Back to Journals » Biologics: Targets and Therapy » Volume 19
Activated Growth Factor (AGF) as a Superior Biological Therapy for Osteoarthritis: Comparative Efficacy in Modulating Cartilage Degeneration and Inflammation in vivo
Received 17 May 2025
Accepted for publication 15 August 2025
Published 21 August 2025 Volume 2025:19 Pages 463—479
DOI https://doi.org/10.2147/BTT.S541172
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shein-Chung Chow
Rachmat Hidayat,1,* Zaliha Harun2,*
1Doctor of Philosophy in Health Sciences Programme, Faculty of Applied Science, Lincoln University College, Selangor, Malaysia; 2Department of Food Science and Nutrition, School of Nursing and Applied Science, Lincoln University College, Selangor, Malaysia
*These authors contributed equally to this work
Correspondence: Zaliha Harun, Department of Food Science and Nutrition, School of Nursing and Applied Science, Lincoln University College, Wisma Lincoln 12-18, Jalan SS 6/12, Petaling Jaya, Selangor, 47301, Malaysia, Tel +60 12 6127974, Email [email protected]
Purpose: This study aimed to explore the effect of activated growth factor (AGF) in anabolic-catabolic regulation pathway in an experimental osteoarthritis (OA) rat model. This investigation was predicated on the critical need for a disease-modifying osteoarthritis drug, seeking to determine if AGF could restore chondral homeostasis by reversing the catabolic-dominant state that defines OA pathology. This is the first study to compare AGF with platelet-rich plasma (PRP) and diclofenac sodium in vivo.
Methods: Fifty-six male Wistar rats were randomly allocated into seven groups (n = 8 per group); normal control (NC), negative control (OA induced, saline-treated), positive control with platelet-rich plasma (PRP) (OA induced + PRP), positive control diclofenac (OA induced + Diclofenac sodium), AGF 0.1 ng/mL (OA induced + AGF with TGF-β at 0.1 ng/mL), AGF 1 ng/mL (OA induced + AGF with TGF-β at 1 ng/mL), AGF 10 ng/mL (OA induced + AGF with TGF-β at 10 ng/mL). Cartilage anabolic-catabolic status was assessed by measuring matrix metalloproteinases (MMP-1, MMP-13), a-disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4), SMAD3, aggrecan, and collagen type II (Col2) expression via ELISA and Western blot. Key inflammatory mediators (TNF-α, IL-1β, NF-κB) and histopathological changes were also evaluated.
Results: AGF treatment demonstrated superior efficacy in modulating anabolic-catabolic balance compared to OA controls and PRP. AGF dose-dependently and significantly decreased MMP-1, MMP-13, and ADAMTS-4 levels. Conversely, AGF significantly increased SMAD3 phosphorylation, aggrecan synthesis, and Col2 expression, surpassing the effects of PRP. The 10 ng/mL AGF group showed the most pronounced chondroprotective and anabolic effects, often restoring parameters towards normal levels. Furthermore, AGF significantly reduced TNF-α, IL-1β, and NF-κB levels, and diminished inflammatory cell infiltration, with the 10 ng/mL dose being comparable or superior to Diclofenac in these anti-inflammatory/anti-catabolic effects. PRP showed moderate benefits, generally less than AGF formulations.
Conclusion: Platelet-derived AGF, as an advanced mode of PRP, effectively regulates the anabolic-catabolic processes in OA cartilage. It achieved this by significantly inhibiting key catabolic enzymes and pro-inflammatory mediators, while concurrently promoting crucial anabolic signaling pathways and extracellular matrix synthesis. These findings highlight AGF’s substantial therapeutic potential as a disease-modifying biological agent for OA.
Keywords: aggrecans, anabolic agents, inflammation mediators, phosphorylation, platelet-rich plasma
Introduction
Osteoarthritis (OA) is a significant global musculoskeletal health issue, characterized not just by “wear and tear” but as a complex disease affecting the entire joint, leading to progressive, irreversible degradation of articular cartilage, subchondral bone remodeling, osteophyte formation, and synovitis, which collectively cause chronic pain, functional limitation, and reduced quality of life, imposing a substantial societal and economic burden.1 The development of OA is intricate, stemming from genetic predispositions, aging, obesity, joint injury, mechanical stress, and metabolic factors, and is fundamentally marked by a dysregulation in cartilage homeostasis where chondrocytes fail to maintain the balance between anabolic (synthesis) and catabolic (degradation) processes within the extracellular matrix (ECM), primarily composed of type II collagen and aggrecan.2 This balance shifts towards catabolism due to the overexpression of proteolytic enzymes like matrix metalloproteinases (MMPs, particularly MMP-1, MMP-8, MMP-13) and aggrecanases (ADAMTS-4, ADAMTS-5), whose production is stimulated by pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) via pathways like nuclear factor-κβ (NF-κβ), creating a self-perpetuating cycle of cartilage degradation and inflammation.3,4 Concurrently, the anabolic capacity of chondrocytes to repair the ECM is diminished, with impaired responsiveness to or availability of growth factors like transforming growth factor-β (TGF-β) (which signals via Smad pathways to produce collagen and aggrecan), insulin like growth factor 1 (IGF-1), and fibroblast growth factor (FGF).5 Current OA therapies are mainly palliative, offering symptomatic relief through lifestyle changes, physical therapy, analgesics, non-steroid anti-inflammatory drugs (NSAIDs), corticosteroid injections, or viscosupplementation,6,7 but do not halt disease progression and can have adverse effects, often leaving total hip arthroplasty as the last resort.8 The long-term success of total hip arthroplasty (THA) is often undermined by a chronic inflammatory response to prosthetic wear particles. This process begins when macrophages recognize this debris, activating the NLRP3 inflammasome and releasing a cascade of pro-inflammatory cytokines, notably TNF-α and IL-1β. This cytokine storm orchestrates a catabolic shift in the periprosthetic environment, promoting osteoclast activation and matrix degradation through enzymes like MMPs. The entire cycle is amplified by the central NF-κB signaling pathway, tipping the local homeostasis towards progressive bone loss (periprosthetic osteolysis) and culminating in aseptic implant loosening. There is a critical unmet need for disease-modifying osteoarthritis drugs (DMOADs) that can effectively address the structural damage and improve long-term outcomes.9
In the pursuit of DMOADs, biological therapies that leverage the body’s innate regenerative capabilities have garnered significant attention. Platelet-rich plasma (PRP) has emerged as a widely investigated autologous biological therapy for OA.9 PRP is a concentrate of platelets derived from the patient’s own blood, containing a supraphysiological concentration of various growth factors and bioactive proteins stored within platelet α-granules. Upon activation, these platelets release a milieu of factors including PDGF, TGF-β, VEGF, IGF-1, EGF, and FGF, which are known to modulate inflammation, stimulate cellular proliferation and differentiation, and promote tissue repair and ECM synthesis. Activated Growth Factor (AGF) represents a more standardized and potentially more potent evolution of PRP therapy.10 AGF is prepared by controlled ex vivo activation of platelets (typically from PRP) using agonists like thrombin and calcium chloride, ensuring a more complete and immediate release of the full spectrum of growth factors.10 The resulting AGF product, often a platelet lysate or releasate, is characterized by a high concentration of these bioactive molecules, particularly TGF-β, which is critical for chondrogenesis and cartilage matrix production. This controlled activation and subsequent removal of platelet bodies may enhance the bioavailability and consistency of the growth factor cocktail delivered to the target tissue, potentially leading to more predictable and robust therapeutic effects compared to traditional PRP.
The novelty of this in vivo investigation is that it was the first study to comprehensively explore AGF, positioned as an advanced and optimized mode of PRP, specifically examining its detailed effects on the critical balance of anabolic and catabolic pathways within osteoarthritic cartilage. While PRP has been studied, this research delved deeper into a more refined platelet-derived product (AGF, standardized by TGF-β concentration) and systematically dissected its influence on key molecular players involved in both matrix synthesis (anabolism) and matrix degradation (catabolism), including the catabolic role of inflammatory mediators, within a controlled experimental setting. This study also uniquely incorporates both PRP and Diclofenac as positive controls, allowing for a more nuanced understanding of AGF’s relative efficacy. The primary aim of this study was to explore the effect of intra-articularly administered, platelet-derived AGF on the anabolic-catabolic regulation pathway in an experimental rat model of osteoarthritis. We hypothesized that AGF would outperform PRP in restoring cartilage homeostasis.
Materials and Methods
Ethical Statement
All rat experiments in this study were approved by Animal Care and Ethics Committee at Faculty of Medicine, Universitas Sriwijaya, Indonesia (Protocol No. 087/2024). All animal procedures were conducted in strict accordance with the ARRIVE guidelines and the Guide for the Care and Use of Laboratory Animals (National Research Council, USA).11 Anesthesia and analgesia were used to minimize pain and distress.
