Back to Journals » Journal of Inflammation Research » Volume 15
Acquired Hemophilia Associated with Rheumatic Diseases: A Case-Based Systematic Review
Authors Tang Q, Liao J ![]() , Xie X
, Xie X ![]()
Received 4 April 2022
Accepted for publication 19 July 2022
Published 3 August 2022 Volume 2022:15 Pages 4385—4393
DOI https://doi.org/10.2147/JIR.S369288
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Qi Tang, Jiafen Liao, Xi Xie
Department of Rheumatology and Immunology, the Second Xiangya Hospital, Central South University, Changsha, People’s Republic of China
Correspondence: Xi Xie, Department of Rheumatology and Immunology, the Second Xiangya Hospital, Central South University, Changsha, 410011, People’s Republic of China, Tel/Fax +86 0731 8529 5255, Email [email protected]
Abstract: To strengthen the understanding of rheumatic diseases (RDs) as the most common underlying conditions associated with acquired hemophilia (AH), a potentially fatal bleeding condition due to the development of autoantibodies or inhibitors to coagulation factor VIII, and rarely to factor IX, here we presented two cases of RDs associated AH to elucidate the disease progression, treatment, and prognosis. The presented 2 cases showed good responses to glucocorticoid (GC) and immunosuppressive agents. And then, a case-based systematic review was conducted to better understand the clinically practiced diagnosis and treatment of RDs associated AH. A total of 14 articles were included in the final literature review. All the identified 14 patients with underlying RDs and AH presented with bleeding symptoms, increased APTT, decreased FVIII activity, and positive FVIII inhibitors. Twelve of the 14 patients (85.7%) started an eradication of autoantibodies treatment with GC and immunosuppressive agents. Among which six patients achieved partial or complete remission, and four patients (28.6%) switched to Rituximab and responded well. Nine of the 14 patients received hemostasis therapy, including recombinant human FVIIa (rFVIIa). Two patients (14.3%) died due to mass bleeding and key organ failure. AH should be highly suspected in patients with RDs presenting spontaneous mucocutaneous or internal bleeding and an isolated prolonged APTT. Given the high morbidity of AH, it is important to facilitate efficient and proper management.
Keywords: acquired hemophilia, systemic lupus erythematosus, Sjogren syndrome, rheumatoid arthritis, connective tissue disease
Introduction
Acquired hemophilia (AH) is a potentially fatal bleeding condition due to the development of autoantibodies or inhibitors to a coagulation factor, usually factor VIII (FVIII), and seldom factor IX (FIX). It occurs with a low incidence rate of approximately 0.2–1.0 per 1 million people per year and with a mortality rate of about 5–10% as a direct result of FVIII inhibitors.1,2 The autoantibodies against specific domains of the FVIII/FIX molecules impair their function of binding, resulting in bleeding into the skin, muscles, gastrointestinal tracts, and other sites.2 The most common underlying conditions associated with AH are malignant tumors, infections, and rheumatic diseases (RDs).1 Although rare, AH has a high fatality, and it poses a great challenge for clinicians to diagnose timely and treat properly. Since autoimmune diseases are among the most common underlying conditions associated with acquired FVIII inhibitors, it is important that the clinician managing patients with RDs be familiar with the diagnosis and treatment of RDs associated AH. Here, we share our own experiences and make a concise and in-depth review to better understand the diagnosis and treatment of RDs associated AH.
