Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Activation of Erk and p53 regulates copper oxide nanoparticle-induced cytotoxicity in keratinocytes and fibroblasts
Authors Luo C, Li Y, Yang L, Zheng Y, Long J, Jia J ![]() , Xiao S
, Xiao S ![]() , Liu J
, Liu J
Received 13 May 2014
Accepted for publication 15 August 2014
Published 10 October 2014 Volume 2014:9(1) Pages 4763—4772
DOI https://doi.org/10.2147/IJN.S67688
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Cheng Luo,1 Yan Li,2 Liang Yang,1 Yan Zheng,3 Jiangang Long,1 Jinjing Jia,3 Shengxiang Xiao,3 Jiankang Liu1
1Center for Mitochondrial Biology and Medicine, The Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology and Frontier Institute of Science and Technology, 2Center for Bioinformatics, The Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi’an Jiaotong University, Xi’an, People’s Republic of China; 3Department of Dermatology, The 2nd Affiliated Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China
Abstract: Copper oxide nanoparticles (CuONP) have attracted increasing attention due to their unique properties and have been extensively utilized in industrial and commercial applications. For example, their antimicrobial capability endows CuONP with applications in dressings and textiles against bacterial infections. Along with the wide applications, concerns about the possible effects of CuONP on humans are also increasing. It is crucial to evaluate the safety and impact of CuONP on humans, and especially the skin, prior to their practical application. The potential toxicity of CuONP to skin keratinocytes has been reported recently. However, the underlying mechanism of toxicity in skin cells has remained unclear. In the present work, we explored the possible mechanism of the cytotoxicity of CuONP in HaCaT human keratinocytes and mouse embryonic fibroblasts (MEF). CuONP exposure induced viability loss, migration inhibition, and G2/M phase cycle arrest in both cell types. CuONP significantly induced mitogen-activated protein kinase (extracellular signal-regulated kinase [Erk], p38, and c-Jun N-terminal kinase [JNK]) activation in dose- and time-dependent manners. U0126 (an inhibitor of Erk), but not SB 239063 (an inhibitor of p38) or SP600125 (an inhibitor of JNK), enhanced CuONP-induced viability loss. CuONP also induced decreases in p53 and p-p53 levels in both cell types. Cyclic pifithrin-α, an inhibitor of p53 transcriptional activity, enhanced CuONP-induced viability loss. Nutlin-3α, a p53 stabilizer, prevented CuONP-induced viability loss in HaCaT cells, but not in MEF cells, due to the inherent toxicity of nutlin-3α to MEF. Moreover, the experiments on primary keratinocytes are in accordance with the conclusions acquired from HaCaT and MEF cells. These data demonstrate that the activation of Erk and p53 plays an important role in CuONP-induced cytotoxicity, and agents that preserve Erk or p53 activation may prevent CuONP-induced cytotoxicity.
Keywords: cell cycle arrest, CuONP, MAPK, nutlin-3α, cyclic pifithrin-α
Introduction
Copper oxide nanoparticles (CuONP) have been widely applied in semiconductors, catalysts, microelectronic materials, and lithium batteries.1 Similar to silver and ZnO nanoparticles, CuONP exhibit excellent antimicrobial efficiency.2–4 Much attention has been paid to the feasible applications of CuONP, such as antimicrobial masks5 and textiles.6,7 However, it is crucial to evaluate their potential effects on the environment and human health before their practical application. Certain reports have revealed that CuONP are toxic to the aquatic macrophyte Lemna gibba8 and Xenopus laevis.9 CuONP were also reported to be cytotoxic to human cells, such as brain,10 lung,11,12 liver,13,14 kidney,15 and skin16 cells. A test using the human lung adenocarcinoma cell line A549 implied that CuONP were more toxic than other metal oxide nanoparticles (TiO2, CuZnFe2O4, Fe3O4, and Fe2O3) and carbon-based nanomaterials (carbon nanopowders and multiwalled carbon nanotubes).17 Organs such as the lung and skin were more vulnerable to nanoparticles due to their direct contact with the external environment. Many studies have focused on CuONP-induced cytotoxicity and corresponding mechanisms in human lung epithelial cells.11,12,17–19 Oxidative stress and autophagy have been reported to play vital roles in CuONP-induced cytotoxicity.12,18 Hanagata et al reported that CuONP exposure upregulated genes involved in mitogen-activated protein kinase (MAPK) pathways while downregulating genes involved in cell cycle progression.11 Semisch et al showed that compared with microscale CuO particles and copper chloride, CuONP were more cytotoxic and induced genotoxicity due to their nanoscale features.20 CuONP were also reported to induce oxidative stress and apoptosis in HaCaT human keratinocytes. However, the decisive factors and the accompanying changes involved in the toxicity of CuONP are unclear.16 Moreover, the underlying mechanism of the toxicity of CuONP to skin-associated cells has not been clarified. In the present work, we examined the cytotoxicity of CuONP to skin-associated cells, keratinocytes (HaCaT) and mouse embryonic fibroblasts (MEF), and investigated the underlying molecular mechanisms.