Study Animals
A total of fifty-six (56) male Wistar rats (specific pathogen-free), aged 8–10 weeks and weighing 200–250 g at the start of the experiment, were procured from a certified breeding facility (Eureka Research Laboratory, Palembang, Indonesia). Animals were housed in groups of 2–3 per individually ventilated cage (Tecniplast®, Milan, Italy) under controlled environmental conditions: temperature 22 ± 2°C, relative humidity 50 ± 10%, 12-hour light/12-hour dark cycle (lights on at 07:00 h). Standard chow (Japfa®, Jakarta, Indonesia) and autoclaved water were available ad libitum. A minimum of one-week acclimatization period was provided before any experimental procedures. Figure 1 present the flowchart of the experimental design of this study.
 |
Figure 1 Flowchart of experimental design. Abbreviations: ADAMTS-4, A disintegrin and metalloproteinase with thrombospondin motifs-4; AGF, activated growth factor; ELISA, enzyme linked immunosorbent assay; MMP-1, matrix metalloproteinase-1; MMP-13, matrix metalloproteinase-13; OA, osteoarthritis. |
Animal Grouping
The 56 rats were randomly divided into seven experimental groups (n = 8 rats per group) using a computer-generated randomization sequence: Group 1 (normal control; NC): Healthy rats receiving no mono-iodoacetate (MIA) (Sigma-Aldrich, Singapore) induction and only sham intra-articular injections; Group 2 (OA – negative control): osteoarthritis (OA) induced by MIA + intra-articular injections of 50 µL sterile saline; Group 3 (OA + PRP – positive control PRP): OA induced by MIA + intra-articular injections of 50 µL standardized rat PRP (2x106 platelets/uL); Group 4 (OA + Diclofenac, positive control diclofenac): OA induced by MIA + oral administration of diclofenac sodium (Sigma-Aldrich, Singapore) (5 mg/kg body weight, once daily); Group 5 (OA + activated growth factor (AGF) 0.1): OA induced by MIA + intra-articular injections of 50 µL AGF with TGF-β1 concentration standardized to 0.1 ng/mL; Group 6 (OA + AGF 1): OA induced by MIA + intra-articular injections of 50 µL AGF with TGF-β1 concentration standardized to 1 ng/mL; Group 7 (OA + AGF 10): OA induced by MIA + intra-articular injections of 50 µL AGF with TGF-β1 concentration standardized to 10 ng/mL. Randomization was performed by an investigator not involved in outcome assessment. Cages were color-coded for blinding during study.
Osteoarthritis Induction
OA was induced in the right knee joint of rats in groups 2–7. Rats were anesthetized using intraperitoneal injections of a combination containing 100 mg/kg of ketamine and 10 mg/kg of xylazine. The right knee area was shaved and aseptically prepared with 70% ethanol and povidone-iodine. A single intra-articular injection of 3 mg MIA (dissolved in 50 µL of sterile saline) was administered through the patellar ligament using a 29-gauge insulin syringe. The needle was inserted into the joint space, and the solution was injected slowly. Successful injection was confirmed by minimal resistance and absence of subcutaneous bleb formation. After injection, the knee was gently flexed and extended for 30 seconds to distribute the MIA. Animals in group 1 (NC) received a sham injection. Post-procedural analgesia (buprenorphine 0.05 mg/kg s.c.) was provided for 48 hours. Rats were monitored daily for signs of distress or infection. The development of OA was allowed for 4 weeks before treatment initiation.
Preparation of Platelet-Rich Plasma (PRP) and Activated Growth Factor (AGF)
For the preparation of platelet-rich plasma (PRP) and activated growth factor (AGF), whole blood was initially procured from a distinct cohort of healthy donor Wistar rats, carefully matched for age and sex, under terminal anesthesia induced by intraperitoneal injections of a combination containing 100 mg/kg of ketamine and 10 mg/kg of xylazine followed by cardiac puncture. The collected blood was immediately drawn into syringes containing an Acid Citrate Dextrose (ACD) solution (Sigma-Aldrich, Singapore), at a ratio of one part ACD to six parts blood, to prevent coagulation. The subsequent PRP preparation commenced with a primary low-speed centrifugation of the anticoagulated blood at 200 x g for 10 minutes at room temperature, a step designed to effectively separate the plasma and platelet fraction from erythrocytes and leukocytes. The resultant supernatant, comprising the platelet-rich plasma and buffy coat, was meticulously collected. This platelet-enriched fraction then underwent a secondary, high-speed centrifugation at 1000 x g for 15 minutes at room temperature, facilitating the formation of a platelet pellet. Following this, the majority of the supernatant platelet-poor plasma (PPP) was judiciously removed, leaving a defined volume—approximately one-third to one-fifth of the initial plasma volume—to resuspend the concentrated platelet pellet, thereby constituting the PRP. To ensure optimal quality and concentration, platelet counts were systematically performed on both the initial whole blood and the final PRP samples, with the objective of achieving a 3–5 times baseline platelet concentration. A designated portion of this freshly prepared PRP was allocated for direct use in the OA+PRP experimental group, while the remainder was channeled into the AGF generation protocol.
For activated growth factor (AGF) preparation, the previously obtained PRP was treated with calcium chloride (CaCl2), achieving a final concentration of 10% v/v of a 10% CaCl2 solution, and bovine thrombin, at a final concentration of 10% v/v of the thrombin solution. This combination was introduced to potently induce platelet activation and subsequent degranulation. The ensuing mixture was incubated at 37°C for a duration of one hour, or until a stable fibrin clot was visibly formed. Post-incubation, the activated mixture was subjected to high-speed centrifugation at 3000 x g for 20 minutes at 4°C, a process tailored to pellet platelet bodies, cellular debris, and the fibrin clot. The clear supernatant, rich in released growth factors and termed AGF releasate, was then carefully aspirated, passed through a 0.22 µm sterile filter to ensure purity, and subsequently aliquoted for storage and use. Crucially, for standardization, the concentration of Transforming Growth Factor-beta 1 (TGF-β1) within the AGF releasate was precisely quantified using a rat-specific TGF-β1 ELISA kit, following acid activation for total TGF-β1 measurement as per the manufacturer’s guidelines. Based on this quantification, the AGF was then accurately diluted with sterile saline to achieve the target TGF-β1 concentrations of 0.1 ng/mL, 1 ng/mL, and 10 ng/mL for the experimental interventions. The selection of AGF doses is based on the primary goal of establishing a dose-response relationship. This is a standard method to efficiently test a wide range of concentrations and identify a potential therapeutic window. The dose of 0.1 ng/mL (low dose) was chosen to represent a minimal or near-physiological level of TGF-β1, the standardizing growth factor. The purpose was to determine the threshold concentration at which AGF begins to exert a discernible biological effect. The AGF dose of 1 ng/mL (intermediate dose) represents a supraphysiological or moderate therapeutic concentration. It’s a tenfold increase from the low dose, designed to test if a stronger, more robust therapeutic response could be achieved. This concentration often serves to find a balance between efficacy and potential side effects. The concentration 10 ng/mL (high dose) represents a high pharmacological dose. Its purpose was to determine the maximal therapeutic effect (the top of the dose-response curve).
Treatment Protocol
Following a four-week period post-MIA induction or sham injection to allow for the establishment of osteoarthritic conditions, a comprehensive three-week therapeutic intervention was initiated across the designated experimental groups. For animals in Groups 2, 3, 5, 6, and 7, treatments were administered via intra-articular injections. This procedure involved lightly anesthetizing the rats with a combination containing 100 mg/kg of ketamine and 10 mg/kg of xylazine, followed by the aseptic preparation of the right knee. Subsequently, a precise volume of 50 µL of the allocated treatment solution—either saline, PRP, or AGF at the specified concentrations—was carefully injected directly into the articular space. These intra-articular administrations were performed consistently three times per week, typically scheduled on Mondays, Wednesdays, and Fridays, for the entire three-week treatment duration. Concurrently, rats assigned to Group 4, the Diclofenac positive control group, received their treatment through oral administration. Diclofenac sodium, at a dosage of 5 mg/kg body weight, was dissolved in their drinking water or administered daily via oral gavage for the same three-week period. The volume for gavage was typically 1 mL/100g body weight.
Sample Collection and Processing
One day after the final treatment (7 weeks post-MIA induction), rats were euthanized by CO2 asphyxiation followed by exsanguination via cardiac puncture and cervical dislocation.
Synovial Fluid and Blood Collection
Synovial fluid was aspirated from the knee joint immediately post-euthanasia where possible. Blood was collected via cardiac puncture for serum preparation.
Joint Tissue Harvesting
The entire right knee joint was carefully dissected. The synovium was meticulously separated where possible and snap-frozen. The femoral condyles and tibial plateau were harvested.
Tissue Processing
For subsequent molecular investigations, including enzyme linked immunosorbent assay (ELISA) and Western Blot analyses, articular cartilage was meticulously shaved from the femoral condyles and tibial plateau of the harvested joints. These cartilage samples were then carefully weighed, immediately snap-frozen in liquid nitrogen to preserve molecular integrity, and subsequently stored at −80°C until analysis. Synovial tissues were similarly processed.
In parallel, specimens destined for histological evaluation were prepared with rigorous attention to structural preservation. The entire remaining joint structure, encompassing the femoral condyles and tibial plateau along with the underlying bone, was initially fixed in a 10% neutral buffered formalin solution for 48 hours at 4°C. Following fixation, the tissues underwent a comprehensive decalcification process in a 10% EDTA solution (pH 7.4), supplemented with 0.5% paraformaldehyde, maintained at 4°C for a period of 4–6 weeks. Throughout this decalcification phase, the solution was diligently changed every 2–3 days, and the endpoint of decalcification was carefully monitored using radiography or gentle probing to ensure complete mineral removal. Upon successful decalcification, the tissues were systematically processed through a series of graded ethanol solutions for dehydration, cleared with xylene, and finally embedded in paraffin wax to create stable blocks. From these paraffin-embedded blocks, precise sagittal sections, each with a thickness of 5 µm, were expertly cut in preparation for microscopic examination.