Case Presentation
Patient 1
A 60-year-old woman complained of ecchymosis over limbs for 10 days. She had repeated attacks of oral ulcers, alopecia, mouth and eye dryness over the past year. Physical examination showed a large amount of subcutaneous hemorrhage. Her right buttock and left thigh were painful with swelling, which was suspected to be a hematoma (Figure 1A). Complete blood count (CBC) showed decreased Hemoglobin (HGB) 44g/L, and Platelet (PLT) 78*10^9/L. Routine urine analysis (RUA) revealed proteinuria (+) and microscopic hematuria (++). Complement 3 (C3) decreased to 0.39 g/L and immunoglobulin ((Ig) G elevated to 19.80g/L. Autoantibodies showed ANA1: 320+, anti-U1RNP+++, anti-SSA+, anti-Ro-52+++, AMA-M2+, anti-3E/BPO+. Coagulation tests revealed prolonged APTT as 108.6 sec, decreased FVIII activity as 0.5% (reference range: 50–150%), and positive Anti-FVIII antibody. Schirmer test 3mm/5 min. Magnetic resonance imaging (MRI) scanning showed occupying lesion (see white arrows) deep under the right gluteus Maximus (Figure 1B). The patient was diagnosed with systemic lupus erythematosus (SLE) (based on 2019 EULAR/ACR classification criteria for SLE), secondary Sjogren Syndrome (SS) and AH. She was treated with 400 mg/kg/day intravenous immunoglobulin (IVIG) for 5 days, 2 mg/kg/day intravenous (IV) methylprednisolone (mPSN), oral hydroxychloroquine (HCQ) 200 mg Bid, cyclosporin A (CsA) 50 mg Bid, and IV cyclophosphamide (CTX) 600 mg in total, accompanied with a supplement of washing red cells (WRCs) and activated prothrombin complex concentrate (APCC). After 15 days of aggressive therapy, she felt better and bleeding regressed, although her lab examinations still demonstrated a lower HGB 42g/L, prolonged APTT 110.7 sec, and decreased FVIII activity of 0.6%. After discharge, she continued to receive oral mPSN gradually tapered from 40mg/d to 8mg/d over six months, with hydroxychloroquine (HCQ) at 200 mg Bid and cyclosporin A (CsA) at 50 mg Bid applied together during the same period. At three months after discharge, she improved significantly with HGB 98g/L, APTT 53.5 sec, and C3 0.56g/L. After six months since discharge, her lab examinations returned to normal range: HGB 110g/L, APTT 26.7 sec, C3 0.77 g/L. There was no relapse during the 2-year follow-up.
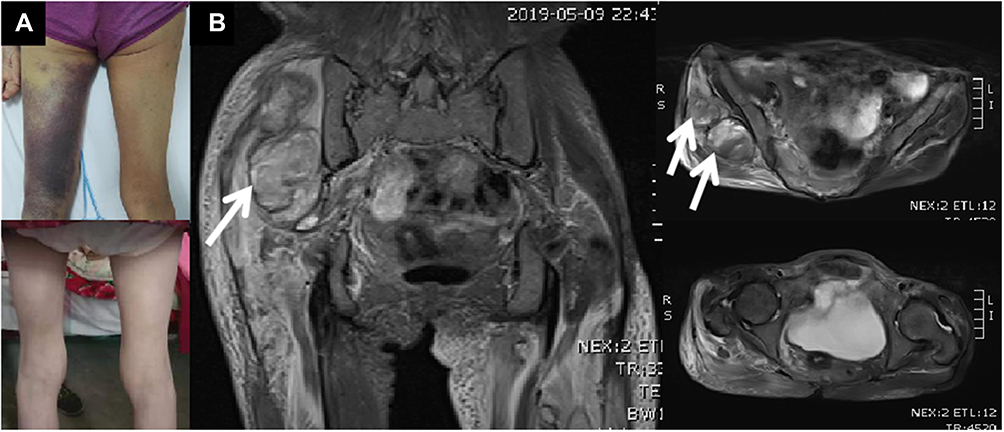 |
Figure 1 (A): The patient’s right buttock and left thigh was obviously enlarged. There was ecchymosis over left thigh (above). The ecchymosis disappeared after 3 months (below). (B): T2 weighted imaging of magnetic resonance imaging showed occupying lesion (probably hematoma, see white arrows) deep under the right gluteus maximus, and the signal of muscles and other soft tissues around the pelvis and femur increased widely. |
Patient 2
A 67-year-old man presented with ecchymosis over limbs for 1 month. His CBC showed a decreased level of HGB 87 g/L. Autoantibodies revealed ANA (1:320)+, anti-SSA++, anti-Ro-52+, anti-Sp100+, anti-gp210+. Coagulation tests revealed prolonged APTT 76.3 sec, decreased FVIII activity 2.5%, and positive anti-FVIII antibody. He was diagnosed with AH associated with undifferentiated connective tissue disease (UCTD). He received IV mPSN 1mg/kg/day and CTX 600 mg as initial therapy, and then continued to receive oral mPSN gradually tapered from 40mg/d to 8mg/d and HCQ at 200 mg Bid as maintenance therapy after discharge. Hemorrhagic manifestations subsided considerably during the 1-year follow-up.