Materials and methods
Materials
CuONP was synthesized as Zhu et al reported,21 and characterized using transmission electron microscope H-7650 (Hitachi Ltd., Tokyo, Japan), a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK), and X-ray Photoelectron Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) (Figure 1). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT), SB 239063, U0126, propidium iodide, SP600125, and anti-actin primary antibody were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS) was purchased from Biological Industries Israel Beit-Haemek Ltd. (Kibbutz Beit-Haemek, Israel). Dulbecco’s Modified Eagle’s Medium (DMEM), penicillin, streptomycin, and trypsin were purchased from Thermo Fisher Scientific. Primary antibodies against p-p53 and phosphorylated extracellular signal-regulated kinase (Erk) were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Primary antibodies against cyclin A, cyclin B1, p53, phosphorylated c-Jun N-terminal kinase (JNK), JNK, p-p38, p38, Erk, and cyclic pifithrin-α (PFT-α) were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). The bicinchoninic acid assay kit and enhanced chemiluminescence Western blotting substrate were obtained from Thermo Fisher Scientific. Cell lysis buffer was purchased from Beyotime Biotechnology (Haimen, Jiangsu, People’s Republic of China). Other chemicals were obtained from local suppliers. Deionized water was used in all experiments.
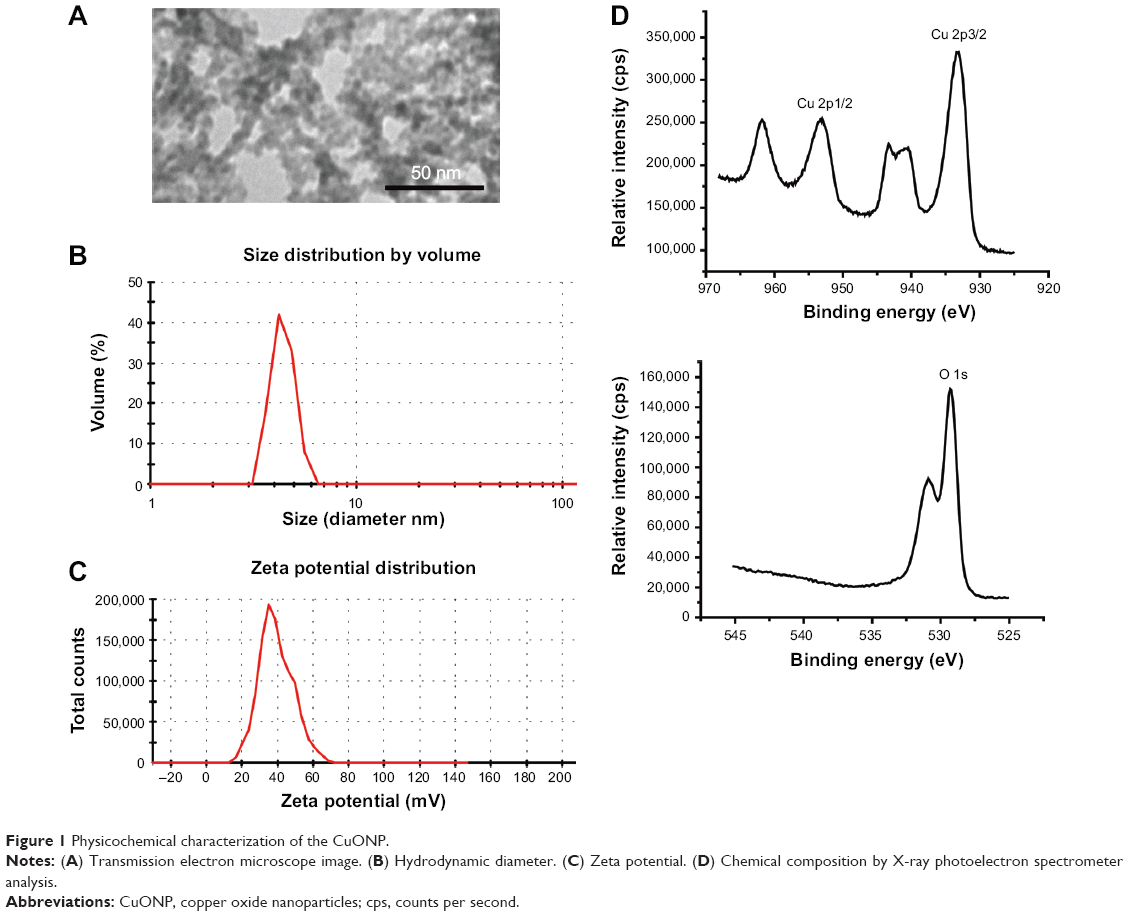 | Figure 1 Physicochemical characterization of the CuONP. |
Cell culture and treatment
Human keratinocytes HaCaT and mouse embryonic fibroblast cells were cultured in DMEM supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (100 μg/mL) in a 5% CO2 humidified incubator at 37°C (Thermo Fisher Scientific). In a dose-dependent manner, HaCaT and MEF cells were treated with the indicated concentrations of CuONP for 24 hours. In a time-dependent manner, HaCaT or MEF cells were treated with 60 or 80 μmol/L CuONP, respectively.
For the MAPK-inhibitor treatments, HaCaT cells were pretreated with 5 μmol/L U0126 for 2 hours and then treated with 20 or 40 μg/mL CuONP for 24 hours; HaCaT cells were pretreated with 20 μmol/L SB 239063 or 20 μmol/L SP600125 for 2 hours and then treated with 20 or 60 μg/mL CuONP for 24 hours; MEF cells were pretreated with 5 μmol/L U0126 for 2 hours and then treated with 40 or 60 μg/mL CuONP for 24 hours; MEF cells were pretreated with 20 μmol/L SB 239063 or 20 μmol/L SP600125 for 2 hours and then treated with 40 or 80 μg/mL CuONP for 24 hours.
