Back to Journals » Journal of Pain Research » Volume 12
Activating transcription factor 3 modulates protein kinase C epsilon activation in diabetic peripheral neuropathy
Authors Chang YS, Kan HW ![]() , Hsieh YL
, Hsieh YL ![]()
Received 15 October 2018
Accepted for publication 20 November 2018
Published 14 January 2019 Volume 2019:12 Pages 317—326
DOI https://doi.org/10.2147/JPR.S186699
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Michael Schatman
Ying-Shuang Chang,1 Hung-Wei Kan,2 Yu-Lin Hsieh1,3
1Department of Anatomy, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan; 2Department of Anatomy and Cell Biology, College of Medicine, National Taiwan University, Taipei 10051, Taiwan; 3Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan
Background: Skin denervation that develops in patients with diabetes mellitus as a neuropathic manifestation is known as diabetic peripheral neuropathy (DPN). Skin denervation is parallel to neuronal injuries that alter intracellular signaling. To date, the correlation between nerve injury and the activation of intracellular responses to neuropathic manifestations has not been elucidated; specifically, whether activating transcription factor 3 (ATF3) is responsible for neuronal injury and a critical molecule that modulates the activation of intracellular protein kinase C epsilon (p-PKCε) and pain development in DPN is a crucial question.
Methods: To address, ATF3 knockout (atf3−/− group, C57/B6 genetic background) and wild-type mice (atf3+/+ group) received a single dose of streptozotocin (200 mg/kg) to generate a mouse model of DPN.
Results: Both atf3+/+ and atf3−/− mice exhibited hyperglycemia and the same pathology of skin denervation at posttreatment month 2, but only atf3+/+ mice developed thermal hyperalgesia (P<0.001) and mechanical allodynia (P=0.002). The atf3+/+ group, but not the atf3−/− group, had preferential ATF3 upregulation on p-PKCε(+) neurons with a ratio of 37.7%±6.1% in p-PKCε(+):ATF3(+) neurons (P<0.001). In addition, B-cell lymphoma-extra large (Bcl-XL), an antiapoptotic Bcl2 family protein, exhibited parallel patterns to p-PKCε (ie, Bcl-XL upregulation was reversed in atf3−/− mice). These two molecules were colocalized and increased by approximately two-fold in the atf3+/+ group compared with the atf3−/− group (30.0%±3.4% vs 13.7% ± 6.2%, P=0.003). Furthermore, linear analysis results showed that the densities of p-PKCε and Bcl-XL had a reverse linear relationship with the degrees of thermal hyperalgesia and mechanical allodynia.
Conclusion: Collectively, this report suggested that ATF3 is a critical upstream molecule that modulates p-PKCε and Bcl-XL expression, which consequently mediated the development of neuropathic manifestation in DPN.
Keywords: activating transcription factor 3, diabetic peripheral neuropathy, protein kinase C epsilon, PKCε, neuropathic pain, B-cell lymphoma-extra large, Bcl-XL
Introduction
Patients with chronic diabetes mellitus often develop diabetic peripheral neuropathy (DPN), which is an epidemiological problem and a clinical challenge that has no satisfactory therapy.1–4 In pathophysiological terms, DPN is the result of damage to peripheral nerves that alters intracellular responses at the neuronal soma level.5–8 Specifically, these nerve injuries result in skin denervation and cause a cascade of responses in neuronal cell bodies; that is, transcription factors are upregulated, leading to the generation of effector molecules that are responsible for maladaptive behaviors in neuropathic pain.
Activating transcription factor 3 (ATF3) is a member of the ATF/CREB transcription factor superfamily9 and is upregulated in dorsal root ganglia (DRG) neurons after nerve injury; for example, spinal nerve ligation10 and small fiber neuropathy with skin denervation markedly increased ATF3 expression in purinergic P2X3 nociceptors11 as well as in antinociceptive-mediating neurons,12 both of which are correlated with neuropathic pain. Accordingly, ATF3 upregulation is not only an indicator of injury but also correlated with the increasing ligand concentration of purinociceptor,11 altering the neurophysiological responses of primary afferent nociceptors. Nevertheless, intracellular responses caused by injured neurons in DPN remain unclear; therefore, they are worthy of investigation. Specifically, ATF3 upregulation coincided with the maladaptive behavior of small fiber neuropathy,11,12 suggesting ATF3 to be an upstream molecule that modulates the changes of intracellular signaling. Protein kinase C epsilon (PKCε) is an intracellular second messenger in primary afferent nociceptors that modulates neuropathic pain,13 and PKCε inhibition alleviated symptoms in several skin denervation-associated neuropathic pain models.14–18 Collectively, these observations raise the following crucial question that requires elucidating: does ATF3 modulate the expression of PKCε, thereby mediating neuropathic manifestations?19
Nerve injury is a major cause of pathology, which leads to painful diabetic neuropathy. B-cell lymphoma-extra large (Bcl-XL), a protein of the antiapoptotic Bcl2 family, plays a role in regulating the cellular energy homeostasis that underlies cellular stress.20 Therefore, Bcl-XL might act as a signaling molecule that responds to nerve injury; in particular, Bcl-XL has received limited attention in terms of pain development,21 which is modulated by ATF3 underneath the nerve injury condition. Collectively, ATF3 upregulation may be correlated with the coexpression of PKCε and Bcl-XL, which mediates pain development. To address the aforementioned problems, this study used ATF3 knockout (atf3−/−) mice and compared them with wild-type mice to investigate whether ATF3 knockout prevents the development of neuropathic manifestations through the downregulation of PKCε(+): Bcl-XL(+) neurons, although underlying the nerve injury condition.
Materials and methods
DPN induction and animal grouping
A mouse model of DPN was established using 8-week-old adult C57/B6 mice as well as age-matched atf3−/− mice22 with a C57/B6 genetic background (a gift from Dr Tsonwin Hai, Ohio State University, Columbus, OH, USA). Briefly, streptozotocin (STZ; Sigma, St. Louis, MO, USA) was dissolved in citrate buffer (pH =5.5; Sigma), and mice were administered a single dose of STZ (200 mg/kg) through intraperitoneal injection. The blood glucose level was examined weekly by using a commercially available glucometer (Accu-Chek Go, Roche Diagnostics GmbH, Mannheim, Germany). Accordingly, our preliminary test showed that hyperglycemia was induced after 1 week of STZ administration with an approximately 92% survival rate (33/36 mice) with this STZ dose. Mice that showed severe hyperglycemia (glucose level >400 mg/dL) at day 7 after STZ administration were included and those that showed mild to moderate hyperglycemia (glucose level <400 mg/dL) were assigned to the non-DPN (nDPN) group. This study had four groups: (1) atf3+/+ (wild-type mice); (2) atf3−/− (atf3−/− mice); (3) nDPN (served as the positive control); and (4) citrate group (a group of mice that received an equal volume of citrate solution, which served as the negative control). Mice were housed in plastic cages in a 12-hour light/12-hour dark cycle with access to water and food ad libitum. All experiments in this report included the behavioral tests and double-labeling immunostaining and were performed in each group during posttreatment month 2 (PTM2) when neuropathic manifestations reached an apex according to our preliminary observations. All procedures were performed in a coded and blinded manner and conducted in accordance with the published ethical guidelines for investigations of experimental pain in conscious animals.23 All efforts were undertaken to reduce animal suffering, and the protocols were approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University.