Systematic Review
In order to obtain a better and deeper understanding of RDs associated with AH, we searched PUBMED and EMBASE from Jan 1st, 2010 to Dec 31, 2021 using the following keywords: acquired hemophilia, connective tissue disease, rheumatic diseases, rheumatoid arthritis, systemic lupus erythematosus. The detailed searching strategies and PRISMA flow chart are seen in Figure 2. Thirty-five articles resulted from PUBMED and 64 from EMBASE. After excluding reviews, duplicates, and articles with too little clinical data, a total of 14 articles were included in the final literature review.3–16 Detailed characteristics were listed in Table 1. SLE (7/14) and rheumatoid arthritis (RA) (4/14) were the most common concurrent RDs. All 14 patients presented with bleeding symptoms, increased APTT, decreased FVIII activity, and positive FVIII inhibitors. Most patients (92.9%) showed negative antiphospholipid antibody and lupus anticoagulant. Twelve of the 14 patients (85.7%) started treatment with GC and immunosuppressive agents (CTX and CsA were the most commonly used) with/without IVIG, with good responses. Four patients (28.6%) switched to B cell-targeted biologic therapy with Rituximab, and one patient (7.1%) switched to IL-6 targeted therapy with tocilizumab. One patient received concomitant plasmapheresis. Nine patients (64.3%) received hemostasis therapy such as recombinant FVIIa (rFVIIa) or APCC. Two patients (14.3%) died due to mass bleeding and multiple organ failures.
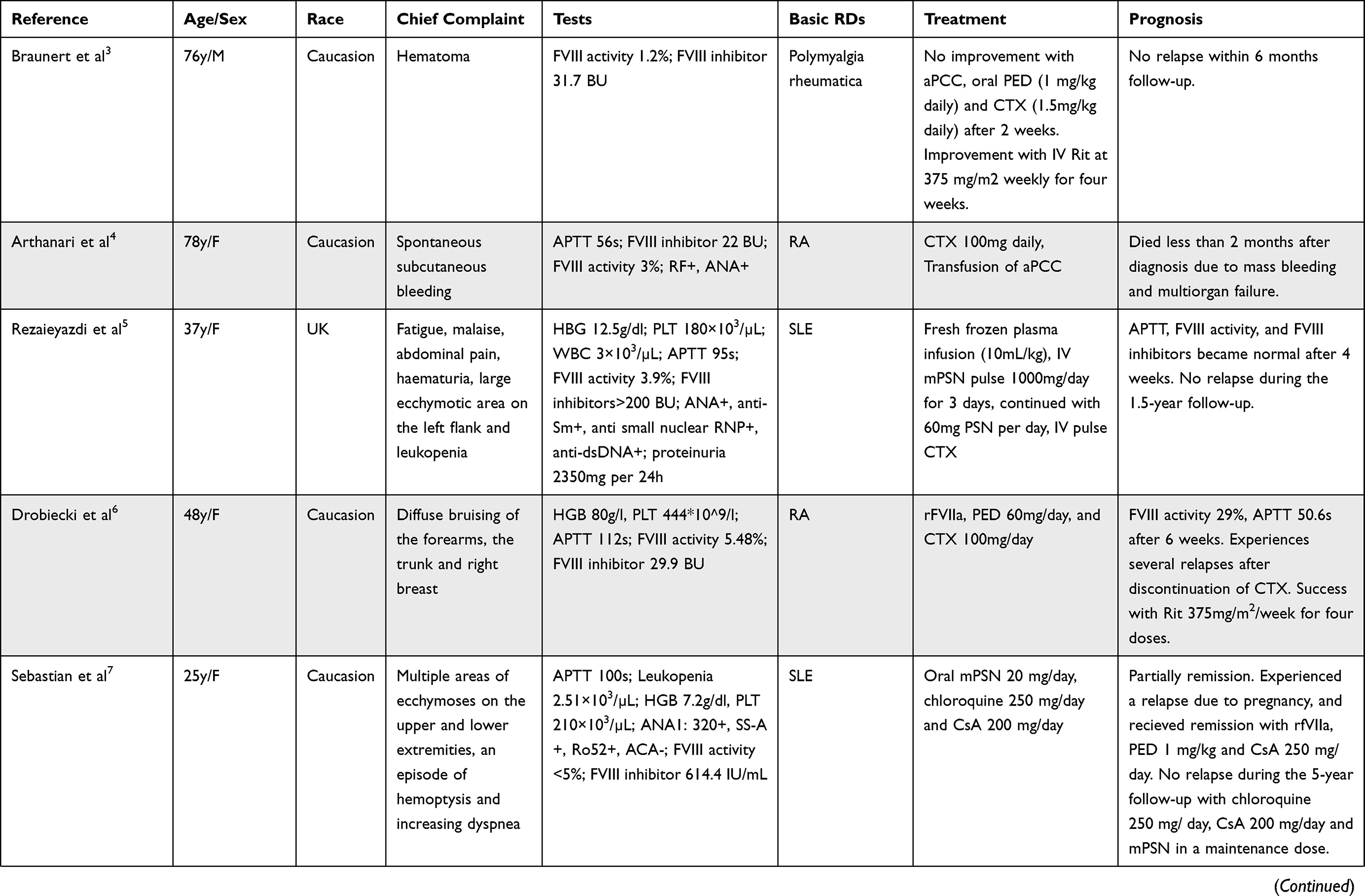 | 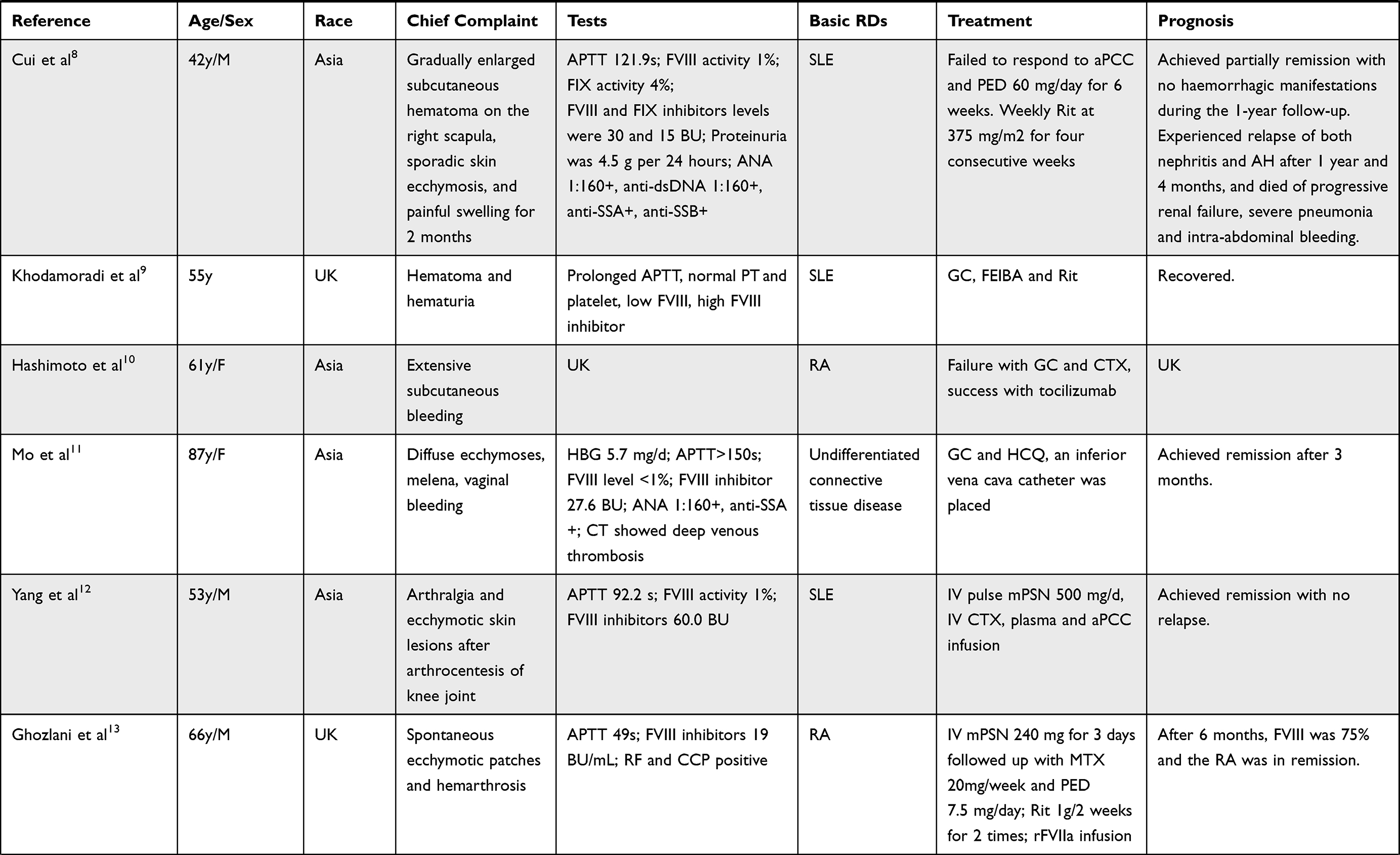 | 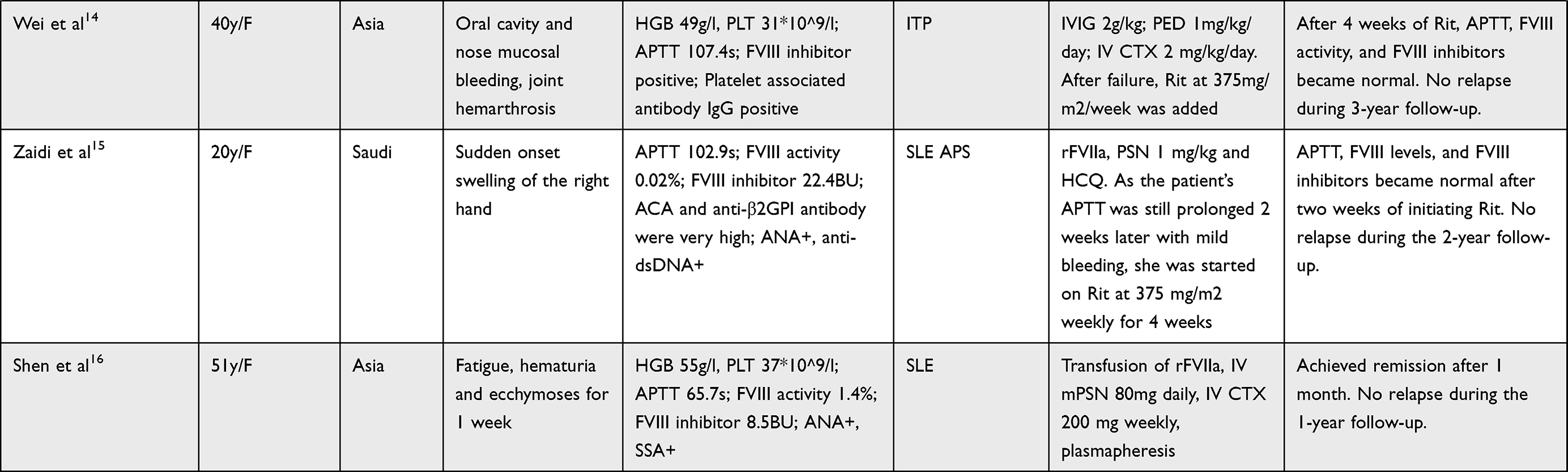 |
Table 1 14 Cases of Acquired Hemophilia Associated with Rheumatic Diseases |
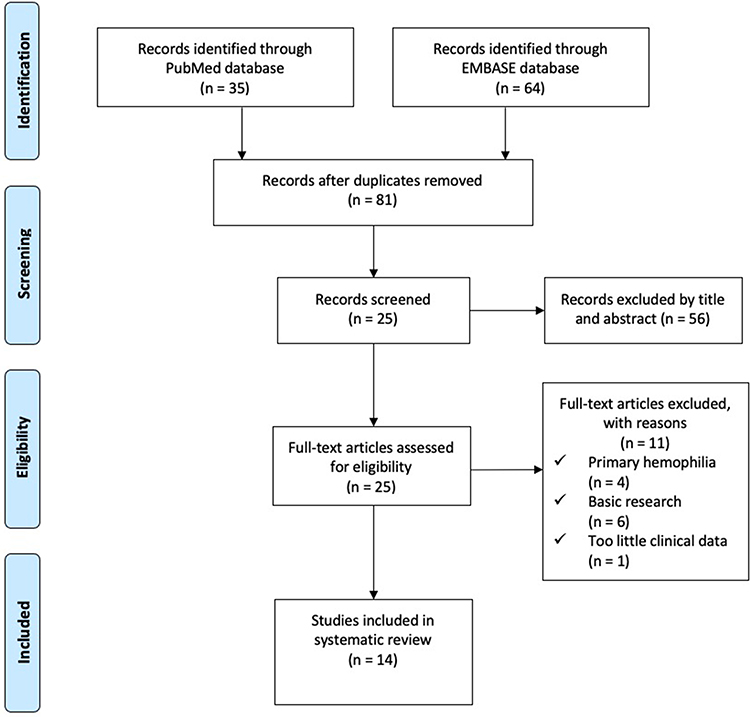 |
Figure 2 PRISMA Flow Diagram. PubMed Searching Strategy: (“acquired hemophilia” OR “secondary hemophilia”) AND (“connective tissue disease” OR “rheumatic