For the p53-transcriptional-inhibitor (cyclic PFT-α) treatment, HaCaT cells were pretreated with 1 or 10 μmol/L cyclic PFT-α for 2 hours and then treated with 20 μg/mL CuONP for 24 hours or with 60 μg/mL CuONP for 6 hours, respectively. MEF cells were pretreated with 1 or 10 μmol/L cyclic PFT-α for 2 hours and then treated with 40 μg/mL CuONP for 24 hours or with 80 μg/mL CuONP for 3 hours, respectively. For p53-stabilizer (nutlin-3α) treatment, HaCaT or MEF cells were pretreated with 30 μmol/L nutlin-3α for 2 hours and then treated with 60 μg/mL or 80 μg/mL CuONP for 24 hours, respectively.
Cellular viability assessment
The cells were washed with FBS-free DMEM once and incubated with 0.5 mg/mL MTT in FBS-free DMEM for 1 hour at 37°C in a CO2 incubator. The formazan generated was dissolved in DMSO and recorded at 490 nm with a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA).22
Migratory assay
The cells were scratched with a 200 μL pipette tip across the center of a plate to create a wound. The detached debris was washed off with FBS-free DMEM. The cells were treated with the indicated concentrations of CuONP. Photographs were taken at 0 and 24 hours after the treatment.23
Cell cycle analysis
The cells were trypsinized and washed with PBS once. The cells were then resuspended in PBS, followed by fixation in ice-cold 70% ethanol and storage at −20°C for at least 24 hours. The fixed cells were collected by centrifugation at 1,000 rpm for 10 minutes and washed with PBS once, followed by incubation with a propidium iodide working solution (0.1% Triton-100, 200 μg/mL RNase A, and 50 μg/mL stock solution) for 15 minutes in the dark and analysis by flow cytometry.24
Western blot analysis
The cells were lysed and centrifuged at 13,000 rpm for 15 minutes at 4°C to collect the supernatant. The protein content in the lysate’s supernatant was quantified with a bicinchoninic acid assay kit. The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 hour at room temperature, probed with primary antibodies against the target proteins overnight at 4°C, and then washed with TBST to remove unbounded antibodies. Next, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature and washed with TBST again. Specific signals on the probed membranes were detected using enhanced chemiluminescence and an X-ray-film exposure system.
Statistical analysis
A one-way analysis of variance, followed by Fisher’s least significant difference test, was used to evaluate the significant differences among the treatments. The data are presented as the mean ± standard deviation (n>3). Statistical significance was indicated as follows: *P<0.05 and **P<0.01.
Results
CuONP induce cytotoxicity in HaCaT and MEF cells
CuONP significantly suppressed the proliferation of HaCaT and MEF cells in a dose-dependent manner in 24 hours’ treatment (Figure 2A and B). Cellular migration capacities were also inhibited in both CuONP-treated cells in scratch-wound assay (Figure 2C and D).
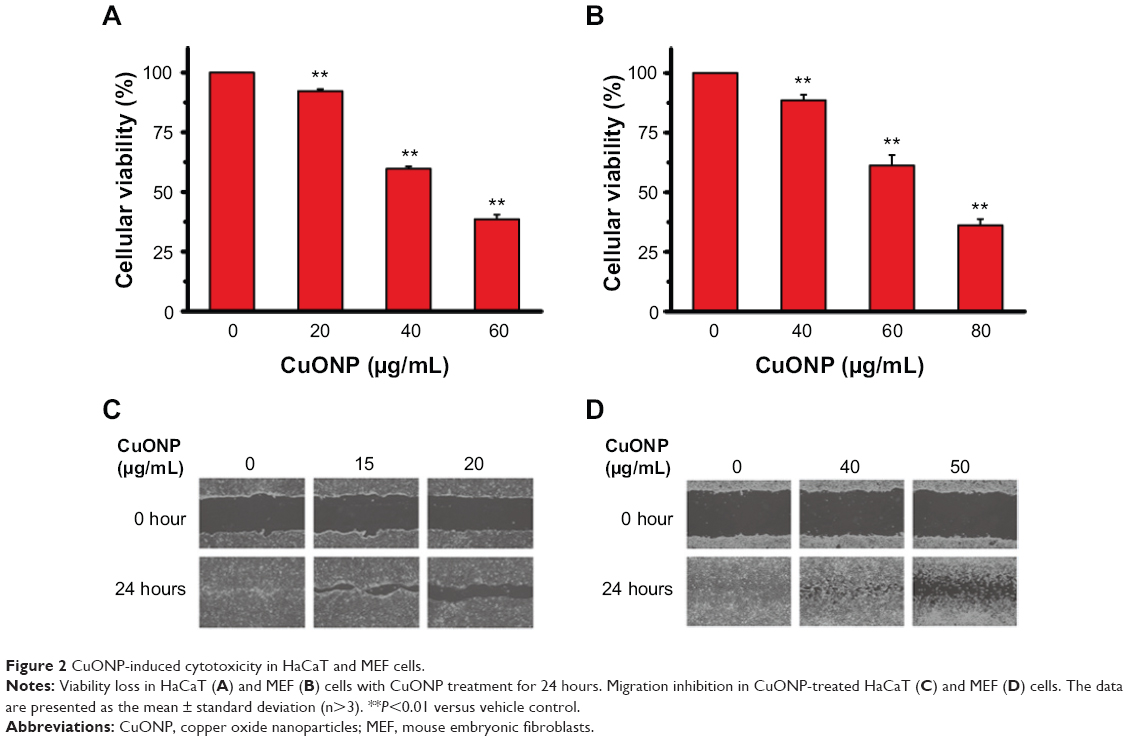 | Figure 2 CuONP-induced cytotoxicity in HaCaT and MEF cells. |
Cell cycle arrest may promote growth and migration inhibition in CuONP-treated HaCaT and MEF cells. To verify this assumption, CuONP-treated cells were subjected to cell cycle analysis with flow cytometry. There was a significant increase in the population of cells in G2/M phase (from 3.9% to 42.8% in HaCaT, or from 1.2% to 32.8% in MEF), with corresponding decreases in the G1 and S phases after 24 hours’ CuONP treatment, which implied that the cell cycle was arrested in G2/M phase in a dose-dependent manner in CuONP-treated cells (Figure 3A and B). There was a dose- and time-dependent decrease in the levels of cyclin A and cyclin B1 in CuONP-treated HaCaT cells (Figure 3C and D) and MEF cells (Figure 3E and F). The downregulation of those G2/M transition-related proteins further revealed that CuONP induced G2/M phase arrest.