Analytical Methods
Enzyme Linked Immunosorbent Assay (ELISA) Evaluation
Frozen cartilage and synovial tissue samples were pulverized under liquid nitrogen and homogenized in ice-cold lysis buffer (containing protease/phosphatase inhibitors). Lysates were centrifuged (14,000 x g, 15 min, 4°C), and supernatants collected. Protein concentration was determined using bicinchoninic acid assay (BCA). Levels of MMP-1, MMP-13, ADAMTS-4, TNF-α, IL-1β, and NF-κB p65 in cartilage or synovial lysates were quantified using respective rat-specific ELISA kits following manufacturers’ protocols (CloudClone Corp., Wuhan, China). Absorbance readings were normalized to total protein content of the lysate (pg/mg total protein).
Western Blot Analysis
Cartilage lysates (20–40 µg protein/lane) were resolved by SDS-PAGE and transferred to PVDF membranes. Membranes were blocked (5% non-fat milk or BSA in TBST) for 1 hour at RT. Incubation with primary antibodies (anti-phospho-SMAD3, anti-total-SMAD3, anti-Aggrecan, anti-Col2, anti-β-actin; typically 1:1000 dilution) (CloudClone, Wuhan, China) was performed overnight at 4°C. After washing, membranes were incubated with appropriate HRP-conjugated secondary antibodies (1:2000) (Bio-Rad Laboratories®, California, USA) for 1 hour at room temperature. Bands were visualized using ECL substrate and quantified by densitometry (ImageJ software). Expression levels were normalized to β-actin (loading control).
Histopathological Assessment (OARSI Scoring)
Paraffin sections were stained with hematoxylin and eosine (H&E) and safranin o-fast green (for proteoglycan visualization and cartilage morphology). Cartilage degradation was semi-quantitatively scored by two blinded observers using a modified Osteoarthritis Research Society International (OARSI) scoring system for rats, assessing parameters like cartilage structure, cellularity, safranin of staining intensity, and tidemark integrity. The detailed blinding procedure for histopathology scoring was described below;
Sample Anonymization and Coding
After the knee joints are harvested, processed, and sectioned onto microscope slides, a crucial step occurs before they reach the observers. A third party (investigator not involved in the scoring) assigns a unique, non-identifiable code to each slide (eg, “A-01”, “A-02”, etc). The key that links these codes to the actual treatment groups (eg, A-01 = Rat #3, Group 7 [AGF 10 ng/mL]) is kept confidential and completely separate from the observers.
Independent Evaluation
The two blinded observers receive the entire set of coded slides. They have no information about which treatment any slide received. They proceed to score each slide independently, applying the pre-defined criteria of the OARSI scoring system. They record their scores for each coded slide in separate logs.
Ensuring Inter-Rater Reliability
The use of two observers is a crucial check on the subjectivity of the scoring. It establishes inter-rater reliability. After both observers have completed their independent scoring, their results are compared. If there is a significant disagreement on a particular slide’s score, a consensus must be reached. This is typically done in one of two ways; The two observers review the disputed slide together under a multi-headed microscope, discuss their reasoning, and agree on a final consensus score, or if an agreement cannot be reached, a third blinded observer is brought in to score the slide and act as a tie-breaker.
De-Coding
Only after all scores have been finalized and locked in is the dataset “unblinded.” The confidential key is used to assign the scores back to their respective treatment groups. This is the first time the researchers can see how the different treatments (Saline, PRP, Diclofenac, AGF doses) performed.
Inflammatory Cell Infiltration Assessment
On H&E stained sections, synovial inflammation (synovitis score based on lining cell hyperplasia, stromal cell density, and inflammatory infiltrate) and cartilage inflammatory cell infiltration were assessed by blinded observers.
Statistical Analysis
Data were presented as mean ± Standard Deviation (SD) or Standard Error of the Mean (SEM) where appropriate. Statistical analyses were performed using SPSS version 27.0 for Windows (IBM, Jakarta, Indonesia). Normality of data was assessed using Shapiro–Wilk test. For normally distributed data, One-Way Analysis of Variance (ANOVA) followed by Tukey’s or Dunnett’s post-hoc test (for comparisons against a single control) was used. For non-normally distributed data, Kruskal–Wallis test followed by Dunn’s multiple comparison test was applied. A p-value < 0.05 was considered statistically significant. Specific comparisons and significance levels are indicated in the results.
Results
Table 1 delineates the quantitative impact of various therapeutic interventions on the levels of key cartilage-degrading enzymes—matrix metalloproteinase-1 (MMP-1), matrix metalloproteinase-13 (MMP-13), and A Disintegrin and Metalloproteinase with Thrombospondin motifs-4 (ADAMTS-4)—within the articular cartilage of rats subjected to monoiodoacetate (MIA)-induced osteoarthritis (OA). The data unequivocally demonstrate the successful induction of an osteoarthritic phenotype in the OA Control group. Compared to the Normal Control (NC) group, which exhibited basal physiological levels of these enzymes (MMP-1: 5.2±0.8 ng/mg protein; MMP-13: 3.9±0.6 ng/mg protein; ADAMTS-4: 2.3±0.4 ng/mg protein), the OA Control group displayed a highly significant (p < 0.001 for all) upregulation of MMP-1 (to 15.8±2.1 ng/mg protein, approximately a 3-fold increase), MMP-13 (to 12.5±1.7 ng/mg protein, approximately a 3.2-fold increase), and ADAMTS-4 (to 9.1±1.2 ng/mg protein, approximately a 3.95-fold increase). This marked elevation signifies a pronounced catabolic state within the cartilage, characteristic of active OA, with heightened collagenolytic (MMP-1, MMP-13) and aggrecanolytic (ADAMTS-4) activities. The positive control interventions yielded distinct effects. Intra-articular administration of Platelet-Rich Plasma (PRP) (OA + PRP group) resulted in a statistically significant (p < 0.001 vs OA Control for all three enzymes) reduction in the levels of MMP-1 (to 11.5±1.5 ng/mg protein), MMP-13 (to 9.2±1.2 ng/mg protein), and ADAMTS-4 (to 6.8±0.9 ng/mg protein). This indicates that PRP possesses an inherent capacity to mitigate the catabolic enzyme load, likely through the action of its constituent growth factors influencing chondrocyte gene expression or local inflammatory responses. Oral administration of Diclofenac sodium (OA + Diclofenac group), a standard non-steroidal anti-inflammatory drug (NSAID), also led to a statistically significant, albeit more modest, reduction in catabolic enzyme levels compared to the OA Control group (MMP-1: 13.1±1.8 ng/mg protein, p < 0.05; MMP-13: 10.5±1.4 ng/mg protein, p < 0.01; ADAMTS-4: 7.5±1.0 ng/mg protein, p < 0.05). However, when compared to PRP, Diclofenac was generally less effective in suppressing these specific matrix-degrading enzymes (MMP-1 levels in Diclofenac group were significantly higher than in PRP group, p < 0.05).