Genotyping was performed using PCR by using genomic DNA extracted from the tail. Total DNA was extracted from tail tissues by using a Wizard Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA). Three primers were used in PCR: 5′-AGAGCTTCAGCAATGGTTTGC-3′ (primer 1), 5′-TGAAGAAGGTAAACACACCGTG3′ (primer 2), and 5′-ATCAGCAGCCTCTGTTCCAC-3′ (primer 3). Single-strand cDNA was amplified by 30 cycles of PCR. The PCR protocol used was as follows: 4 minutes of heating followed by 30 seconds of denaturation at 95°C; 30 seconds of annealing at 60°C; and 30 seconds of extension at 72°C. PCR products were electrolyzed using 1.5% agarose gel, and the PCR product was of 236 bp for the atf3-/- group and 329 bp for the wild-type mice.
Behavioral evaluation
The behavioral evaluation included thermal and mechanical responses, which were tested using the hot plate test and von Frey hair test, respectively.
Hot plate test
Animal was placed on a 52°C hot plate (IITC, Woodland Hills, CA, USA) enclosed in a transparent Plexiglas cage. The withdrawal latencies of the hind paw to thermal stimulations of hot plate were determined at an accuracy of 0.1 second. Each test session included three trials at 30-minute intervals. Withdrawal criteria included licking, shaking, or jumping on the hot plate. The mean latency was defined as an individual animal’s response duration to the thermal stimulation.
von Frey hair test
The changes in the mechanical threshold of animal were evaluated using the up-and-down method by different calibers of von Frey hairs (Somedic Sales AB, Hörby, Sweden) in accordance with our established protocol.11,24 Briefly, a series of hairs was applied to the plantar of the hind paw. If paw withdrawal occurred, a hair with a smaller caliber was applied. In case of the absence of paw withdrawal, a hair with a larger caliber was applied. Four additional applications of hairs with various calibers were applied according to previous preceding responses, and mechanical thresholds of each animal were calculated using a previously published formula.25
Evaluation and quantitation of protein gene product (PGP) 9.5(+) intraepidermal nerve fibers (IENFs)
Immunohistochemistry of PGP 9.5(+) IENFs
Skin innervation was evaluated using a pan-axonal marker, PGP 9.5, with the immunohistochemical studies. For evaluation of PGP 9.5(+) IENFs, the intracardiac perfusion was performed on animal with 0.1 M phosphate buffer (PB), followed by 4% paraformaldehyde (4P) in 0.1 M PB. After perfusion, footpad skin was postfixed for another 6 hours and stored in PB. For cryosection, footpad skin was cryoprotected with 30% sucrose in PB overnight and sectioned perpendicular to the epidermis in a 30 µm thickness on an HM440E sliding microtome (Microm, Walldorf, Germany). For adequate sampling, every third section of each footpad and six sections were chosen, and briefly, the sections were quenched with 1% H2O2 in methanol and blocked with 0.5% nonfat dry milk and 0.1% TritonX-100 in 0.5M Tris buffer (Tris). Footpad sections were incubated with anti-PGP 9.5 (1:1,000, UltraClone, Isle of Wight, UK) antiserum overnight at 4°C. After rinsing with Tris, the footpad sections were then incubated with biotinylated secondary antiserum (Vector, Burlingame, CA, USA) and with avidin–biotin complex (Vector). The reaction product was demonstrated using 3,3′-diaminobenzidine (Sigma), and the sections were mounted on gelatin-subbed slides for further analyses.
Quantitation of PGP 9.5(+) IENFs
PGP 9.5(+) IENFs were counted under 400× magnification (Axiophot microscope, Carl Zeiss, Oberkochen, Germany) and following established criteria in a coded fashion.26,27 IENFs with branching points within the epidermis were calculated as a single IENF, and those IENF with branching points in the dermis were counted as single IENF. The length in the lower border of the stratum corneum was defined as the epidermal length and was determined with ImageJ version 1.44d (National Institutes of Health, Bethesda, MD, USA). The IENF density was defined as the number of IENF divided by the epidermal length (fibers/mm).
Double-labeling immunofluorescence staining of DRG neurons
The fourth and fifth lumbar (L4 and L5, respectively) DRGs were postfixed for another 6 hours. The DRG tissues were cryoprotected with same protocol of footpad skin, and cryosections were cut in 8 µm thickness using a cryostat (CM1850; Leica, Wetzlar, Germany). To ensure adequate sampling, five to eight sections per DRG tissue and both L4 and L5 DRG tissues per mouse were immunostained. Primary antisera included anti-ATF3 (rabbit, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), antiphosphorylated PKCε (p-PKCε, goat, 1:200; Santa Cruz Biotechnology), and anti-Bcl-XL (rabbit, 1:250; Cell Signaling, Danvers, MA, USA). There were two combinations of primary antisera: (1) ATF3:p-PKCε and (2) Bcl-XL:p-PKCε. Briefly, sections were incubated with primary antisera combinations overnight, followed with Texas Red and fluorescein isothiocyanate (FITC)-conjugated secondary antisera (1:100; Jackson ImmunoResearch, West Grove, PA, USA) corresponding to appropriate primary antisera for 1 hour.
Quantification of different phenotypic DRG neurons
To quantify the different phenotypic DRG neurons, each DRG section was systematically photographed at 200× under a fluorescence microscope (Carl Zeiss) with appropriate filters. A montage of the entire DRG section was obtained according to our established protocols.11,24,27 Ganglia neurons were gathered together and were larger than other cell types, such as fibroblasts, in a DRG section. To identify labeled neurons, optical intensities between immunoreactive and background neurons were determined. On a 0–255 scale, a preliminary analysis showed the optical intensities of FITC to be in the range of 130–255, and similarly, the optical intensities of Texas Red were shown to be in the range of 121–252. Each signal of a fluorochrome below these ranges acted as the background. To avoid bias, only neurons with a clear nuclear profile and an intensity threshold that met the criteria were counted, and only areas containing ganglia neurons were measured using ImageJ version 1.44d. In addition, the ratios of p-PKCε(+) and Bcl-XL(+) neurons in whole DRG neurons were calculated by dividing the total numbers of DRG neurons, which were obtained using a Nomarski differential interference contrast filter (Figure S1).
Statistical analyses
To reduce individual variations, each group composed five to eight animals, and coding information was masked during behavioral tests and all quantification procedures. All data are expressed as the mean ± SD of the mean, and a t-test was performed for data with a Gaussian distribution. Data without a Gaussian distribution were analyzed using the nonparametric Mann–Whitney U test. In behavioral studies and physiological examinations, one-way repeated-measures ANOVA was performed, followed by Tukey’s post hoc test when a significance level of P<0.05 was obtained.