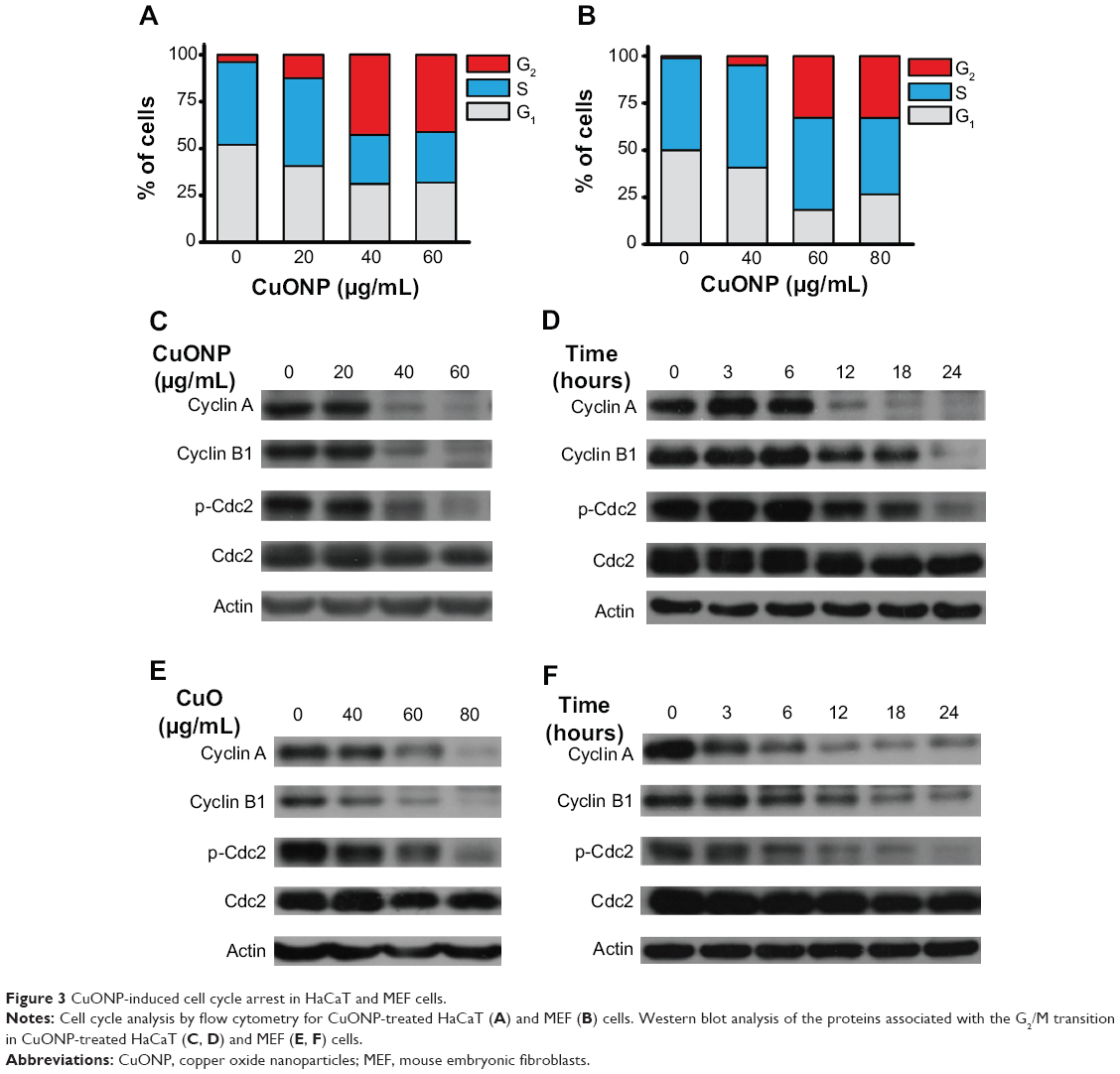 | Figure 3 CuONP-induced cell cycle arrest in HaCaT and MEF cells. |
Erk activation may prevent CuONP-induced growth inhibition in HaCaT and MEF cells
The activation of Erk, p38, and JNK was significantly induced in dose- and time-dependent manners in CuONP-treated HaCaT cells (Figure 4A and B) and MEF cells (Figure 4C and D). The inhibition of Erk activation by pretreatment with U0126 significantly promoted growth inhibition relative to treatment with CuONP alone in HaCaT cells (Figure 4E) and MEF cells (Figure 4F). However, pretreatment with SB 239063 (an inhibitor of p38 activation) and SP600125 (an inhibitor of JNK activation) exhibited little or no effect on the viabilities of CuONP-treated HaCaT cells (Figure 4G) and MEF cells (Figure 4H). These data implied that Erk plays a vital role in CuONP-induced cytotoxicity, whereas the changes in p38 and JNK were incidental.