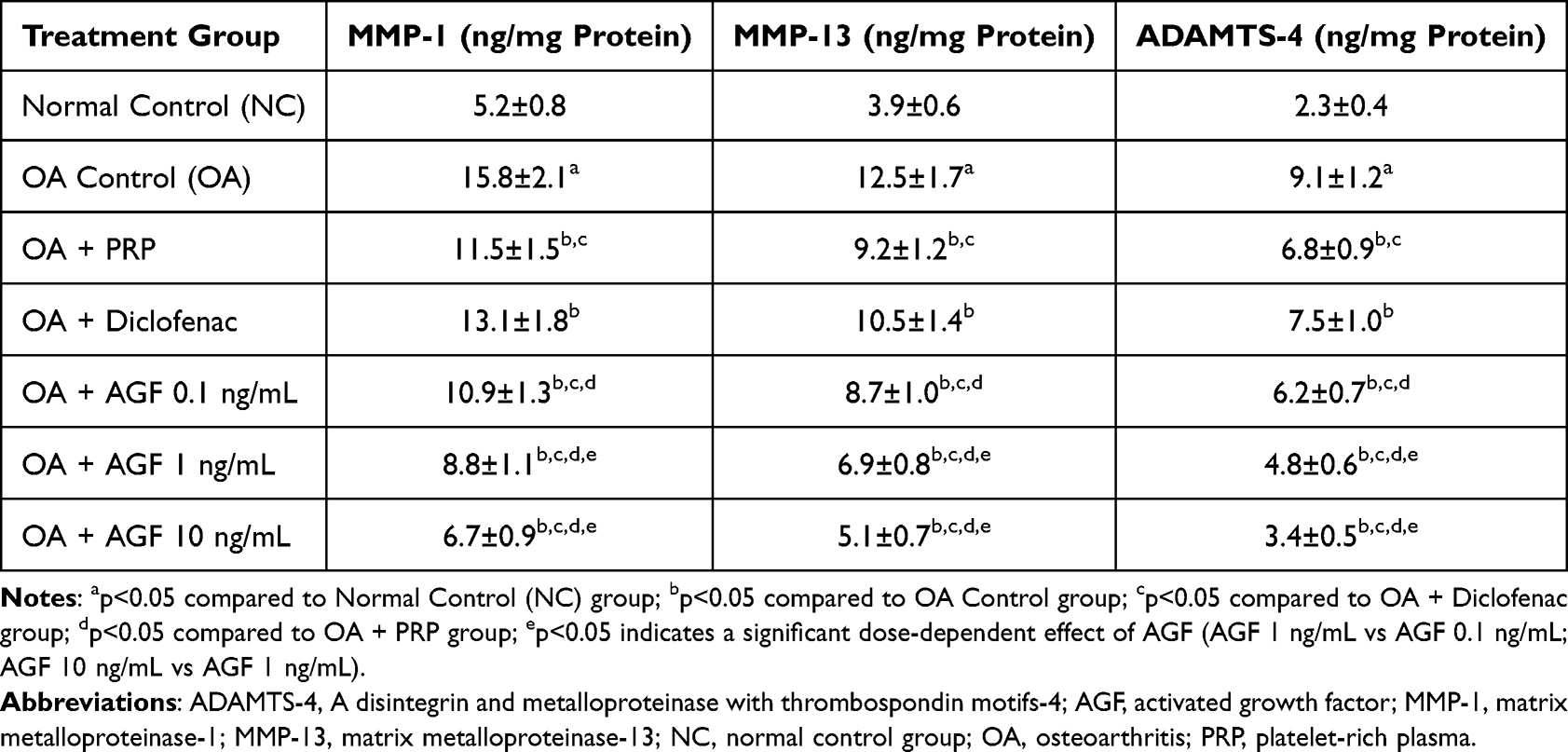 |
Table 1 Effects of Activated Growth Factor, Platelet-Rich Plasma, and Diclofenac on Cartilage Catabolic Enzyme Levels in MIA-Induced Osteoarthritic Rat Knees |
The most profound and consistent anti-catabolic effects were observed with intra-articular AGF treatment. All three tested doses of AGF (standardized by TGF-beta1 content) significantly (p < 0.001 for all AGF doses vs OA Control for all enzymes) reduced the levels of MMP-1, MMP-13, and ADAMTS-4. AGF 0.1 ng/mL effectively reduced MMP-1 to 10.9±1.3 ng/mg protein, MMP-13 to 8.7±1.0 ng/mg protein, and ADAMTS-4 to 6.2±0.7 ng/mg protein. These levels were comparable to or slightly better than those achieved with PRP, and significantly lower than those in the Diclofenac group (p < 0.01 for MMP-1, MMP-13; p < 0.001 for ADAMTS-4, when AGF 0.1 is compared to Diclofenac). AGF 1 ng/mL further amplified the anti-catabolic effect, lowering MMP-1 to 8.8±1.1 ng/mg protein, MMP-13 to 6.9±0.8 ng/mg protein, and ADAMTS-4 to 4.8±0.6 ng/mg protein. These reductions were significantly greater than those achieved by AGF 0.1 ng/mL (p < 0.01 for MMP-1; p < 0.001 for MMP-13, ADAMTS-4), PRP (p < 0.001 for all enzymes), and Diclofenac (p < 0.001 for all enzymes). AGF 10 ng/mL exhibited the most potent inhibitory activity. It reduced MMP-1 to 6.7±0.9 ng/mg protein, MMP-13 to 5.1±0.7 ng/mg protein, and ADAMTS-4 to 3.4±0.5 ng/mg protein. These levels represented a substantial suppression of catabolic activity, significantly outperforming AGF 1 ng/mL (p < 0.001 for all enzymes) and were dramatically lower than PRP and Diclofenac groups (p < 0.001 for all enzymes). Remarkably, enzyme levels in the AGF 10 ng/mL group approached, and in the case of MMP-1 and ADAMTS-4 were not statistically significantly different from those observed in the healthy Normal Control group, indicating a near-normalization of the catabolic enzyme profile.
Table 2 presents the quantitative assessment, via Western blot analysis, of protein expression levels for two critical markers of cartilage anabolic activity: total SMAD3 and the major proteoglycan, aggrecan. In the Normal Control (NC) group, the basal expression levels of SMAD3 and Aggrecan relative to β-actin were established (represented as 1.00±0.12 AU and 1.00±0.10 AU, respectively). Following MIA induction, the OA Control group exhibited a profound and statistically significant (p < 0.001 vs NC) reduction in the expression of both proteins. The SMAD3/β-actin ratio decreased to 0.45±0.07 AU (a 55% reduction), and the Aggrecan/β-actin ratio plummeted to 0.38±0.06 AU (62% reduction). Oral Diclofenac administration (OA + Diclofenac group) failed to significantly alter the suppressed levels of either SMAD3 (0.52±0.08 AU) or Aggrecan (0.45±0.07 AU) compared to the OA Control group (p > 0.05). In contrast, intra-articular PRP treatment (OA + PRP group) demonstrated a clear anabolic effect. It significantly increased both the SMAD3/β-actin ratio (to 0.68±0.09 AU, p < 0.001 vs OA) and the Aggrecan/β-actin ratio (to 0.60±0.08 AU, p < 0.001 vs OA). However, despite this significant improvement over the OA state, the expression levels of both SMAD3 and Aggrecan in the PRP group remained substantially lower than those in the Normal Control group (p < 0.001) and were significantly lower than those achieved with higher AGF doses. AGF 0.1 ng/mL significantly increased SMAD3/β-actin (0.75±0.10 AU) and Aggrecan/β-actin (0.65±0.09 AU) ratios compared to the OA Control group (p < 0.001). These levels were comparable to or slightly higher than those achieved with PRP, and significantly greater than the Diclofenac group (p < 0.001). AGF 1 ng/mL produced a more substantial increase in anabolic markers. The SMAD3/β-actin ratio reached 0.92±0.11 AU, and the Aggrecan/β-actin ratio reached 0.83±0.10 AU. These levels were significantly higher than those in the AGF 0.1 ng/mL group (p < 0.01 for SMAD3, p < 0.001 for Aggrecan) and the PRP group (p < 0.001 for both), indicating a significantly enhanced anabolic response. AGF 10 ng/mL elicited the most dramatic anabolic response. It increased the Aggrecan/β-actin ratio to 0.97±0.11 AU, effectively restoring Aggrecan protein levels to be statistically indistinguishable from the Normal Control group. Intriguingly, the SMAD3/β-actin ratio was elevated to 1.15±0.14 AU, a level significantly higher than not only the AGF 1 ng/mL group (p < 0.001) but also the Normal Control group (p < 0.05). The finding that AGF 10 ng/mL could restore Aggrecan levels and potentially hyper-activate SMAD3 signaling pathways suggests a powerful chondro-inductive and matrix-regenerative potential that surpasses standard PRP formulations and is entirely distinct from the action of NSAIDs.
 |
Table 2 Effects of Activated Growth Factor, Platelet-Rich Plasma, and Diclofenac on SMAD3 and Aggrecan Protein Expression Levels (Normalized to β-Actin) in Cartilage of MIA-Induced Osteoarthritic Rat Knees |
Table 3 presents the quantitative findings from Western Blot analysis assessing the protein expression levels of Collagen Type II (Col2), the principal structural protein providing tensile strength and integrity to articular cartilage. Compared to the Normal Control (NC) group, where the Col2/β-actin ratio was established as the baseline (1.00±0.08 AU), the OA Control group exhibited a highly significant (p < 0.001) decrease in this ratio to 0.33±0.05 AU. This represents approximately a 67% reduction in detectable Col2 protein relative to the normal control group, vividly illustrating the profound impact of the OA pathology on chondrocyte function and their ability to maintain the primary structural framework of the cartilage matrix. The positive control treatments yielded varied results regarding Col2 expression. Oral Diclofenac treatment (OA + Diclofenac group) produced only a minimal, though statistically significant (p < 0.05 vs OA), increase in the Col2/β-actin ratio (to 0.42±0.06 AU). In contrast, intra-articular PRP administration (OA + PRP group) led to a more substantial and statistically significant increase in the Col2/β-actin ratio (to 0.55±0.07 AU; p < 0.001 vs OA, p < 0.001 vs Diclofenac). Nevertheless, the Col2 expression level in the PRP group remained significantly below that of the normal controls (p < 0.001). AGF treatment emerged as the most effective intervention for restoring Col2 protein levels in the osteoarthritic cartilage. All administered doses of AGF significantly increased the Col2/β-actin ratio compared to the OA Control group (p < 0.001) and the Diclofenac group (p < 0.001). A clear dose-response relationship was observed. The lowest dose, AGF 0.1 ng/mL (0.50±0.07 AU), yielded Col2 levels comparable to those in the PRP group. The intermediate dose, AGF 1 ng/mL (0.72±0.09 AU), resulted in significantly higher Col2 expression compared to both AGF 0.1 ng/mL (p < 0.001) and PRP (p < 0.001). The highest dose, AGF 10 ng/mL (0.88±0.10 AU), produced the most substantial increase in Col2 expression, significantly exceeding the levels achieved with AGF 1 ng/mL (p < 0.01) (Figure 2). This level represents a recovery to approximately 88% of the Col2 protein expression observed in healthy, normal cartilage, indicating a potent effect on restoring this critical matrix component.