Results
ATF3 knockout prevented neuropathic manifestation of STZ-induced DPN
Compared with the citrate and nDPN groups, STZ induced hyperglycemia in the atf3+/+ group at 1 week after STZ administration until PTM2 (Figure 1A). Hyperglycemia was not reversed in the aft3−/− group, and body weight changes in the aft3−/− group were comparable with those in the atf3+/+ group (Figure 1B). The development of thermal hyperalgesia (Figure 1C) and mechanical allodynia (Figure 1D) was antiparallel to hyperglycemia; for example, thermal latencies (5.4±0.8 vs 9.8±1.9 seconds, P<0.001) and mechanical thresholds (424.1±75.3 vs 720.0±138.5 mg, P=0.002) decreased in the atf3+/+ group compared with the citrate and nDPN groups (8.2±1.2 seconds, P=0.001 and 779.0±133.4 mg, P=0.007, respectively). Intriguingly, thermal hyperalgesia (7.3±0.5 seconds, P=0.02) and mechanical allodynia (740.9±227.8 mg, P=0.01) were prevented in the aft3−/− group, although under a hyperglycemic condition (Figure 1C, D).
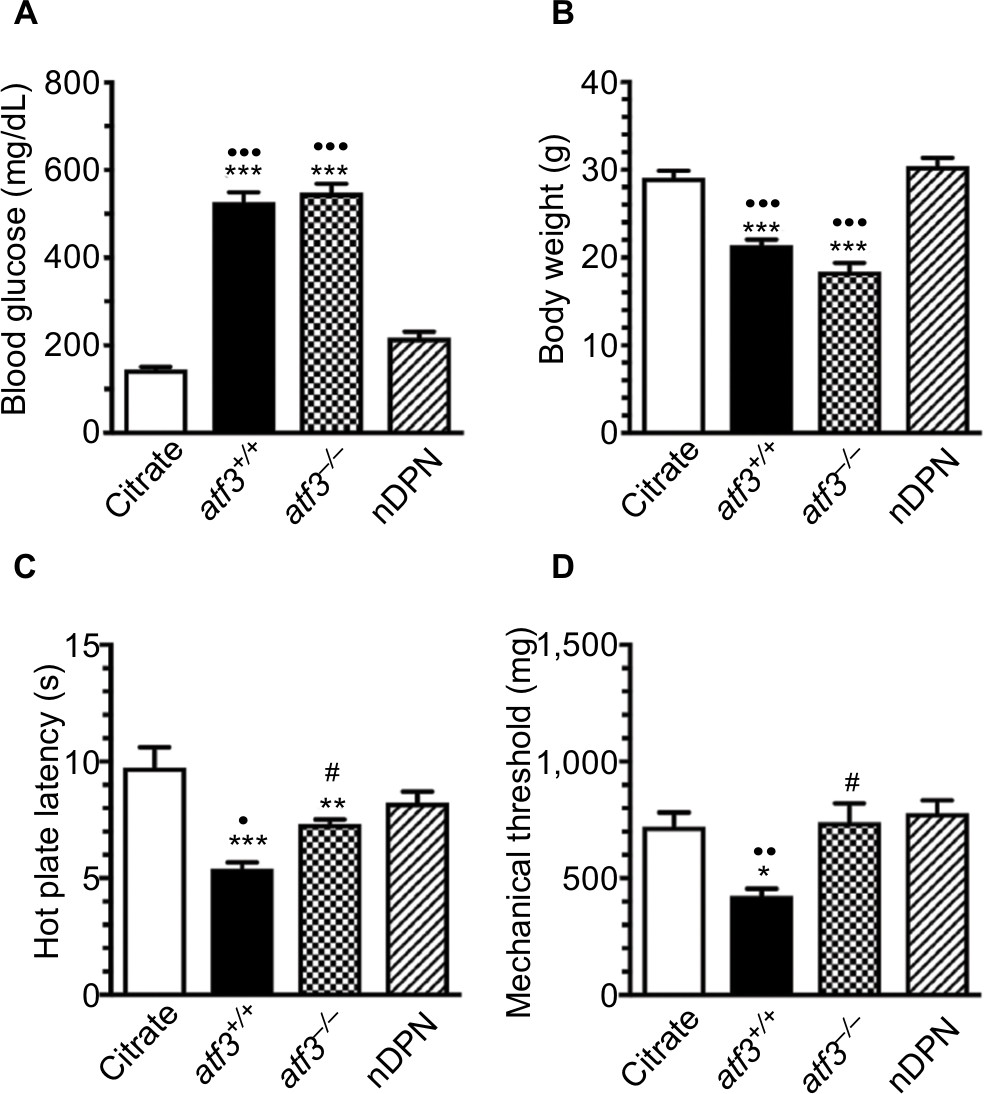 | Figure 1 Different neuropathic manifestation outcomes after activating transcription factor 3 knockout (atf3−/−) in streptozotocin (STZ)-induced diabetic peripheral neuropathy (DPN). Notes: A mouse model of DPN was generated through intraperitoneal injection of STZ (200 mg/kg) to measure changes in (A) blood glucose, (B) body weight, (C) neuropathic manifestation evaluated using the hot plate test, and (D) mechanical thresholds evaluated using the von Frey hair test in citrate (opened bar), atf3+/+ (wild-type mice with blood glucose >400 mg/dL, filled bar), atf3−/− (ATF3 knockout mice with blood glucose >400 mg/dL, dotted bar), and nDPN (blood glucose <400 mg/dL, slashed bar) groups, respectively, as described in the Materials and methods section. (A, B) The graphs show the changes in blood glucose (A) and body weight (B) for each group. (C, D) The graphs show the changes in thermal latency (B) and mechanical threshold (C) of each group. ATF3 knockout relieved the degree of neuropathic manifestation. *P<0.05, **P<0.01, ***P<0.001: atf3+/+, atf3−/− groups compared with the citrate group. •P<0.05, ••P<0.01, •••P<0.001: atf3+/+, atf3−/− groups compared with the nDPN group. #P<0.05: atf3+/+ vs atf3−/− group. |
Skin denervation after STZ-induced DPN
Skin denervation was associated with thermal hyperalgesia (Figure 2). PGP9.5(+) IENFs arose from the subepidermal plexus with a varicose appearance in the citrate and nDPN group (Figure 2A, D), and these PGP9.5(+) IENFs were markedly decreased in the atf3+/+ (10.8±3.9 vs 2.6±2.0 fibers/mm; P<0.001) and aft3−/− groups (2.9±1.7 fibers/mm; P<0.001) (Figure 2B, C, E). Paradoxically, the results of linear analyses showed that the densities of PGP9.5(+) IENFs were correlated with their thermal latencies (r=0.64, P=0.006; Figure 2F), but no relationship was revealed with the mechanical thresholds (r=0.46, P=0.07; Figure 2G).
