI3K-Akt pathway.32 Furthermore, activated Notch can protect against apoptosis in physiological and pathological conditions by activating Akt signaling.33,34 In the current study, we explored the anti-apoptotic effect of Notch signaling in MSCs in conditions simulating the CS microenvironment in vitro. In addition, inhibition of the Akt pathway caused an increase in cellular apoptosis and a reduction in anti-apoptotic proteins.
The Bcl-2 protein family is highly involved in apoptosis. An increase in the Bax/Bcl-2 ratio and the level of cleaved caspase-3 induces cell apoptosis.35 Previous studies reported that Notch1 down-regulation decreased Bcl-2 and Bcl-X (L) protein expression via inactivation of Akt, thereby contributing to apoptosis.36,37 In addition, Notch inhibition was associated with a decrease in Akt, which controls proliferation and/or cell death, and targets caspases.38 Our data suggest that inhibition of the Bax/caspase-3 axis could be a consequence of Notch activation, leading to the prevention of MSC apoptosis, which may help preserve MSC function. In hematopoietic stem cells, Notch signaling can induce growth arrest and apoptosis.39,40 This phenomenon could be explained by the fact that activation of the Notch pathway has various effects on proliferation and survival, which are highly cell-type specific, although these cell-specific mechanisms have not been elucidated in most systems.41 Nevertheless, accumulating evidence highlighted that MSCs therapeutic effects vary in response to local environmental cues.4 Moreover, activation of Notch signaling increased the p-Akt level, and PI3K inhibitors partially blocked the anti-apoptotic effect of Notch signaling in MSCs, making PI3K/Akt signaling regulation an attractive alternative to targeting the Notch signaling pathway regulating MSC apoptosis.
In the present study, Notch was an upstream activator of the PI3K/Akt pathway, and Akt inhibition partially blocked the promoting effects of N1ICD on proliferation, migration, and apoptosis. Many studies have indicated that the accumulation of secreted frizzled-related protein 242 in MSCs, induced by Akt activation, increases MSC self-renewal by inhibiting both Wnt and bone morphogenetic protein43 and prevents the apoptosis of MSCs by triggering cytoplasmic β-catenin translocation into the nucleus.44 Therefore, the activation of the PI3K/Akt pathway is probably involved in MSC proliferation and apoptosis. As reported, Akt activation also causes a dramatic increase in CXCR4 expression45 and MSC migratory behavior.46 Additionally, inhibiting Akt by LY294002 could not completely suppress the promoting effects of Notch signaling on MSCs, suggesting that other pathways may be involved in the action of Notch. Unfortunately, we have only revealed that N1ICD overexpression alleviates dysfunction of MSC induced by CSE in vitro. Whether N1ICD overexpression is involved in the enhanced proliferation, migration, and survival of MSCs in vivo remains to be determined.
In conclusion, these findings suggest that the Notch-PI3K/Akt signaling pathway may improve the biological function of MSCs by effectively reversing the limited proliferation, excessive apoptosis, and weak migration capacity induced by CSE. Therefore, approaches to activate the Notch pathway could potentially increase the efficiency of MSC-based transplantation therapies in the future.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81470232) (www.nsfc.gov.cn). The study was also supported by the Natural Science Foundation of Shanghai (No. 17ZR1418400) and (No. 14401970500).
Disclosure
The authors report no conflicts of interest in this work.
References
Caplan AI. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol. 2007;213(2):341–347. | ||
Kern S, Eichler H, Stoeve J, Kluter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. 2006;24(5):1294–1301. | ||
Lee JW, Gupta N, Serikov V, Matthay MA. Potential application of mesenchymal stem cells in acute lung injury. Expert Opin Biol Ther. 2009;9(10):1259–1270. | ||
Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med. 2013;45:e54. | ||
Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. 2008;294(4):L612–L631. | ||
Jafari N, Hintzen RQ. The association between cigarette smoking and multiple sclerosis. J Neurol Sci. 2011;311(1–2):78–85. | ||
Antunes MA, Abreu SC, Cruz FF, et al. Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir Res. 2014;15:118. | ||
Chen YB, Lan YW, Chen LG, et al. Mesenchymal stem cell-based HSP70 promoter-driven VEGFA induction by resveratrol alleviates elastase-induced emphysema in a mouse model. Cell Stress Chaperones. 2015;20(6):979–989. | ||
Janssen WJ, Yunt ZX, Muldrow A, et al. Circulating hematopoietic progenitor cells are decreased in COPD. COPD. 2014;11(3):277–289. | ||
Palange P, Testa U, Huertas A, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. 2006;27(3):529–541. | ||
Zhou Y, Gan Y, Taylor HS. Cigarette smoke inhibits recruitment of bone-marrow-derived stem cells to the uterus. Reprod Toxicol. 2011;31(2):123–127. | ||
LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem. 2003;278(36):34427–34437. | ||