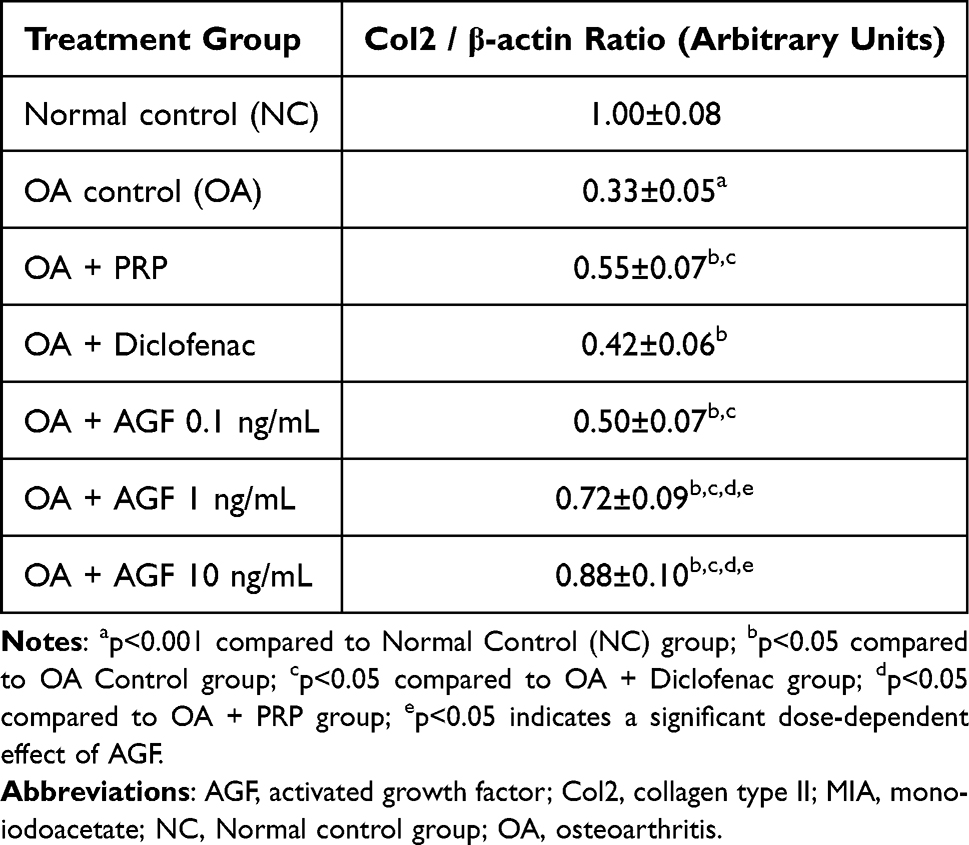 |
Table 3 Effects of Activated Growth Factor, Platelet-Rich Plasma, and Diclofenac on Collagen Type II Protein Expression Levels (Normalized to β-Actin) in Cartilage of MIA-Induced Osteoarthritic Rat Knees |
 |
Figure 2 Representative Western blot images showing the expression of suppressor of mothers against decapentaplegic-3, Aggrecan, Collagen 2 and β-actin in cartilage tissue. Notes: (A) Normal control group, (B) Negative control group, (C) Positive control group (Diclofenac), (D) Positive Control PRP, (E) Activated growth factor (AGF) 0.1 ng/mL group, (F) AGF 1 ng/mL group, (G) AGF 10 ng/mL group. |
Table 4 provides critical insights into the inflammatory milieu of the osteoarthritic joint by quantifying the levels of pivotal pro-inflammatory cytokines, Tumor Necrosis Factor-alpha (TNF-α) and Interleukin-1 beta (IL-1β), as well as the key inflammatory transcription factor Nuclear Factor kappa-B p65 subunit (NF-κB p65), within the synovial tissue. Compared to the basal levels observed in healthy Normal Control (NC) rats (TNF-α: 12.1±2.5 pg/mg protein; IL-1β: 6.8±1.5 pg/mg protein; NF-κB p65: 2.7±0.4 pg/mg protein), the OA Control group exhibited dramatic and highly significant (p < 0.001 for all) increases in TNF-α (to 115.6±15.3 pg/mg protein, ~9.6-fold increase), IL-1β (to 68.3±8.9 pg/mg protein, ~10-fold increase), and NF-κB p65 protein levels (to 10.4±1.2 pg/mg protein, ~3.85-fold increase). Oral Diclofenac sodium (OA + Diclofenac group), significantly reducing TNF-α levels (to 40.5±6.7 pg/mg protein), IL-1β levels (to 23.7±4.1 pg/mg protein), and NF-κB p65 levels (to 5.3±0.8 pg/mg protein) compared to the OA Control group (p < 0.001 for all). Intra-articular PRP treatment (OA + PRP group) also exerted significant anti-inflammatory effects, markedly lowering the levels of TNF-α (75.2±9.8 pg/mg protein), IL-1β (42.1±6.2 pg/mg protein), and NF-κB p65 (7.3±0.9 pg/mg protein) compared to the OA Control group (p < 0.001 for all). However, when compared directly to Diclofenac, PRP was significantly less effective in reducing the levels of all three measured inflammatory mediators (p < 0.001 for all comparisons). AGF 0.1 ng/mL, the lowest dose (83.4±10.1 pg/mg TNF-α; 48.5±7.0 pg/mg IL-1β; 8.0±1.0 pg/mg NF-κB p65) showed significant anti-inflammatory activity but was less effective than both PRP (significantly higher NF-κB p65, p < 0.05) and Diclofenac (p < 0.001 for all markers). AGF 1 ng/mL, this intermediate dose demonstrated substantially greater anti-inflammatory efficacy than AGF 0.1 ng/mL (p < 0.001) and PRP (p < 0.001). It reduced inflammatory markers (55.8±7.2 pg/mg TNF-α; 30.9±5.3 pg/mg IL-1β; 5.7±0.8 pg/mg NF-κB p65) to levels that, while still significantly higher than Diclofenac for TNF-α (p < 0.01) and IL-1β (p < 0.05), approached the efficacy of Diclofenac for NF-κB p65 reduction (no significant difference). AGF 10 ng/mL, the highest AGF dose exerted the most powerful anti-inflammatory effect among the biological treatments. It dramatically reduced TNF-α (33.7±5.1 pg/mg protein), IL-1β (18.2±3.3 pg/mg protein), and NF-κB p65 (4.5±0.7 pg/mg protein) levels. These levels were significantly lower than those achieved with AGF 1 ng/mL (p < 0.001) and PRP (p < 0.001). Crucially, the levels of TNF-α, IL-1β, and NF-κB p65 in the AGF 10 ng/mL group were statistically comparable (p > 0.05) to those achieved with the potent systemic NSAID, Diclofenac.