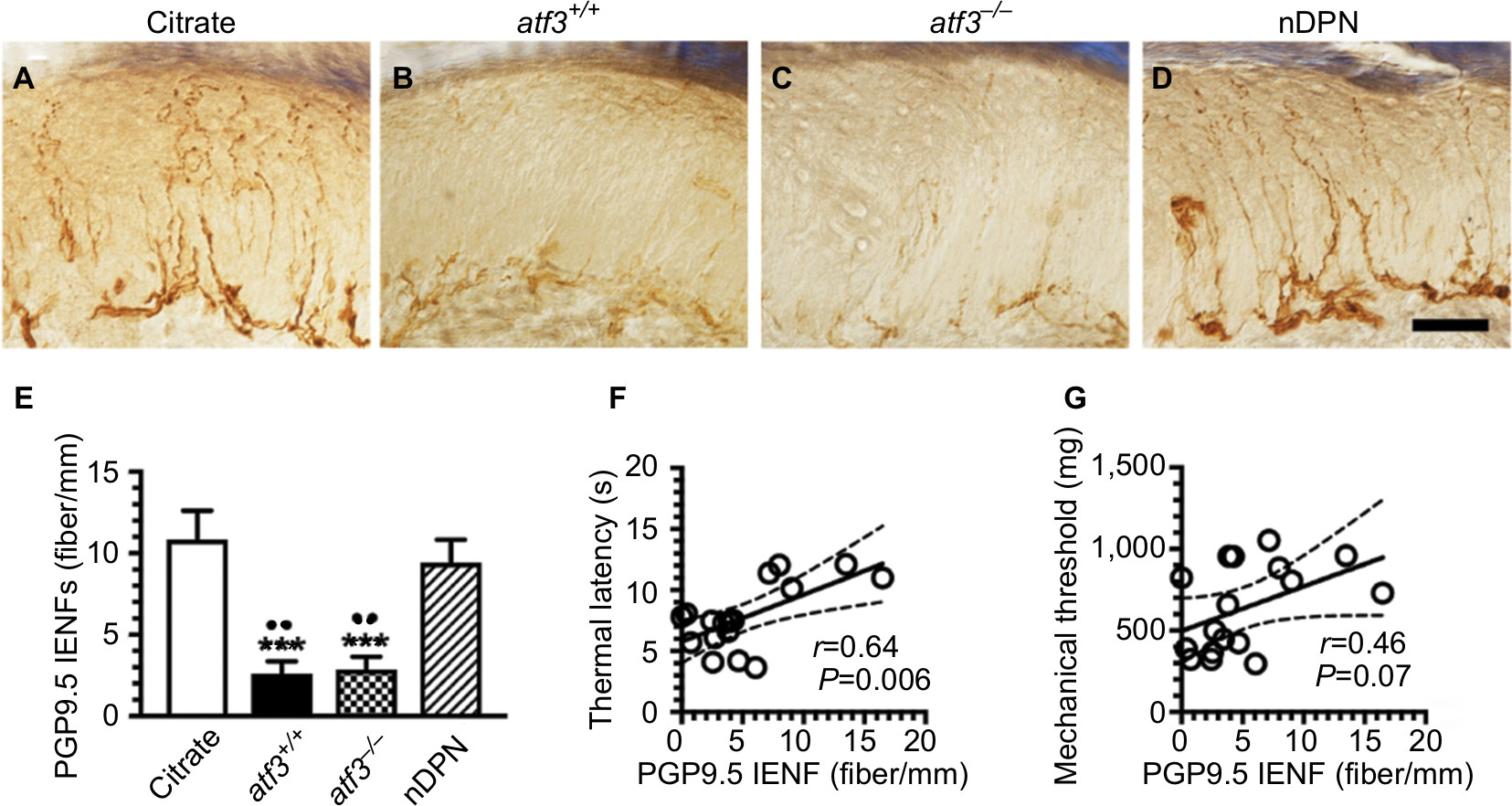 | Figure 2 Changes in skin innervation after activating transcription factor 3 knockout (atf3−/−) in streptozotocin-induced diabetic peripheral neuropathy (DPN). Notes: (A–D) The intraepidermal nerve fibers (IENFs) of the skin were visualized using the pan-axonal marker, protein gene product 9.5 (PGP9.5) in citrate (A), atf3+/+ (wild-type mice with blood glucose >400 mg/dL) (B), atf3−/− (ATF3 knockout mice with blood glucose >400 mg/dL) (C), and nDPN (blood glucose <400 mg/dL) (D) groups, respectively, as described in the Materials and methods section. (E) The graphs show changes in the density of PGP9.5(+) IENFs according to Figure 2A–D. PGP9.5 IENFs were markedly reduced in the atf3+/+ and atf3−/− groups. (F, G) The diagrams show that the densities of PGP9.5(+) IENFs were linear to the thermal latencies (F), but no linear relationship existed with the mechanical threshold (G). ***P<0.001: atf3+/+, atf3−/− groups compared with the citrate group. ••P<0.01: atf3+/+, atf3−/− groups compared with the nDPN group. Bar, 50 µm. |
Upregulation of p-PKCε:ATF3 in small-diameter neurons after STZ-induced DPN
We examined the profiles of p-PKCε(+):ATF3(+) neurons by performing double-labeling immunostaining to investigate whether p-PKCε was activated by injured neurons that were parallel to skin denervation (Figure 3). In the citrate group, PKCε(+) neurons belonged to small-diameter neurons and no ATF3 upregulation occurred (Figure 3A, E, I). Notably, ATF3 was upregulated (27.0±10.7 vs 2.9±2.9 neurons/mm2) in the atf3+/+ group (Figure 3B, F, J, M). Moreover, these ATF3(+) neurons were colocalized with p-PKCε(+) neurons at ratios of 37.7% ± 6.1% vs 0.2% ± 0.4% (Figure 3N). Predictably, no ATF3 expression occurred in the atf3−/− group (Figure 3C, G, K), and only limited ATF3 upregulation occurred in the nDPN group (Figure 3D, H, L). Furthermore, the results of linear analyses showed the ratios of p-PKCε(+):ATF3(+) neurons to be antiparallel to the thermal latencies (Figure 3O) and mechanical thresholds (Figure 3P).