diseases” OR “rheumatoid arthritis” OR “systemic lupus erythematosus”) AND (“2010/01/01”[Date - Publication]: “2021/12/31”[Date - Publication]) AND English[Language] EMBASE Searching Strategy: (“acquired hemophilia”: ab,ti OR “secondary hemophilia”: ab,ti) AND (“connective tissue disease”: ab,ti OR ‘rheumatic diseases’: ab,ti OR ‘rheumatoid arthritis’: ab,ti OR “systemic lupus erythematosus”: ab,ti) AND English: la AND (2010: py OR 2011: py OR 2012: py OR 2013: py OR 2014: py OR 2015: py OR 2016: py OR 2017: py OR 2018: py OR 2019: py OR 2020: py OR 2021: py). |
Discussion
AH is a rare but fatal autoimmune disorder based on antibody-mediated depletion of coagulation FVIII/FIX. AH is easily ignored or mistaken as other hemorrhagic diseases. The incidence of AH was estimated to be 1–4 per million based on results from the European Acquired Haemophilia Registry,17 although the incidence is increasing. A large survey in the 1980s suggested a mortality rate of 22%;18 however, lower mortality rate <10% was reported in two recent studies, which may be attributed to improved hemostatic agents.19,20 AH is a disorder driven by autoimmune B cells producing FVIII-specific antibodies, which are capable of hydrolyzing FVIII into smaller fragments and resulting in severe, spontaneous, life-threatening bleeding.21
From our experience. When patients with RDs present with bleeding symptoms, platelet count and clotting screen tests should be performed as soon as possible. If APTT is prolonged, further tests should be undertaken to evaluate FVIII/FIX activity, acquired FVIII/FIX inhibitors, and lupus anticoagulant. After the diagnosis of AH is confirmed based on decreased FVIII/IX activity and positive FVIII/IX inhibitors, immediate interventions should be taken to stop the bleeding and to reduce inhibitors (Figure 3). In AH, FVIII inhibitors are most common, and FIX inhibitors are extraordinarily rare.