 | Figure 4 Erk was involved in CuONP-induced cytotoxicity in HaCaT and MEF cells. |
Loss of p53 accelerates CuONP-induced growth inhibition in HaCaT and MEF cells
There was a remarkable decrease in the protein levels of p53 and p-p53 in a dose-dependent manner in CuONP-treated HaCaT cells (Figure 5A) and MEF cells (Figure 5B). Pretreatment with cyclic PFT-α, a p53 transcriptional inhibitor, promoted growth inhibition relative to treatment with CuONP alone in HaCaT cells (Figure 5C and D) and MEF cells (Figure 5E and F). Briefly, pretreatment with 1 μmol/L cyclic PFT-α promoted growth inhibition in both cells with low doses of CuONP for 24 hours’ treatment (Figure 5C and E), and pretreatment with 10 μmol/L cyclic PFT-α promoted growth inhibition in high doses of CuONP for 6 hours’ (for HaCaT) or 3 hours’ (for MEF) treatment (Figure 5D and F). Moreover, pretreatment with nutlin-3α, a murine double minute-2 (MDM2)/p53 inhibitor that stabilizes p53, prevented high dose of CuONP-induced viability loss in HaCaT cells (Figure 5G). Nutlin-3α itself was toxic to MEF cells, so no inhibition of the viability loss induced by CuONP was observed (Figure 5H).
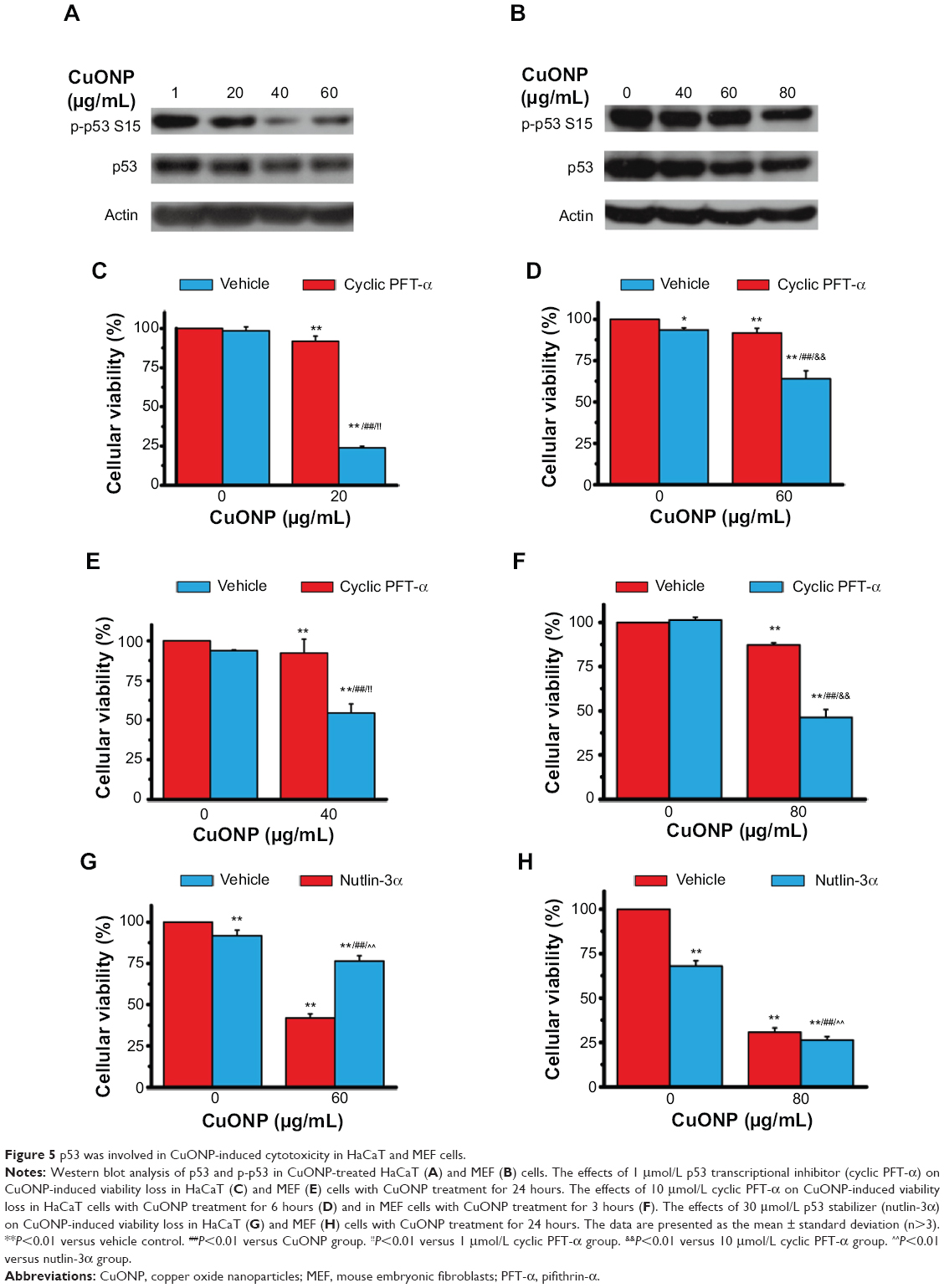 | Figure 5 p53 was involved in CuONP-induced cytotoxicity in HaCaT and MEF cells. |
CuONP-induced cytotoxicity in primary keratinocytes
Primary rat keratinocytes were isolated from newborn rat skins according to a report from Albarenque and Doi.25 CuONP induced dose-dependent viability loss in primary keratinocytes (Figure 6A). Erk inhibitor (U0126) and p53 transcriptional inhibitor (cyclic PFT-α) promoted CuONP-induced viability loss (Figure 6B). Nutlin-3α was toxic to primary keratinocytes, which is the same as the MEF; no inhibition of the viability loss induced by CuONP was observed.