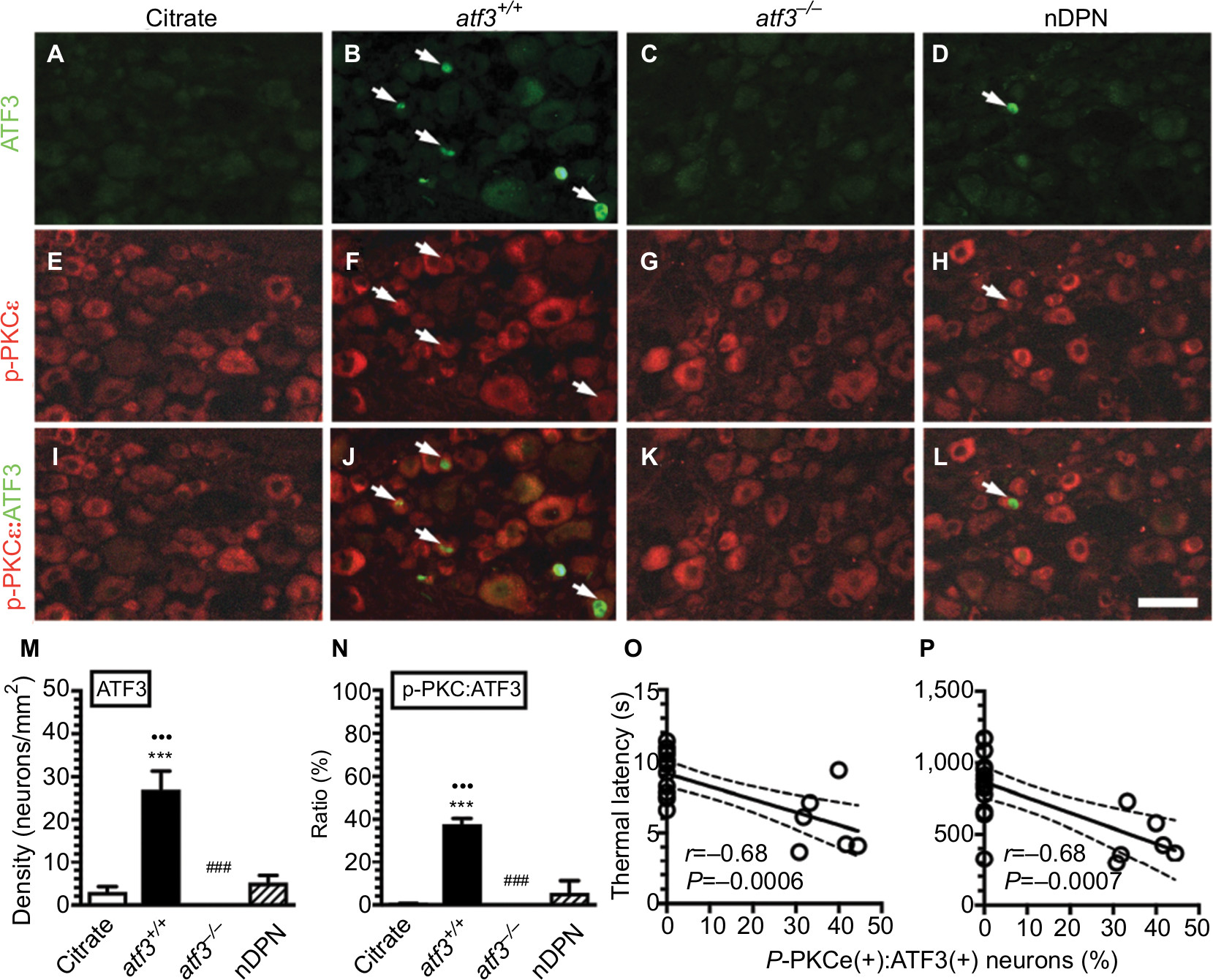 | Figure 3 Activating transcription factor 3 knockout (atf3−/−) prevents phosphorylated protein kinase C epsilon (p-PKCε) upregulation in streptozotocin (STZ)-induced diabetic peripheral neuropathy (DPN). Notes: (A–L) Double-labeling immunofluorescence staining performed using anti-ATF3 (A–D) and anti-p-PKCε (E–H) on the dorsal root ganglia sections of the citrate (A), atf3+/+ (wild-type mice with blood glucose >400 mg/dL) (B), atf3−/− (ATF3 knockout mice with blood glucose >400 mg/dL) (C), and nDPN (blood glucose <400 mg/dL) (D) groups. (I–L) Merged images of ATF3 and p-PKCε for the analysis of the colocalized ratio of each group from Figure 3A–H. Arrows indicate ATF3(+) neurons for each group. (M, N) The graphs show the changes of (M) ATF3(+) neuronal densities and (N) the p-PKCε:ATF3 ratio according to Figure 3A–L. ***P<0.001: atf3+/+, atf3−/− groups compared with the citrate group. •••P<0.001: atf3+/+, atf3−/− groups compared with the nDPN group. ###P<0.01: atf3+/+ vs atf3−/− group. Bar, 50 µm. (O, P) The behavioral changes were assessed using the hot plate test (O) and von Frey hair test (P) using the up-and-down algorithm. The thermal latencies and mechanical thresholds were antiparallel and linear to the ratios of p-PKCε(+):ATF3(+) neurons. |
Co-upregulation of p-PKCε(+) and Bcl-XL(+) neurons was correlated with neuropathic manifestation after STZ-induced DPN
The results of double-labeling studies indicated that Bcl-XL(+) (Figure 4A–D) and p-PKCε(+) neurons (Figure 4E–H) were coexpressed on small-diameter neurons (Figure 4I–L), and these p-PKCε(+):Bcl-XL(+) neurons were co-upregulated in the atf3+/+ group (302.4±49.8 vs 192.8±22.4 neurons/mm2, P<0.001 for p-PKCε and 336.8±53.6 vs 206.2±22.1 neurons/mm2, P<0.001 for Bcl-XL) (Figure 4B, F, J, M). Intriguingly, this co-upregulation was ameliorated in the atf3−/− group; for example, the densities of p-PKCε were normalized and comparable with those in the citrate group (205.6±45.6 neurons/mm2, P>0.05) (Figure 4G, K, M). Similarly, the densities of Bcl-XL(+) neurons decreased in parallel (265.5±28.2 neurons/mm2, P<0.05) (Figure 4M). In addition, no density changes occurred in the total DRG neurons among each group, and the colocalized ratios of p-PKCε:Bcl-XL neurons increased by approximately two-fold in the atf3+/+ group compared with the atf3−/− group (30.0% ± 3.4% vs 13.7% ± 6.2%, P=0.003) (Figure S1).
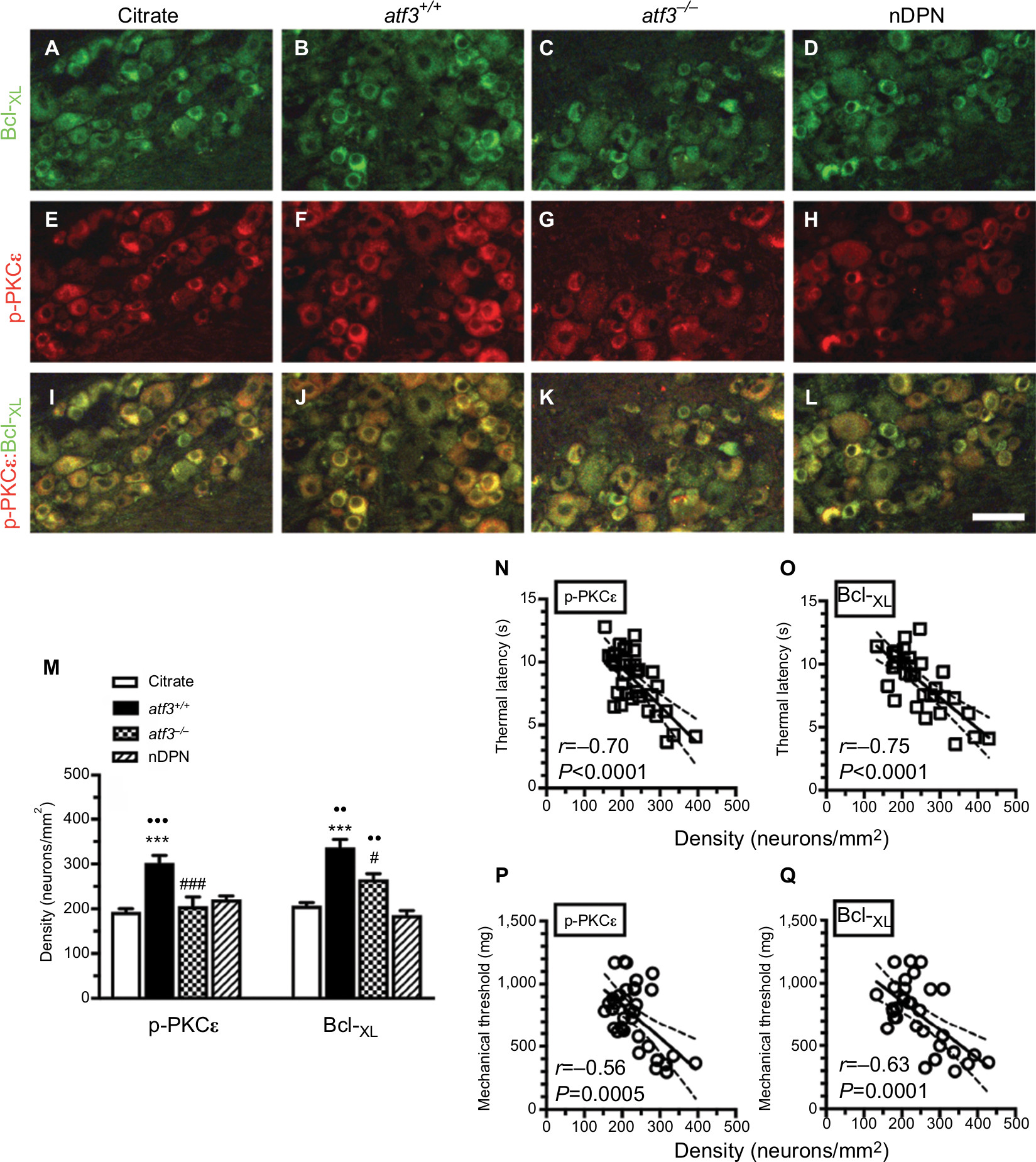 | Figure 4 Effect of activating transcription factor 3 knockout (atf3−/−) on the expression of B-cell lymphoma-extra large (Bcl-XL) and phosphorylated protein kinase C epsilon (p-PKCε) in streptozotocin (STZ)-induced diabetic peripheral neuropathy (DPN). Notes: (A–L) Double-labeling immunofluorescence staining performed using anti-Bcl-XL (A–D) and anti-p-PKCε (E–H) on the dorsal root ganglia sections of the citrate (A), atf3+/+ (wild-type mice with blood glucose >400 mg/dL) (B), atf3−/− (ATF3 knockout mice with blood glucose >400 mg/dL) (C), and nDPN (blood glucose <400 mg/dL) (D) groups. (I–L) Merged images of Bcl-XL and p-PKCε for analysis of the colocalized ratio of each group from Figure 4A–H. (I–L) Photos of Bcl-XL and PKCε were merged for analyzing colocalization patterns. (M) The graphs show changes in the density of p-PKCε (left panel) and Bcl-XL (right panel) according to Figure 4A–H. Opened bar, citrate; filled bar, atf3+/+; dotted bar, atf3−/−; and slashed bar, nDPN groups. ***P<0.001: atf3+/+, atf3−/− groups compared with the citrate group. ••P<0.01, •••P<0.001: atf3+/+, atf3−/− groups compared with the nDPN group. #P<0.05, ###P<0.001: atf3+/+vs atf3−/− group. Bar, 50 µm. (N–Q) The behavioral changes were assessed using the hot plate test (N and O) and von Frey hair tests (P and Q) with the up-and-down algorithm. The densities of (N) p-PKCε(+) and (O) Bcl-XL(+) neurons were antiparallel to the thermal latencies. (P, Q) Similar outcomes were obtained in (P) p-PKCε(+) and (Q) Bcl-XL(+) neurons to the changes of mechanical thresholds. |
The densities of p-PKCε(+) (Figure 4N) and Bcl-XL (+) neurons (Figure 4O) had a reverse linear relationship with thermal latencies. Similar relationships existed with mechanical thresholds (Figure 4P, Q). Collectively, the molecular significance of the upregulation of p-PKCε and Bcl-XL reflected the degree of neuropathic manifestations after STZ-induced DPN.