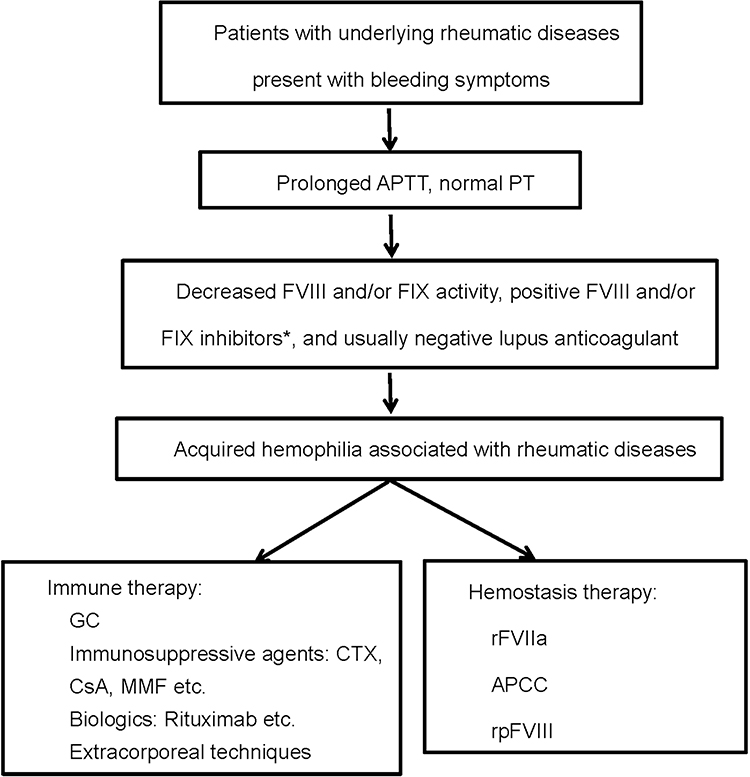 |
Figure 3 Diagnosis and treatment flow chart of rheumatic diseases associated acquired hemophilia. *FVIII inhibitors are most common in AH, and FIX inhibitors are extraordinarily rare. |
Based on the case series, GC and immunosuppressive agents presented to be highly cost-effective as the preferred treatment to reduce inhibitors. As for the patients who did not respond to or could not tolerate treatment with GC and immunosuppressive agents, biologics, most commonly Rituximab, seemed to be a good choice. This result is almost in accordance with a study of 331 patients from the prospective EACH2 registry,22 suggesting that GC combined with CTX resulted in more stable complete remission than GC alone or Rituximab-based regimens. This study also showed that Rituximab-based regimens required a longer time to achieve complete remission compared with GC with or without CTX. However, the case series in our review showed GC, and immunosuppressive agents took around 4–8 weeks, and Rituximab took around 2–4 weeks to show therapeutic effects. Patients with fatal and quick progressive bleeding should also be administered with bypassing agents, including APCC and rFVIIa, to control bleeding episodes. If bleeding persists, recombinant porcine FVIII (rpFVIII) can be used.23 When patients with RDs associated AH are accompanied by immune thrombocytopenia, they are at higher risk of life-threatening hemorrhage. Exploratory surgery or arterial punctures for angiograms should be avoided in case of severe blood loss and uncontrollable hemorrhage.24
Conclusion
AH should be highly suspected in patients with RDs presenting with spontaneous bleeding and an isolated prolonged APTT. Given the high morbidity of AH, it is important to facilitate efficient and proper management. GC combined with immunosuppressive agents is a first-line treatment. After failure, Rituximab seems to be a good choice. Patients with fatal and quick progressive bleeding should be administered with APCC and rFVIIa.
Abbreviations
AH, acquired hemophilia; FVIII, coagulation factor VIII; FIX, coagulation factor IX; RDs, rheumatic diseases; BU, Bethesda units; APCC, activated prothrombin complex concentrate; PED, prednisone; Rit, Rituximab; CTX, cyclophosphamide; MMF, mycophenolate mofetil; IVIG, intravenous immunoglobulin; UK, unknown; Anti-Sm, anti-Smith; Anti-RNP, anti ribonucleoproteins; Anti dsDNA, anti double-stranded DNA; mPSN, methylprednisolone; PSN, prednisolone; rFVIIa, recombinant FVIIa; GC, glucocorticoid; FEIBA, factor VIII inhibitor bypass activity; HCQ, hydroxychloroquine; ITP, immune thrombocytopenia; ACA, anticardiolipin antibodies; anti-β2GPI, anti beta 2-glycoprotein I; APS, antiphospholipid syndrome.
Ethics and Consent
The ethics committee of the Second Xiangya Hospital of Central South University approved this study. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Acknowledgments
Thank Dr. Jinshen He, for his editorial contributions to the manuscript.
Funding
The study is supported by the National Natural Science Foundation of China (No. 81701552, No. 81873882).
Disclosure
The authors report no conflicts of interest in this work.