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. | ||
Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194(3):237–255. | ||
Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13(9):654–666. | ||
Shapiro SD. Smoke gets in your cells. Am J Respir Cell Mol Biol. 2004;31(5):481–482. | ||
Huh JW, Kim SY, Lee JH, et al. Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L255–L266. | ||
Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol. 2006;35(6):681–688. | ||
Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31(10):890–896. | ||
Sun X, Gao X, Zhou L, Sun L, Lu C. PDGF-BB-induced MT1-MMP expression regulates proliferation and invasion of mesenchymal stem cells in 3-dimensional collagen via MEK/ERK1/2 and PI3K/AKT signaling. Cell Signal. 2013;25(5):1279–1287. | ||
Ishida S, Huang E, Zuzan H, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21(14):4684–4699. | ||
Zhang WG, He L, Shi XM, et al. Regulation of transplanted mesenchymal stem cells by the lung progenitor niche in rats with chronic obstructive pulmonary disease. Respir Res. 2014;15:33. | ||
Carnevali S, Petruzzelli S, Longoni B, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L955–L963. | ||
Vujovic S, Henderson SR, Flanagan AM, Clements MO. Inhibition of gamma-secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell Prolif. 2007;40(2):185–195. | ||
Na T, Liu J, Zhang K, Ding M, Yuan BZ. The notch signaling regulates CD105 expression, osteogenic differentiation and immunomodulation of human umbilical cord mesenchymal stem cells. PLoS One. 2015;10(2):e0118168. | ||
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3(3):245–252. | ||
Brauninger S, Bialleck H, Thorausch K, Felt T, Seifried E, Bonig H. Allogeneic donor peripheral blood “stem cell” apheresis: prospective comparison of two apheresis systems. Transfusion. 2012;52(5):1137–1145. | ||
Wang YC, Hu XB, He F, et al. Lipopolysaccharide-induced maturation of bone marrow-derived dendritic cells is regulated by notch signaling through the up-regulation of CXCR4. J Biol Chem. 2009;284(23):15993–16003. | ||
Li Y, Hiroi Y, Ngoy S, et al. Notch1 in bone marrow-derived cells mediates cardiac repair after myocardial infarction. Circulation. 2011;123(8):866–876. | ||
Harada M, Qin Y, Takano H, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11(3):305–311. | ||
Wang L, Wang YC, Hu XB, et al. Notch-RBP-J signaling regulates the mobilization and function of endothelial progenitor cells by dynamic modulation of CXCR4 expression in mice. PLoS One. 2009;4(10):e7572. | ||
Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66(9):4715–4724. | ||
Sade H, Krishna S, Sarin A. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Bioll Chem. 2004;279(4):2937–2944. | ||
Nair P, Somasundaram K, Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J Virol. 2003;77(12):7106–7112. | ||
Lau GJ, Godin N, Maachi H, et al. Bcl-2-modifying factor induces renal proximal tubular cell apoptosis in diabetic mice. Diabetes. 2012;61(2):474–484. | ||
Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5(3):483–493. | ||
Wang Z, Li Y, Banerjee S, et al. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J Cell Biochem. 2010;109(4):726–736. | ||
Luo HR, Hattori H, Hossain MA, et al. Akt as a mediator of cell death. Proc Natl Acad Sci U S A. 2003;100(20):11712–11717. | ||
Carlesso N, Aster JC, Sklar J, Scadden DT. Notch1-induced delay of human hematopoietic progenitor cell differentiation is associated with altered cell cycle kinetics. Blood. 1999;93(3):838–848. | ||
Chadwick N, Nostro MC, Baron M, Mottram R, Brady G, Buckle AM. Notch signaling induces apoptosis in primary human CD34+ hematopoietic progenitor cells. Stem Cells. 2007;25(1):203–210. | ||
Baldi A, De Falco M, De Luca L, et al. Characterization of tissue specific expression of Notch-1 in human tissues. Biol Cell. 2004;96(4):303–311. | ||
Mirotsou M, Zhang Z, Deb A, et al. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci U S A. 2007;104(5):1643–1648. | ||
Alfaro MP, Vincent A, Saraswati S, et al. sFRP2 suppression of bone morphogenic protein (BMP) and Wnt signaling mediates mesenchymal stem cell (MSC) self-renewal promoting engraftment and myocardial repair. J Biol Chem. 2010;285(46):35645–35653. | ||
Gehmert S, Sadat S, Song YH, Yan Y, Alt E. The anti-apoptotic effect of IGF-1 on tissue resident stem cells is mediated via PI3-kinase dependent secreted frizzled related protein 2 (Sfrp2) release. Biochem Biophys Res Commun. 2008;371(4):752–755. | ||
Liu H, Xue W, Ge G, et al. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1alpha in MSCs. Biochem Biophys Res Commun. 2010;401(4):509–515. | ||
Bulj Z, Duchi S, Bevilacqua A, et al. Protein kinase B/AKT isoform 2 drives migration of human mesenchymal stem cells. Int J Oncol. 2013;42(1):118–126. |
Supplementary material
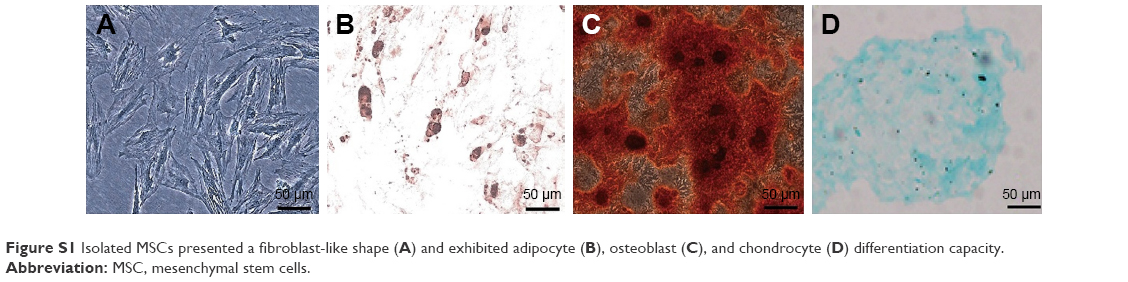 | Figure S1 Isolated MSCs presented a fibroblast-like shape (A) and exhibited adipocyte (B), osteoblast (C), and chondrocyte (D) differentiation capacity. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.