Discussion
This study discovered a new molecular mechanism underlying pain development: ATF3 is an upstream modulator that regulates the expression of p-PKCε: Bcl-XL, which is correlated with the degree of maladaptive neuropathic behavior after DPN.
STZ-induced DPN shared pathological characteristics with small fiber neuropathy
A decreased density of IENFs on the hind paw skin is known as skin denervation, which is a remarkable pathological characteristic of small fiber neuropathy. Traditionally, PGP9.5 is widely used as a pan-axonal marker to label IENFs, and the evaluation of PGP9.5(+) IENFs densities using skin biopsy is a reliable clinical procedure for evaluating maladaptive neuropathic behaviors.28,29 For example, the densities of PGP9.5(+) IENFs were correlated with the degree of thermal hyperalgesia after DPN. Paradoxically, PGP9.5(+) IENFs were not linear to the thresholds of mechanical allodynia after DPN in this study. These findings imply that the injured neuronal soma required intracellular molecules, which could reflect neuropathic manifestations. ATF3 is a candidate molecule; it was traditionally considered a stress sensor and also correlated to neuropathic behavior by ATF3 expressed by the different phenotypic small-diameter neurons.11,12 This study further strengthens this speculation according to hyperglycemia and PGP9.5(+) IENF reductions, which were significant in both the atf3+/+ and atf3−/− groups; however, only the atf3+/+ group exhibited maladaptive neuropathic behaviors. ATF3 acted on glucose homeostasis30 and tolerance31 in opposite effects, and accordingly, ATF3 is suggested an epiregulator that follows the downstream of intracellular signaling alternations. In this study, the mouse model of DPN mimicked patients with DPN who exhibit major characteristics of small fiber neuropathy, such as pathological skin denervation and altered intracellular molecular responses, and ATF3 was determined to be a potential molecule that mediates maladaptive neuropathic behavior.
ATF3 is an upstream modulator of maladaptive neuropathic behavior after DPN
DPN mice exhibited an antiparallel fashion of IENF reduction and ATF3 upregulation; that is, DPN extensively affected the neuronal soma and their peripheral terminals. ATF3 upregulation has been associated with increased ATP, which sensitized the P2X3 purinociceptor,11 and reduced adenosine, which mediated the antinociceptive effect.12,32 The susceptible small-diameter neurons have created further challenges in distinguishing the contributions made by changing intracellular signaling molecules. Although ATF3 upregulation was injury-inducible33 and cell type specific,33,34 the responses of the neuronal soma in patients with DPN, which contributed to neuropathic pain through modulated intracellular signaling molecules, remain ambiguous. The co-upregulation of ATF3 and p-PKCε determined the degrees of neuropathic behavior, and the linear analyses strengthened the molecular significance of p-PKCε. Intriguingly, the ratio of p-PKCε(+):ATF3(+) neurons was limited, implying the existence of a signaling enhancement that is modulated by p-PKCε-dependent signaling cascades. For example, Bcl-XL, a neuroprotective molecule,20 may be a downstream candidate molecule.
This report suggested that the co-upregulation of p-PKCε(+) and Bcl-XL(+) neurons was neuronal injury dependent, which was reversed in the atf3−/− group. Collectively, this report suggests that ATF3, a stress-inducible gene, modulated the profile expression of p-PKCε and Bcl-XL, which was correlated with the development of neuropathic behavior. Paradoxically, recent reports have indicated that ATF2 – but not ATF3 – is responsible for neuropathic pain.35 Our previous studies reported ATF3 preferentially on small-diameter DRG neurons,11,12 and in those cases, we suggested that the role of ATF3 is more convincing in small-diameter neuropathy. However, the colocalization of p-PKCε(+):ATF3(+) neurons and p-PKCε(+):Bcl-XL(+) neurons provided limited evidence of molecular interactions. Therefore, these topics require further investigation.