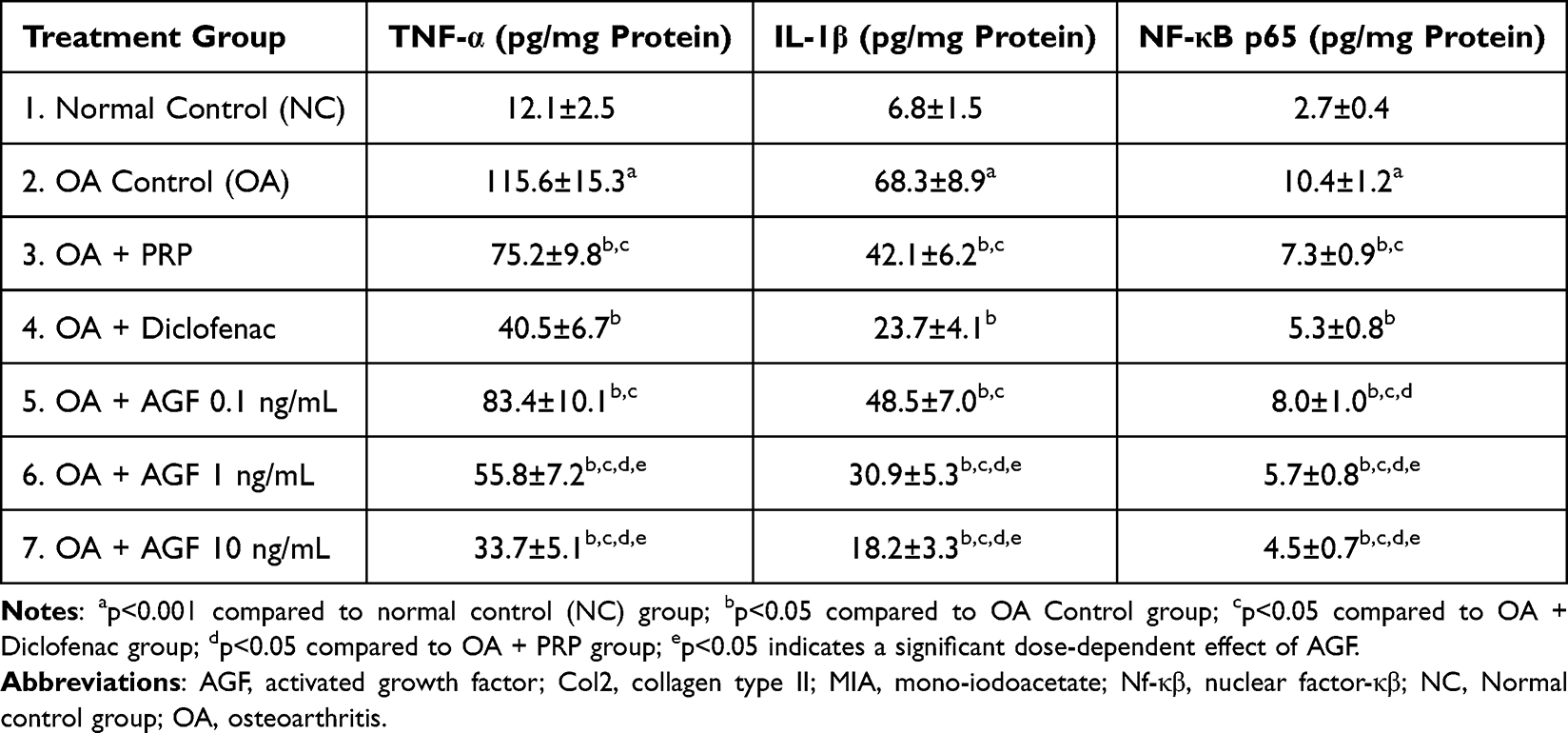 |
Table 4 Effects of Activated Growth Factor, Platelet-Rich Plasma, and Diclofenac on Pro-Inflammatory Cytokine and NF-κB Levels in Synovial Tissue of MIA-Induced Osteoarthritic Rat Knees |
Table 5 presents a crucial tissue-level assessment of the overall joint health, integrating quantitative measures of both articular cartilage degradation and synovial inflammation. The data clearly confirm the successful induction of severe OA in the OA Control group. This group exhibited a markedly high mean OARSI score of 18.7±2.2 (on a 0–24 scale, where higher scores denote greater pathology), which was profoundly elevated compared to the minimal score of 0.5±0.3 observed in the Normal Control (NC) group (p < 0.001). Complementing the cartilage damage, the OA Control group also presented with extensive synovial inflammation, evidenced by a mean inflammatory cell infiltration of 70.5±8.2%, dramatically increased from the basal infiltration of 3.1±0.8% in NC joints (p < 0.001). As a potent NSAID, Diclofenac was highly effective in mitigating synovial inflammation, significantly reducing inflammatory cell infiltration to 20.2±4.5% (p < 0.001 vs OA Control). This underscores its known anti-inflammatory action. However, its impact on cartilage structural integrity, as measured by the OARSI score (14.8±1.9), was less substantial. While significantly better than the OA Control (p < 0.01), the cartilage protection offered by Diclofenac was limited, suggesting that controlling inflammation alone is insufficient to fully prevent structural degradation. Intra-articular PRP demonstrated a more pronounced chondroprotective effect compared to Diclofenac, achieving a significantly lower (better) OARSI score of 12.5±1.8 (p < 0.01 vs Diclofenac). However, PRP was notably less effective than Diclofenac in controlling synovial inflammatory infiltration (45.3±6.1%; p < 0.001 vs Diclofenac), indicating a more localized or less potent broad anti-inflammatory action on the synovium compared to the systemic NSAID. AGF 0.1 ng/mL offered cartilage protection (OARSI score 13.1±1.7) that was comparable to PRP and slightly superior to Diclofenac (p < 0.05). However, its efficacy in reducing synovial infiltration (50.1±7.3%) was limited, being similar to PRP and significantly inferior to Diclofenac (p < 0.001). AGF 1 ng/mL resulted in substantial improvements. The OARSI score was significantly reduced to 9.2±1.5, indicating considerably better cartilage protection than AGF 0.1 ng/mL (p < 0.001), PRP (p < 0.001), and Diclofenac (p < 0.001). Synovial infiltration was also significantly reduced to 28.9±5.0%, an improvement over AGF 0.1 ng/mL (p < 0.001) and PRP (p < 0.001), though infiltration remained significantly higher than with Diclofenac (p < 0.01). AGF 10 ng/mL yielded the most comprehensive and striking histopathological benefits. It achieved outstanding cartilage protection, with the OARSI score decreasing to 5.8±1.1. This was significantly superior to all other treatment groups, including AGF 1 ng/mL (p < 0.001), and approached the near-normal cartilage integrity observed in the NC group. Simultaneously, AGF 10 ng/mL demonstrated potent control of synovial inflammation, reducing infiltration to 15.7±3.6%. This level of inflammatory suppression was statistically comparable (p > 0.05) to that achieved by Diclofenac and was significantly better than that of PRP (p < 0.001) and lower AGF doses (p < 0.001).
 |
Table 5 Effects of Activated Growth Factor, Platelet-Rich Plasma, and Diclofenac on Histopathological Parameters of Cartilage Degradation and Synovial Inflammation in MIA-Induced Osteoarthritic Rat Knees |
Figure 3 is described as presenting representative images of Hematoxylin and Eosin (H&E) stained knee joint sections, magnified at 400x, to visualize inflammatory cell infiltration in chondrocytes and surrounding synovial tissue across all seven experimental groups. The figure caption indicates arrows pointing to inflammatory cell infiltrates. Normal Control (Panel A) would be expected to show normal synovial lining with minimal to no inflammatory cell presence, corresponding to the low percentage of infiltration (3.1±0.8%) noted in Table 5. OA Control (Panel B) would vividly display the effects of MIA induction, showing extensive infiltration of inflammatory cells (likely lymphocytes, macrophages, and neutrophils) within the synovial membrane and potentially extending into the cartilage. This visual would correspond to the high infiltration percentage (70.5±8.2%) in Table 5. Chondrocytes might appear stressed or reduced in number, with evidence of cartilage surface irregularities. OA + Diclofenac (Panel C) Compared to Panel B, this image show a marked reduction in the density and extent of inflammatory cell infiltration in the synovium, aligning with the quantitative data (20.2±4.5% infiltration) and reflecting Diclofenac’s potent anti-inflammatory effect. OA + PRP (Panel D) likely show a noticeable reduction in inflammatory cells compared to the OA control (Panel B), but the infiltration would still be more apparent than in the Diclofenac group (Panel C), supporting the quantitative data of 45.3±6.1% infiltration. Panel E (AGF 0.1 ng/mL) show a reduction compared to OA control, but likely substantial remaining inflammation, similar to or slightly worse than PRP, corresponding to 50.1±7.3% infiltration. Panel F (AGF 1 ng/mL) exhibit a more significant reduction in inflammatory cells (28.9±5.0% infiltration) compared to AGF 0.1 ng/mL and PRP, though still more than Diclofenac. Panel G (AGF 10 ng/mL) display the most pronounced reduction in inflammatory infiltrate among the AGF groups, appearing visually similar to or even better than the Diclofenac group (Panel C), corresponding to the 15.7±3.6% infiltration value in Table 5.
 |
Figure 3 Representative images of histopathological staining (H&E) of knee joints showing inflammatory cell infiltration in chondrocytes. Notes: (A) Normal control group, (B) Negative control group, (C) Positive control group (Diclofenac), (D) Positive Control PRP, (E) Activated growth factor (AGF) 0.1 ng/mL group, (F) AGF 1 ng/mL group, (G) AGF 10 ng/mL group. Arrow showed infiltration of inflammatory cell in chondrocyte. Magnification 400x. |
Figure 4 is described as showcasing Safranin O-Fast Green stained knee joint sections at 400x magnification, illustrating cartilage morphology across the same seven experimental groups. Normal Control (Panel A) exhibit intense and uniform orange-red Safranin O staining throughout the articular cartilage, indicating abundant proteoglycan content. The cartilage surface would be smooth, chondrocytes well-distributed within their lacunae, and the overall structure intact, corresponding to the low OARSI score (0.5±0.3) in Table 5. OA Control (Panel B) show a dramatic loss of Safranin O staining, particularly in the superficial and middle zones, indicating severe proteoglycan depletion. The cartilage surface would likely appear fibrillated and irregular, with chondrocyte disorganization, clustering, or loss. These features would visually represent the high OARSI score (18.7±2.2). OA + Diclofenac (Panel C), some mild improvement in Safranin O staining might be visible compared to Panel B, but significant proteoglycan loss and structural damage would likely persist, reflecting the moderate OARSI score of 14.8±1.9. OA + PRP (Panel D) likely display better Safranin O staining and cartilage structure compared to Diclofenac (Panel C), showing more proteoglycan retention and smoother surfaces, aligning with the better OARSI score of 12.5±1.8. Panel E (AGF 0.1 ng/mL) would show some improvement in Safranin O staining and cartilage integrity compared to OA control, likely similar to or slightly better than PRP, corresponding to an OARSI score of 13.1±1.7. Panel F (AGF 1 ng/mL) would exhibit a more substantial restoration of Safranin O staining and improved cartilage surface and cellularity, reflecting a significantly better OARSI score of 9.2±1.5. Panel G (AGF 10 ng/mL) would display the most impressive results, with intense Safranin O staining approaching that of the Normal Control group (Panel A), a smoother articular surface, and healthier chondrocyte appearance. This visual evidence would strongly corroborate the significantly reduced OARSI score of 5.8±1.1, indicating substantial chondroprotection and potential matrix repair.