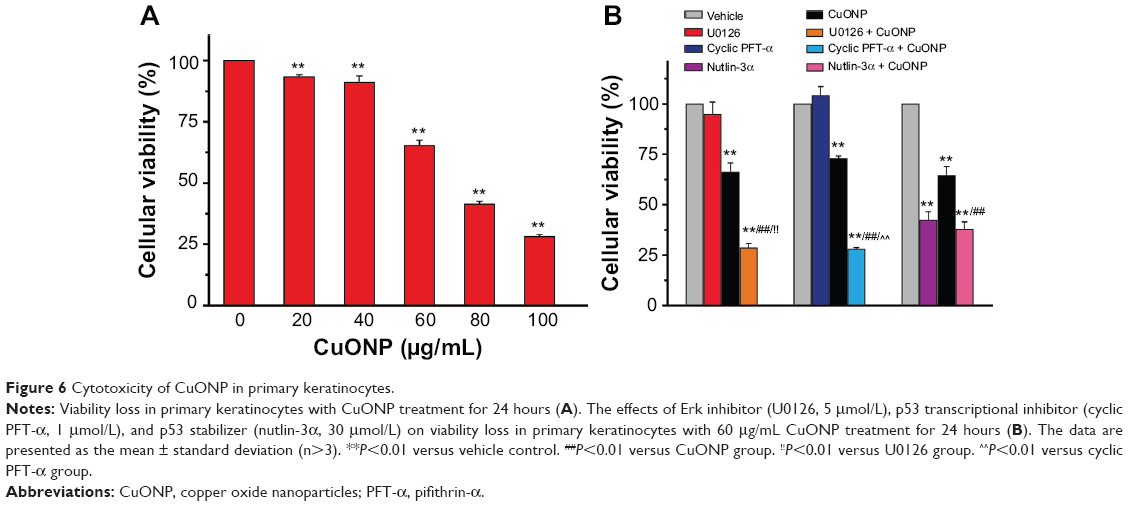 | Figure 6 Cytotoxicity of CuONP in primary keratinocytes. |
Discussion
CuONP have been applied in various industrial and medical areas, such as heat-transfer systems, gas sensors, electronic chips, and biocides.1 The skin, respiratory tract, and gastrointestinal tract are the most susceptible to the adverse effects of hazardous materials outside the human body due to their direct exposure to the environment. The skin is the largest among these organs, which increases the risk of being affected. Moreover, potent medical applications utilizing the antibacterial activity of CuONP give rise to direct contact between CuONP and the skin.6,7 Gauze and bandages containing CuONP could protect wounds against microbial infection. However, there is a risk in using CuONP application as a microbicide for covering the skin surface: ie, harm to skin-associated cells while killing microorganisms. Keratinocytes and fibroblasts, cooperating to facilitate skin repair, will be affected by CuONP. The redox activities and nanoscale properties of CuONP may promote cytotoxicity to these cells. CuONP exposure has been reported to induce a significant decrease in cellular viability and glutathione levels, and an increase in oxidative stress, apoptosis, and DNA damage in HaCaT cells.16 However, the underlying mechanisms are not well clarified. The present work reveals that cell cycle arrest occurred in CuONP-treated keratinocytes and fibroblasts and that the activation of Erk and p53 protected both cell types from cytotoxicity.
The proliferation and migration of keratinocytes and fibroblasts are important stages of wound repair and skin development.26 A loss of skin integrity renders the body vulnerable to invasion by foreign pathogens. Viability loss restrains the proliferation and migration capacity of both cell types, which will undermine cutaneous wound healing and lead to inflammation and infection. Cell cycle progression dysfunction is another pivotal factor affecting proliferation. Generally, one cell divides into two daughter cells when one cell cycle completes, and those progressions are under the control of checkpoints.27 Combinations of cyclins and cyclin-dependent kinases, the main components of checkpoints, vary in different stages of the cell cycle and regulate cell cycle progression. The cell cycle can be blocked in certain stages due to cellular damage and stress signaling, which makes the checkpoints defective. If the protein levels of cyclin A and B do not increase after G1 phase, cell progression cannot pass through G2/M phase and complete mitosis.28 In the present work, following CuONP treatment, the deregulation of cyclin A and B dynamics led to blockage of G2/M transitions and further promoted viability loss (Figures 2 and 3).
Erk, JNK, and p38 MAPK compose a large family of MAPKs that regulates various cellular functions and activities, such as proliferation, differentiation, mitosis, survival, and death.29 The activation of Erk, p38, and JNK was significantly induced in dose- and time-dependent manners in CuONP-treated HaCaT cells (Figure 4A and B) and MEF cells (Figure 4C and D). The activation of JNK and p38 MAPK has been reported to promote cell death.30 However, neither the inhibition of p38 activation nor the inhibition of JNK activation prevented viability loss in CuONP-treated HaCaT and MEF cells (Figure 4G and H). These data imply that the activation of p38 and JNK under CuONP exposure was incidental. Strong Erk activation was induced and sustained for several hours (Figure 4A–D). There are two opposing functions of Erk activation in cell proliferation and cell death.30 When Erk activation was suppressed by inhibitors (eg, U0126), cell death due to harmful reagents was promoted or suppressed, corresponding to Erk’s prosurvival and prodeath functions, respectively. Martin and Pognonec described both functions of Erk in cadmium toxicity, depending on different experimental conditions (eg, the cadmium treatment and cell type).31 It was necessary to inhibit Erk activation to uncover the roles that Erk played in CuONP-induced cytotoxicity. The inhibition of Erk activation promoted viability loss in cells treated with both low and high concentrations of CuONP, which implies a prosurvival function for Erk in this context. Several studies reported that CuONP induced reactive oxygen species (ROS) generation in different cells,10,13,18,32 including keratinocytes16 and fibroblasts.33 For example, Alarifi et al reported that CuONP induced dose-dependent cell death and ROS generation in keratinocytes.16 Considering that ROS generation would activate Erk,29,34,35 the dose-dependent Erk activation in the present work might be similarly due to CuONP-induced ROS generation.