Modulation of the expression of p-PKCε(+):Bcl-XL(+) signaling molecules is a new direction for pain control in diabetes mellitus-induced DPN.
DPN-associated maladaptive neuropathic behavior remains a clinical challenge, and current therapeutic interventions are merely satisfactory in clinics. This study indicated that the densities of p-PKCε(+) and Bcl-XL(+) neurons were linear to the degree of neuropathic behavior, which explains that p-PKCε and/or Bcl-XL are potential targets for relieving pain associated with ATF3 upregulation; this molecule could not be genetically depleted under normal conditions. For example, PKCε inhibition alleviated symptoms in several skin denervation-associated neuropathic pain models.14–18,36,37 The antiapoptotic effect of Bcl-XL has been addressed in relevant studies, but this is the first report to indicate that the neuropathological profile of Bcl-XL is correlated with maladaptive neuropathic behavior. A report demonstrated that ATF3 possessed transcriptional repression of Bcl-XL activity,38 which suggested that Bcl-XL activity is correlated with ATF3, although this report still lacks direct evidence linking these two molecules. In addition, the linkage of injury to the activation of p-PKC and Bcl-XL remains unclear; for example, whether Bcl-XL is the downstream effector of PKCε-dependent signaling cascades as well as the cascade sequence between p-PKCε and Bcl-XL require further investigation.
Collectively, the current report revealed that ATF3 is an intracellular upstream molecule that modulates the development of neuropathic manifestations in DPN.
Acknowledgments
We sincerely thank Professor Tsonwin Hai of Ohio State University who kindly provided us with ATF3 knockout mice. This study was supported by grants from the Ministry of Science and Technology (107-2320-B037-022), Chi-Mei Medical Center, and Kaohsiung Medical University Research Foundation (107CM-KMU-09), Taiwan.
Disclosure
The authors report no conflicts of interest in this work.
References
Ziegler D. Current concepts in the management of diabetic polyneuropathy. Curr Diabetes Rev. 2011;7(3):208–220. | ||
Knopp M, Srikantha M, Rajabally YA. Insulin neuritis and diabetic cachectic neuropathy: a review. Curr Diabetes Rev. 2013;9(3):267–274. | ||
Boulton AJ, Kempler P, Ametov A, Ziegler D. Whither pathogenetic treatments for diabetic polyneuropathy? Diabetes Metab Res Rev. 2013;29(5):327–333. | ||
Smith HS, Argoff CE. Pharmacological treatment of diabetic neuropathic pain. Drugs. 2011;71(5):557–589. | ||
Vincent AM, Callaghan BC, Smith AL, Feldman EL. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol. 2011;7(10):573–583. | ||
Crown ED. The role of mitogen activated protein kinase signaling in microglia and neurons in the initiation and maintenance of chronic pain. Exp Neurol. 2012;234(2):330–339. | ||
Mishra RK, Alokam R, Sriram D, Yogeeswari P. Potential role of Rho kinase inhibitors in combating diabetes-related complications including diabetic neuropathy – a review. Curr Diabetes Rev. 2013;9(3):249–266. | ||
Hur J, Sullivan KA, Pande M, et al. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain. 2011;134(Pt 11):3222–3235. | ||
Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273(1):1–11. | ||
Fukuoka T, Yamanaka H, Kobayashi K, et al. Re-evaluation of the phenotypic changes in L4 dorsal root ganglion neurons after L5 spinal nerve ligation. Pain. 2012;153(1):68–79. | ||
Hsieh YL, Chiang H, Lue JH, Hsieh ST. P2X3-mediated peripheral sensitization of neuropathic pain in resiniferatoxin-induced neuropathy. Exp Neurol. 2012;235(1):316–325. | ||
Wu CH, Ho WY, Lee YC, Lin CL, Hsieh YL. Express: NGF-trkA signaling modulates the analgesic effects of prostatic acid phosphatase in resiniferatoxin-induced neuropathy. Mol Pain. 2016;12:1744806916656846. | ||
Maiya RP, Messing RO. Peripheral systems: neuropathy. Handb Clin Neurol. 2014;125:513–525. | ||
He Y, Wang ZJ. Nociceptor beta II, delta, and epsilon isoforms of PKC differentially mediate paclitaxel-induced spontaneous and evoked pain. J Neurosci. 2015;35(11):4614–4625. | ||
Alvarez P, Ferrari LF, Levine JD. Muscle pain in models of chemotherapy-induced and alcohol-induced peripheral neuropathy. Ann Neurol. 2011;70(1):101–109. | ||
Norcini M, Vivoli E, Galeotti N, Bianchi E, Bartolini A, Ghelardini C. Supraspinal role of protein kinase C in oxaliplatin-induced neuropathy in rat. Pain. 2009;146(1–2):141–147. | ||
Dina OA, Gear RW, Messing RO, Levine JD. Severity of alcohol-induced painful peripheral neuropathy in female rats: role of estrogen and protein kinase (A and C epsilon). Neuroscience. 2007;145(1):350–356. | ||