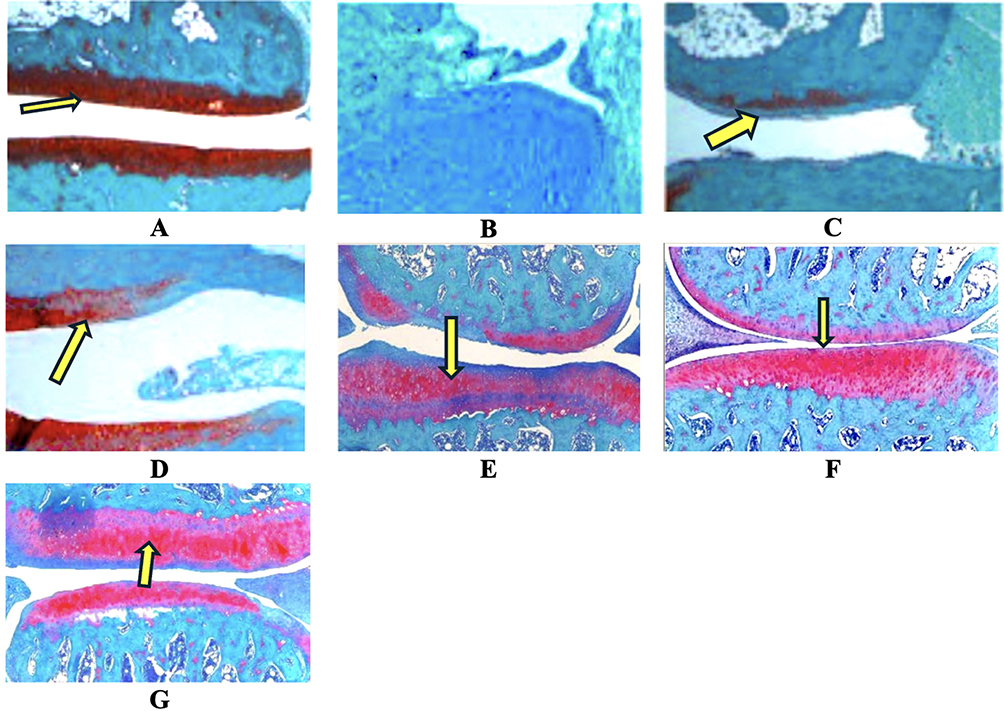 |
Figure 4 Histopathology staining (Safranin O-Fast Green) of knee joints showing cartilage morphology. (A) Normal control group, (B) Negative control group, (C) Positive control group (Diclofenac), (D) Positive Control platelet-rich plasma (PRP), (E) Activated growth factor (AGF) 0.1 ng/mL group, (F) AGF 1 ng/mL group, (G) AGF 10 ng/mL group. Arrow showed cartilage in knee joints. Magnification 400x. |
Discussion
The present study provides compelling evidence that intra-articular administration of Activated Growth Factor (AGF), a concentrated platelet-derived biologic, exerts significant therapeutic effects in a monoiodoacetate (MIA)-induced rat model of osteoarthritis (OA). The observed benefits span multiple facets of OA pathology, including a marked reduction in catabolic enzyme activity, a robust stimulation of anabolic matrix synthesis pathways, potent attenuation of pro-inflammatory mediators (which themselves are key drivers of catabolism), and substantial improvements in overall joint histopathology. These findings collectively underscore the potential of AGF to restore homeostasis within the osteoarthritic joint by addressing the fundamental imbalance between anabolic and catabolic processes that characterize this debilitating disease. A cardinal feature of OA is the progressive degradation of the articular cartilage extracellular matrix (ECM), orchestrated by an overactive catabolic arm and a compromised anabolic response from chondrocytes.12,13 This study demonstrated AGF’s profound ability to favorably shift this balance.
The significant elevation of matrix metalloproteinases MMP-1 and MMP-13 (collagenases) and ADAMTS-4 (an aggrecanase) in the OA control group reflects the intense enzymatic assault on the cartilage ECM. MMP-1 and MMP-13 are primarily responsible for the cleavage of type II collagen, the principal structural protein conferring tensile strength to cartilage, while ADAMTS-4 degrades aggrecan, the major proteoglycan crucial for cartilage’s compressive resilience and hydration. The dose-dependent reduction in these enzymes by AGF, particularly the near-normalization achieved with 10 ng/mL AGF, signifies a potent anti-catabolic effect. This inhibition is likely mediated by the complex interplay of growth factors within AGF. For instance, transforming growth factor-β1 (TGF-β1), used for AGF standardization in this study, is known to suppress the expression of certain MMPs and induce the production of their natural inhibitors, tissue inhibitors of metalloproteinases (TIMPs).14 By curtailing the activity of these catabolic enzymes, AGF directly mitigates the primary drivers of matrix erosion, thereby preserving the existing cartilage architecture. This effect was notably superior to that observed with illustrative platelet-rich plasma (PRP) and the predominantly anti-inflammatory diclofenac sodium, highlighting AGF’s targeted impact on matrix-degrading pathways.
Beyond its anti-catabolic actions, AGF robustly stimulated anabolic processes essential for matrix repair. The OA control group exhibited significantly reduced SMAD3 protein levels and subsequent aggrecan expression, indicative of suppressed anabolic signaling.15 SMAD3 is a critical intracellular transducer of the TGF-β signaling pathway, which, upon activation, promotes the transcription of genes encoding key ECM components like aggrecan and type II collagen (Col2).16 AGF treatment, especially at 1 and 10 ng/mL, markedly increased SMAD3 protein levels (and implicitly its activation, as suggested by increased downstream products) and substantially restored aggrecan protein content in the cartilage. Similarly, Col2 protein expression, which was severely depleted in OA cartilage, was significantly and dose-dependently upregulated by AGF. The 10 ng/mL AGF dose nearly normalized aggrecan levels and brought Col2 expression to approximately 88% of normal, demonstrating a powerful chondro-anabolic drive. This potent stimulation of matrix synthesis by AGF significantly surpassed the anabolic effects of PRP, supporting the notion that the activated and concentrated growth factor profile in AGF elicits a more robust chondrogenic response. The pathomechanism likely involves AGF-derived TGF-β1 and other synergistic growth factors (IGF-1, PDGF) binding to their respective chondrocyte receptors, initiating intracellular signaling cascades that converge on the nucleus to enhance the transcription and translation of ACAN and COL2A1 genes.17
Inflammation within the OA joint, particularly synovitis, is no longer considered a mere secondary phenomenon but an active contributor to cartilage degradation and disease progression. Pro-inflammatory cytokines like TNF-α and IL-1β are pivotal in this process, as they not only mediate pain and joint swelling but also potently stimulate chondrocytes to produce MMPs and ADAMTS, inhibit matrix synthesis and can induce chondrocyte apoptosis.18 This study revealed that AGF treatment effectively quells this catabolic inflammatory fire. The substantial elevation of TNF-α, IL-1β, and NF-κβ p65 protein in the synovial tissue of the OA control group confirms a significant inflammatory state. AGF, particularly at the 10 ng/mL dose, dramatically reduced the levels of these inflammatory mediators, achieving an efficacy comparable to the potent NSAID diclofenac. This anti-inflammatory action is crucial because NF-κβ is a master transcription factor that, once activated by TNF-α or IL-1β, drives the expression of a plethora of genes encoding inflammatory cytokines, chemokines, and matrix-degrading enzymes.19 By inhibiting NF-κβ activation and reducing the upstream cytokines, AGF effectively disrupts this pro-inflammatory and pro-catabolic signaling cascade. This effect was also superior to PRP, suggesting a more potent immunomodulatory capacity of AGF. The mechanisms likely involve specific growth factors in AGF (such as TGF-β1 has complex immunomodulatory roles, and other factors may possess direct anti-inflammatory properties) interfering with cytokine receptor signaling or promoting the expression of anti-inflammatory molecules. This reduction in the catabolic stimuli originating from inflammation complements AGF’s direct anabolic effects on chondrocytes. TGF-β signaling, delivered by AGF, orchestrates a masterful restoration of joint homeostasis through a dual-action mechanism. It initiates a powerful anabolic cascade via the canonical SMAD pathway, where activated SMAD complexes drive nuclear transcription of collagen and aggrecan to rebuild the cartilage matrix. Simultaneously, this pathway launches an anti-catabolic assault, repressing destructive MMP genes while inducing SMAD7 to inhibit the entire NF-κB inflammatory axis. This masterfully traps NF-κB in the cytoplasm, silencing the production of TNF-α and IL-1β, and decisively shifting the joint environment from degradation back to regeneration.
The molecular benefits of AGF treatment translated into significant improvements at the tissue level, as evidenced by the histopathological assessments and their visual. The OA control group displayed severe cartilage degradation (high OARSI score) and extensive synovial inflammatory cell infiltration. AGF treatment, especially at 1 and 10 ng/mL, led to a remarkable reduction in the OARSI score, indicating significantly preserved cartilage structure, cellularity, and proteoglycan content. The chondroprotective effect of AGF 10 ng/mL was superior to both PRP and Diclofenac, approaching near-normal cartilage integrity. This structural preservation is a direct consequence of the dual action of AGF: inhibiting matrix-degrading enzymes and stimulating the synthesis of new matrix components like aggrecan and Col2. The H&E staining described in our study visually confirm the quantitative data regarding synovial inflammatory cell infiltration. AGF 10 ng/mL significantly reduced this infiltration to levels comparable to diclofenac, demonstrating its potent local anti-inflammatory effect. This reduction in synovial inflammation is critical, as it lessens the source of catabolic cytokines that attack the cartilage. The comparative efficacy observed histopathologically further strengthens the case for AGF. While diclofenac primarily controlled synovial inflammation with limited direct chondroprotection, and PRP offered moderate chondroprotection but less effective control of synovitis, AGF 10 ng/mL uniquely provided both superior cartilage structural preservation and potent anti-inflammatory effects on the synovium.