p53 is a transcription factor that regulates the expression of hundreds of genes. Those genes are involved in metabolic homeostasis, antioxidant defense, DNA repair, growth arrest, senescence, apoptosis, autophagy, and other processes. The stability of p53 protein is mainly regulated by interaction with MDM2. The p53/MDM2 complex promotes p53 degradation by ubiquitylation and blocks the transactivation domain.36 Nutlin-3α can stabilize p53 levels and the transcriptional activity by inhibiting the MDM2–p53 interaction.37 The phosphorylation of p53 at serine 15 stimulates its transactivation.38,39 Cyclic PFT-α, a cyclic analog of PFT-α, inhibits p53-dependent gene transcription.40 Surprisingly, in the current study, p53 and p-p53 levels decreased in CuONP-treated cells (Figure 5A and B), which implies that p53 did not have a prodeath function. Cyclic PFT-α and nutlin-3α were used to further evaluate whether p53 had a prosurvival function. Figure 5C–H show that cyclic PFT-α significantly promoted CuONP-induced viability loss in both HaCaT and MEF cells, whereas nutlin-3α treatment inhibited the cellular viability loss of CuONP-treated HaCaT cells. These data indicate that p53 is involved in CuONP-induced cytotoxicity and plays a prosurvival role. There was no viability-loss inhibition observed following nutlin-3α treatment of CuONP-treated MEF cells, which may have resulted from the cytotoxicity of nutlin-3α to MEF cells. The different responses of HaCaT and MEF cells to nutlin-3α treatment might have been due to the different p53 statuses of these two types of cells: there are p53 mutations in HaCaT cells.41 Tovar et al reported that nutlin-3 treatment induced growth inhibition in p53-wildtype fibroblasts.42 However, no literature has reported similar phenomena in HaCaT cells. These reports suggest that HaCaT cells may be more tolerant to high p53 levels than MEF cells are. Therefore, pretreatment with nutlin-3α stabilized p53 and increased its levels in MEF cells, resulting in cytotoxicity.
Conclusion
In conclusion, we have investigated the cytotoxic effects of CuONP on skin-associated HaCaT cells, MEF cells, and primary keratinocytes, as well as the underlying mechanisms. The present study showed that CuONP is toxic to skin-associated cells and that Erk and p53 may be the key factors regulating this cytotoxicity. These results suggest that contact with CuONP may cause damage to the skin and that the activation of Erk and p53 might be a possible way to prevent the potential toxicity of CuONP to the skin.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81071299, 81371732) and by projects 985 and 211 of Xi’an Jiaotong University and was partially supported by NCET and the Fundamental Research Funds for the Central Universities, and Changjiang Scholars Innovative Research Team in University (PCSIRT:1171).
Disclosure
The authors report no conflicts of interest in this work.
References
Bondarenko O, Juganson K, Ivask A, Kasemets K, Mortimer M, Kahru A. Toxicity of Ag, CuO and ZnO nanoparticles to selected environmentally relevant test organisms and mammalian cells in vitro: a critical review. Arch Toxicol. 2013;87(7):1181–1200. | ||
Ren G, Hu D, Cheng EW, Vargas-Reus MA, Reip P, Allaker RP. Characterisation of copper oxide nanoparticles for antimicrobial applications. Int J Antimicrob Agents. 2009;33(6):587–590. | ||
Applerot G, Lellouche J, Lipovsky A, et al. Understanding the antibacterial mechanism of CuO nanoparticles: revealing the route of induced oxidative stress. Small. 2012;8(21):3326–3337. | ||
Hans M, Erbe A, Mathews S, Chen Y, Solioz M, Mucklich F. Role of copper oxides in contact killing of bacteria. Langmuir. 2013;29(52): 16160–16166. | ||
Borkow G, Zhou SS, Page T, Gabbay J. A novel anti-influenza copper oxide containing respiratory face mask. PLoS One. 2010;5(6):e11295. | ||
Zatcoff RC, Smith MS, Borkow G. Treatment of tinea pedis with socks containing copper-oxide impregnated fibers. Foot (Edinb). 2008; 18(3):136–141. | ||
Anita S, Ramachandran T, Rajendran R, Koushik CV, Mahalakshmi M. A Study of the Antimicrobial Property of Encapsulated Copper Oxide Nanoparticles on Cotton Fabric. Textile Research Journal. 2011;81(10): 1081–1088. | ||
Perreault F, Popovic R, Dewez D. Different toxicity mechanisms between bare and polymer-coated copper oxide nanoparticles in Lemna gibba. Environ Pollut. 2014;185:219–227. | ||
Thit A, Selck H, Bjerregaard HF. Toxicity of CuO nanoparticles and Cu ions to tight epithelial cells from Xenopus laevis (A6): effects on proliferation, cell cycle progression and cell death. Toxicol In Vitro. 2013;27(5):1596–1601. | ||
Bulcke F, Thiel K, Dringen R. Uptake and toxicity of copper oxide nanoparticles in cultured primary brain astrocytes. Nanotoxicology. 2014;8(7):775–785. | ||