Dina OA, Messing RO, Levine JD. Ethanol withdrawal induces hyperalgesia mediated by PKCepsilon. Eur J Neurosci. 2006;24(1):197–204. | ||
Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113(1–2):185–190. | ||
Gottlieb E, Armour SM, Thompson CB. Mitochondrial respiratory control is lost during growth factor deprivation. Proc Natl Acad Sci U S A. 2002;99(20):12801–12806. | ||
de Novellis V, Siniscalco D, Galderisi U, et al. Blockade of glutamate mGlu5 receptors in a rat model of neuropathic pain prevents early over-expression of pro-apoptotic genes and morphological changes in dorsal horn lamina II. Neuropharmacology. 2004;46(4):468–479. | ||
Hartman MG, Lu D, Kim ML, et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24(13):5721–5732. | ||
Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. | ||
Lin CL, Fu YS, Hsiao TH, Hsieh YL. Enhancement of purinergic signalling by excessive endogenous ATP in resiniferatoxin (RTX) neuropathy. Purinergic Signal. 2013;9(2):249–257. | ||
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. | ||
Hsieh YL, Lin WM, Lue JH, Chang MF, Hsieh ST. Effects of 4-methylcatechol on skin reinnervation: promotion of cutaneous nerve regeneration after crush injury. J Neuropathol Exp Neurol. 2009;68(12):1269–1281. | ||
Hsieh YL, Chiang H, Tseng TJ, Hsieh ST. Enhancement of cutaneous nerve regeneration by 4-methylcatechol in resiniferatoxin-induced neuropathy. J Neuropathol Exp Neurol. 2008;67(2):93–104. | ||
Mellgren SI, Nolano M, Sommer C. The cutaneous nerve biopsy: technical aspects, indications, and contribution. Handb Clin Neurol. 2013;115:171–188. | ||
Hsieh ST. Pathology and functional diagnosis of small-fiber painful neuropathy. Acta Neurol Taiwan. 2010;19(2):82–89. | ||
Zmuda EJ, Viapiano M, Grey ST, Hadley G, Garcia-Ocaña A, Hai T. Deficiency of Atf3, an adaptive-response gene, protects islets and ameliorates inflammation in a syngeneic mouse transplantation model. Diabetologia. 2010;53(7):1438–1450. | ||
Zmuda EJ, Qi L, Zhu MX, Mirmira RG, Montminy MR, Hai T. The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic beta-cell dysfunction. Mol Endocrinol. 2010;24(7):1423–1433. | ||
Kan HW, Chang CH, Lin CL, Lee YC, Hsieh ST, Hsieh YL. Downregulation of adenosine and adenosine A1 receptor contributes to neuropathic pain in resiniferatoxin neuropathy. Pain. 2018;159(8):1580–1591. | ||
Rau KK, Hill CE, Harrison BJ, et al. Cutaneous tissue damage induces long-lasting nociceptive sensitization and regulation of cellular stress- and nerve injury-associated genes in sensory neurons. Exp Neurol. 2016;283(Pt A):413–427. | ||
Bráz JM, Basbaum AI. Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain. 2010;150(2):290–301. | ||
Salinas-Abarca AB, Velazquez-Lagunas I, Franco-Enzástiga Ú, Torres-López JE, Rocha-González HI, Granados-Soto V. ATF2, but not ATF3, participates in the maintenance of nerve injury-induced tactile allodynia and thermal hyperalgesia. Mol Pain. 2018;14:174480691878742. | ||
Alvarez P, Levine JD, Green PG. Eccentric exercise induces chronic alterations in musculoskeletal nociception in the rat. Eur J Neurosci. 2010;32(5):819–825. | ||
Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30(13):4660–4666. | ||
Chüeh AC, Tse JWT, Dickinson M, et al. ATF3 repression of BCL-XL determines apoptotic sensitivity to HDAC inhibitors across tumor types. Clin Cancer Res. 2017;23(18):5573–5584. |
Supplementary material
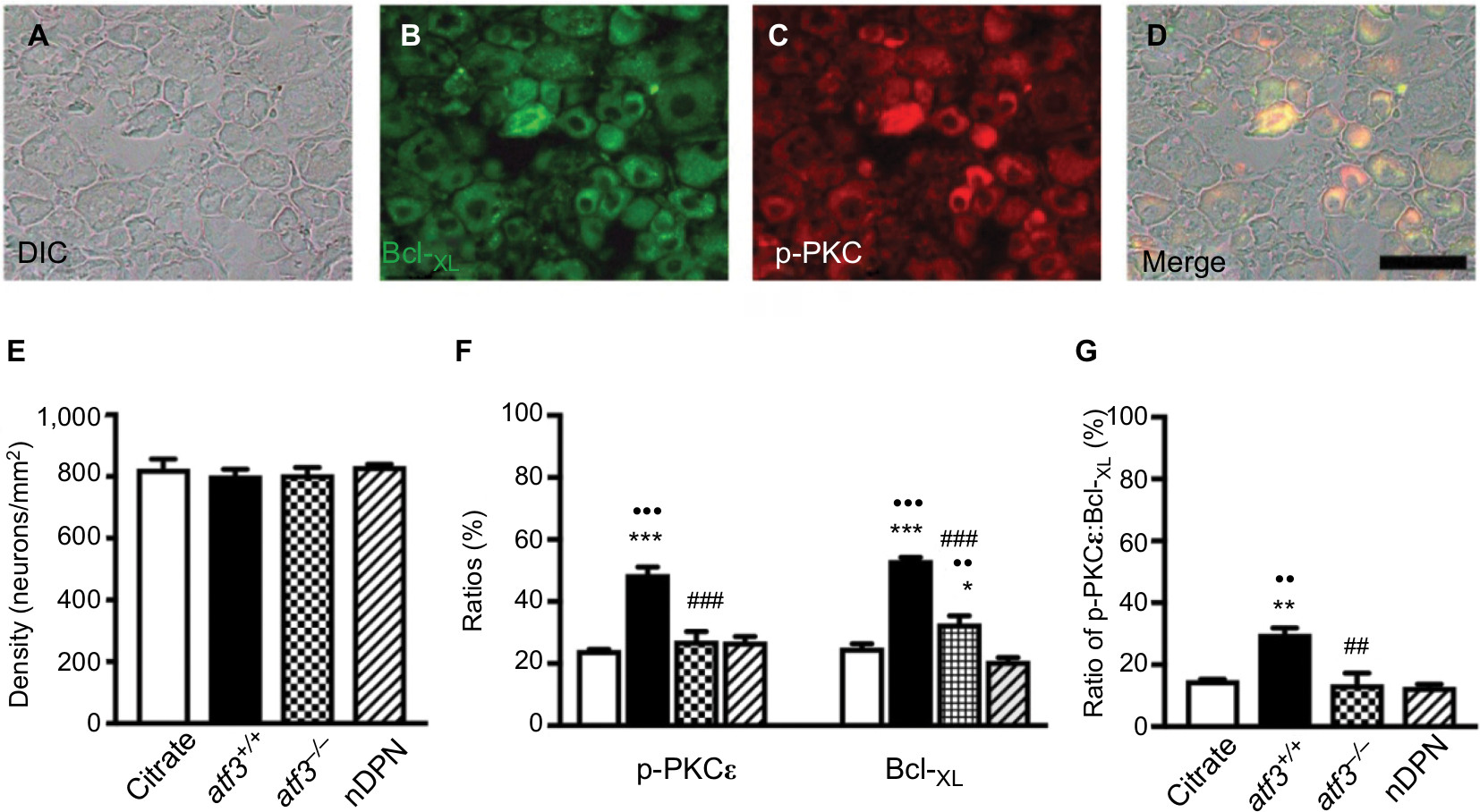 | Figure S1 Profiles of B-cell lymphoma-extra large (Bcl-XL)(+) and phosphorylated protein kinase C epsilon (p-PKCε)(+) neurons on dorsal root ganglion (DRG) neurons in streptozotocin (STZ)-induced diabetic peripheral neuropathy (DPN). Notes: (A–D) Neurons in DRG were detected using a Nomarski differential interference contrast (DIC) filter (A, D) and compared with the fluorescence Bcl-XL(+) (B in green) and p-PKCε(+) neurons (C in red). (D) Merged DIC and fluorescence image of Bcl-XL(+) (B) and p-PKCε(+) neurons (C) according to Figure S1A–C. (E–G) Graphs show the density changes of DRG neurons (E: DIC image), and (F) ratio changes of p-PKCε(+) (left panel) and Bcl-XL(+) neurons (right panel) according to Figure S1A–C. (G) Graph indicating the ratio changes of colocalized p-PKCε(+):Bcl-XL(+) neurons on total DRG neurons according to Figure S1D. Opened bar, citrate; filled bar, atf3+/+; dotted bar, atf3−/−; and slashed bar, nDPN groups. *P<0.05, **P<0.01, ***P<0.001: atf3+/+, atf3−/− groups compared with the citrate group. ••P< 0.01, •••P< 0.001: atf3+/+, atf3−/− groups compared with the nDPN group. ##P< 0.01, ###P< 0.001: atf3+/+ vs atf3−/− group. Bar, 50 µm. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.