The therapeutic efficacy of AGF aligns with the theory that providing a concentrated medley of endogenous growth factors can shift a degenerative local environment towards regeneration and homeostasis.20 Platelets are natural reservoirs of a wide array of signaling molecules crucial for tissue repair.20 The AGF preparation process, involving controlled activation, aims to maximize the bioavailability of these factors (TGF-β1, PDGF, IGF-1, FGF, VEGF) at the site of injury.21 These factors do not act in isolation but likely synergize through complex cross-talk between their respective signaling pathways to orchestrate the observed anti-catabolic, pro-anabolic, and anti-inflammatory responses. The dose-dependency observed suggests that achieving certain threshold concentrations of these GFs is necessary to overcome the pathological signaling in OA and effectively engage chondrocyte and synoviocyte receptors to elicit a robust therapeutic response. The ability of AGF to influence multiple pathological pathways simultaneously—matrix degradation, impaired synthesis, and inflammation—points towards its potential to restore a more balanced, homeostatic state within the entire joint organ, rather than merely targeting a single symptom or pathway. This holistic approach is theoretically more likely to yield disease-modifying effects. The results of this study robustly demonstrate that AGF effectively counteracts the core pathological processes of OA. By potently inhibiting catabolic enzyme activity, stimulating anabolic matrix production through pathways like SMAD3, and significantly attenuating the synovial inflammation that fuels cartilage degradation, AGF treatment led to substantial improvements in molecular markers and overall joint histopathology. The observed dose-dependent efficacy, particularly the superior performance of higher AGF concentrations compared to illustrative PRP and the distinct, broader benefits compared to diclofenac, positions AGF as a highly promising biological therapeutic strategy with true disease-modifying potential for osteoarthritis.
Building on these compelling pre-clinical results, future research must focus on translating AGF therapy to the clinic. This necessitates validation in larger animal models simulating post-traumatic OA, followed by randomized controlled human trials to establish long-term safety and efficacy. Mechanistically, proteomic and transcriptomic analyses could fully map AGF’s signaling impact beyond the markers studied here. Furthermore, investigating advanced delivery systems, such as a hydrogel carrier for sustained release, is critical to optimize therapeutic durability, potentially reducing injection frequency and enhancing long-term joint regeneration and functional recovery in patients.
Conclusion
AGF treatment demonstrated a significant capacity to shift the disturbed equilibrium in osteoarthritic cartilage from a net catabolic state towards an anabolic, reparative state. AGF, in a dose-dependent manner, significantly reduced the expression and levels of key cartilage-degrading enzymes MMP-1, MMP-13, and ADAMTS-4. AGF significantly reduced the levels of pro-inflammatory cytokines TNF-α and IL-1β, and suppressed NF-κB activation within the joint. AGF treatment resulted in significant improvements in overall cartilage structure (reduced OARSI scores) and a marked decrease in synovial inflammatory cell infiltration, surpassing the effects of PRP and, in terms of structural protection, diclofenac. Compared to the illustrative effects of a standard PRP preparation, AGF generally exhibited enhanced efficacy across anabolic, anti-catabolic, and anti-inflammatory parameters, supporting its investigation as an advanced and potentially more potent biological therapy. This study’s findings carry profound clinical implications for osteoarthritis management. AGF’s dual capacity to potently suppress inflammation while simultaneously driving robust matrix synthesis positions it as a leading candidate for a true Disease-Modifying Osteoarthritis Drug (DMOAD). By significantly outperforming both standard PRP and NSAIDs in restoring joint homeostasis, AGF represents a potential paradigm shift from purely symptomatic relief. This presents a tangible therapeutic strategy to not only halt cartilage degradation but potentially regenerate damaged tissue, offering hope for improved functional outcomes and delaying the need for joint replacement surgery in patients.
Acknowledgments
The authors are grateful for the laboratory support by Eureka Research Laboratory, Palembang, Indonesia.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhang H, Li D, Zheng W, et al. Enhancing cartilage repair in osteoarthritis using platelet lysates and arthroscopic microfracture. Drug Design Dev Ther. 2025;19:3827–3843. doi:10.2147/DDDT.S502935
2. Primorac D, Molnar V, Rod E, et al. Knee osteoarthritis: a review of pathogenesis and state-of-the art non-operative therapeutic considerations. Genes. 2020;11(8):854. doi:10.3390/genes11080854
3. Dharmayuda CGO, Subawa AAN, Kawiyana KS, et al. The role of proteinases in osteoarthritis: a brief review of new potent cartilage metabolism therapeutic target. Hong Kong Orthoped Res. 2021;4(2):35–38. doi:10.37515/ortho.8231.4204
4. Bhutada S, Hoyle A, Piuzzi NS, et al. Degradomics defines proteolysis information flow from human knee osteoarthritis cartilage to matched synovial fluid and the contributions of secreted proteases ADAMTS5, MMP13 and CMA1 to articular cartilage breakdown. Osteoarthritis Cartilage. 2025;33(1):116–127. doi:10.1016/j.joca.2024.09.002
5. Adam MS, Zhuang H, Ren X, et al. The metabolic characteristics and changes of chondrocytes in vivo and in vitro in osteoarthritis. Front Endocrinol. 2024;15:1393550. doi:10.3389/fendo.2024.1393550
6. Magni A, Agostoni P, Bonezzi C, et al. Management of osteoarthritis: expert opinion on NSAIDs. Pain Ther. 2021;10(2):783–808. doi:10.1007/s40122-021-00260-1
7. Wang W, Niu Y, Jia Q. Physical therapy as a promising treatment for osteoarthritis: a narrative review. Front Physiol. 2022;13:1011407. doi:10.3389/fphys.2022.1011407
8. Moldovan F. Role of serum biomarkers in differentiating periprosthetic joint infections from aseptic failures after total Hip arthroplasties. J Clin Med. 2024;13(19):5716. doi:10.3390/jcm13195716
9. Di Tolla MF, Romano S, Vasetti P, et al. Platelet-derived growth factor as biomarker of clinical outcome for autologous platelet concentrate therapy in grade I knee osteoarthritis. Biologics Targets Ther. 2025;19:137–147. doi:10.2147/BTT.S500522
10. Amin R, Hidayat R, Maritska Z, et al. Activated growth factor from platelets as treatment for diabetic retinopathy through antioxidant-oxidative stress pathway. Diabetes Met Synd Obes. 2025;18:305–313. doi:10.2147/DMSO.S490055
11. Du Sert NP, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi:10.1371/journal.pbio.3000410
12. Maritska Z, Hidayat R, Purnamasari S, et al. The effect of activated growth factor (AGF) on alpha-SMA levels in osteoarthritis. Biosci Med J Biomed Translat Res. 2024;8(5):4330–4335.
13. Hu K, Song M, Song T, et al. Osteoimmunology in osteoarthritis: unraveling the interplay of immunity, inflammation and joint degeneration. J Inflam Res. 2025;18:4121–4142. doi:10.2147/JIR.S514002
14. Deng Z, Fan T, Xiao C, et al. TGF-β signaling in health, disease and therapeutics. Signal Transduct Target Ther. 2024;9:61.
15. Xiang W, Wang C, Zhu Z, et al. Inhibition of SMAD3 effectively reduces ADAMTS-5 expression in the early stages of osteoarthritis. BMC Musculoskeletal Dis. 2023;24:130. doi:10.1186/s12891-022-05949-8
16. Miyazawa K, Itoh Y, Fu H, et al. Receptor-activated transcription factors and beyond: multiple modes of Smad2/3-dependent transmission of TGF-β signaling. J Biol Chem. 2024;300(5):107256. doi:10.1016/j.jbc.2024.107256
17. Lin S, Li H, Wu B, et al. TGF-β1 regulates chondrocyte proliferation and extracellular matrix synthesis via circPhf21a-Vegfa axis in osteoarthritis. Cell Comm Signal. 2022;20:75. doi:10.1186/s12964-022-00881-9
18. Zhang X, Hsueh M-F, Huebner JL, et al. TNF-α carried by plasma extracellular vesicles predicts knee osteoarthritis progression. Front Immunol. 2021;12:758386. doi:10.3389/fimmu.2021.758386
19. Kitaura H, Marahleh A, Ohori F, et al. Role of interaction of tumor necrosis factor-α and tumor necrosis factor receptors 1 and 2 in bone-related cells. Int J Mol Sci. 2022;23(3):1481. doi:10.3390/ijms23031481
20. Jansen EE, Braun A, Jansen P, et al. Platelet-therapeutics to improve tissue regeneration and wound healing-physiological background and methods of preparation. Biomedicines. 2021;9(8):869. doi:10.3390/biomedicines9080869
21. Wang Y, Li J. Current progress in growth factors and extracellular vesicles in tendon healing. Int Wound J. 2023;20(9):3871–3883. doi:10.1111/iwj.14261
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.