Hanagata N, Zhuang F, Connolly S, Li J, Ogawa N, Xu M. Molecular responses of human lung epithelial cells to the toxicity of copper oxide nanoparticles inferred from whole genome expression analysis. ACS Nano. 2011;5(12):9326–9338. | ||
Sun T, Yan Y, Zhao Y, Guo F, Jiang C. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One. 2012;7(8):e43442. | ||
Piret JP, Jacques D, Audinot JN, et al. Copper(II) oxide nanoparticles penetrate into HepG2 cells, exert cytotoxicity via oxidative stress and induce pro-inflammatory response. Nanoscale. 2012;4(22): 7168–7184. | ||
Cuillel M, Chevallet M, Charbonnier P, et al. Interference of CuO nanoparticles with metal homeostasis in hepatocytes under sub-toxic conditions. Nanoscale. 2014;6(3):1707–1715. | ||
Xu J, Li Z, Xu P, Xiao L, Yang Z. Nanosized copper oxide induces apoptosis through oxidative stress in podocytes. Arch Toxicol. 2013;87(6): 1067–1073. | ||
Alarifi S, Ali D, Verma A, Alakhtani S, Ali BA. Cytotoxicity and genotoxicity of copper oxide nanoparticles in human skin keratinocytes cells. Int J Toxicol. 2013;32(4):296–307. | ||
Karlsson HL, Cronholm P, Gustafsson J, Möller L. Copper oxide nanoparticles are highly toxic: a comparison between metal oxide nanoparticles and carbon nanotubes. Chem Res Toxicol. 2008;21(9): 1726–1732. | ||
Ahamed M, Siddiqui MA, Akhtar MJ, Ahmad I, Pant AB, Alhadlaq HA. Genotoxic potential of copper oxide nanoparticles in human lung epithelial cells. Biochem Biophys Res Commun. 2010;396(2):578–583. | ||
Cronholm P, Karlsson HL, Hedberg J, et al. Intracellular uptake and toxicity of Ag and CuO nanoparticles: a comparison between nanoparticles and their corresponding metal ions. Small. 2013;9(7):970–982. | ||
Semisch A, Ohle J, Witt B, Hartwig A. Cytotoxicity and genotoxicity of nano – and microparticulate copper oxide: role of solubility and intracellular bioavailability. Part Fibre Toxicol. 2014;11:10. | ||
Zhu J, Li D, Chen H, Yang X, Lu L, Wang X. Highly dispersed CuO nanoparticles prepared by a novel quick-precipitation method. Mater Lett. 2004;58(26):3324–3327. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. | ||
Darzynkiewicz Z, Juan G, Bedner E. Determining cell cycle stages by flow cytometry. Curr Protoc Cell Biol. 2001;Chapter 8:Unit 8.4. | ||
Albarenque SM, Doi K. T-2 toxin-induced apoptosis in rat keratinocyte primary cultures. Exp Mol Pathol. 2005;78(2):144–149. | ||
Bielefeld KA, Amini-Nik S, Alman BA. Cutaneous wound healing: recruiting developmental pathways for regeneration. Cell Mol Life Sci. 2013;70(12):2059–2081. | ||
Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology. 2002;181–182:475–481. | ||
Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. 2013;5(3):a008904. | ||
Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58(11):621–631. | ||
Subramaniam S, Unsicker K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience. 2006;138(4): 1055–1065. | ||
Martin P, Pognonec P. ERK and cell death: cadmium toxicity, sustained ERK activation and cell death. FEBS J. 2010;277(1):39–46. | ||
Siddiqui MA, Alhadlaq HA, Ahmad J, Al-Khedhairy AA, Musarrat J, Ahamed M. Copper oxide nanoparticles induced mitochondria mediated apoptosis in human hepatocarcinoma cells. PLoS One. 2013; 8(8):e69534. | ||
Akhtar MJ, Ahamed M, Fareed M, Alrokayan SA, Kumar S. Protective effect of sulphoraphane against oxidative stress mediated toxicity induced by CuO nanoparticles in mouse embryonic fibroblasts BALB 3T3. J Toxicol Sci. 2012;37(1):139–148. | ||
Keshari RS, Verma A, Barthwal MK, Dikshit M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J Cell Biochem. 2013; 114(3):532–540. | ||
Ortuño-Sahagún D1, González RM, Verdaguer E, et al. Glutamate excitotoxicity activates the MAPK/ERK signaling pathway and induces the survival of rat hippocampal neurons in vivo. J Mol Neurosci. 2014; 52(3):366–377. | ||
Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–758. | ||
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303(5659):844–848. | ||
Faour WH, He Q, Mancini A, Jovanovic D, Antoniou J, Di Battista JA. Prostaglandin E2 stimulates p53 transactivational activity through specific serine 15 phosphorylation in human synovial fibroblasts. Role in suppression of c/EBP/NF-kappaB-mediated MEKK1-induced MMP-1 expression. J Biol Chem. 2006;281(29):19849–19860. | ||
Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18(24):7002–7010. | ||
Barchéchath SD, Tawatao RI, Corr M, Carson DA, Cottam HB. Inhibitors of apoptosis in lymphocytes: synthesis and biological evaluation of compounds related to pifithrin-alpha. J Med Chem. 2005;48(20): 6409–6422. | ||
Lehman TA, Modali R, Boukamp P, et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14(5):833–839. | ||
Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA. 2006;103(6):1888–1893. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.