References
1. O’Connor CR. Systematic review of the presentation of coagulation factor VIII inhibitors in rheumatic diseases: a potential cause of life-threatening hemorrhage. Semin Arthritis Rheum. 2015;44(6):695–709. doi:10.1016/j.semarthrit.2014.11.008
2. Boggio LN, Green D. Acquired hemophilia. Rev Clin Exp Hematol. 2001;5(4):389–431. doi:10.1046/j.1468-0734.2001.00049.x
3. Braunert L, Bruegel M, Pfrepper C, et al. Rituximab in der Behandlung der erworbenen Hämophilie A bei einem patienten mit polymyalgia rheumatica [Rituximab in the treatment of acquired haemophilia A in a patient with polymyalgia rheumatica]. Hamostaseologie. 2010;30(Suppl 1):S40–S43. doi:10.1055/s-0037-1619069
4. Arthanari S, Ahmad H, Nisar M. Fatal acquired hemophilia A in a patient with rheumatoid arthritis treated with Adalimumab. J Clin Rheumatol. 2012;18(1):50–51. doi:10.1097/RHU.0b013e31823ee3cd
5. Rezaieyazdi Z, Sharifi-Doloui D, Hashemzadeh K, et al. Acquired haemophilia A in a woman with autoimmune hepatitis and systemic lupus erythematosus: review of literature. Blood Coagul Fibrinolysis. 2011;22(8):738–741. doi:10.1097/MBC.0b013e32834a5c8e
6. Drobiecki A, Pasiarski M, Hus I, et al. Acquired hemophilia in the patient suffering from rheumatoid arthritis: case report. Blood Coagul Fibrinolysis. 2013;24(8):874–880. doi:10.1097/MBC.0b013e3283646635
7. Sebastian A, Misterska-Skóra M, Podolak-Dawidziak M, et al. Pregnancy exacerbates complications of acquired hemophilia in a patient with systemic lupus erythematosus. Postepy Dermatol Alergol. 2015;32(3):235–238. doi:10.5114/pdia.2014.40964
8. Cui QY, Wu TQ, Shen HS, et al. Refractory lupus nephropathy and acquired factor VIII and IX deficiencies in a patient with systemic lupus erythematosus treated with Rituximab. Haemophilia. 2017;23(6):e504–e506. doi:10.1111/hae.13232
9. Khodamoradi Z, Nazarinia MA, Bazdar S. Acquired hemophilia as initial presentation in a patient with systemic lupus erythematosus. Curr Rheumatol Rev. 2017;13(3):236–238. doi:10.2174/1573397113666170519121952
10. Hashimoto A, Takafuta T, Kido M, et al. Successful management of recurrent bleeding with tocilizumab in an acquired hemophilia A patient with rheumatoid arthritis. Rinsho Ketsueki. 2017;58(7):738–742. doi:10.11406/rinketsu.58.738
11. Mo L, Bao GC. Acquired factor VIII deficiency: two case reports and a review of literature. Exp Hematol Oncol. 2017;6:8. doi:10.1186/s40164-017-0068-3
12. Yang F, Zhou YS, Jia Y. Systemic lupus erythematosus with acquired hemophilia A: a case report. Beijing Da Xue Xue Bao Yi Xue Ban. 2018;50(6):1108–1111.
13. Ghozlani I, Mounach A, Ghazi M, et al. Targeting acquired hemophilia a with rheumatoid arthritis by a rituximab shot: a case report and review of the literature. Am J Case Rep. 2018;19:582–588. doi:10.12659/AJCR.908854
14. Wei F. Successful treatment of acquired hemophilia A associated with immune thrombocytopenia and joint hemarthrosis: a case report and literature review. Medicine. 2018;97(38):e12044. doi:10.1097/MD.0000000000012044
15. Zaidi ARZ, AlSheef M, Motabi IH, et al. Systemic lupus erythematosus presenting as hematoma of the hand due to acquired inhibitors to factor VIII: early and prolonged remission achieved with upfront rituximab. Cureus. 2019;11(5):e4786. doi:10.7759/cureus.4786
16. Shen P, Li J, Tu S, et al. Acquired hemophilia A in a woman with systemic lupus erythematosus: a case report and review of literature. Medicine. 2020;99(43):e22926. doi:10.1097/MD.0000000000022926
17. Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
18. Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost. 1981;45(3):200–203. doi:10.1055/s-0038-1650169
19. Schep SJ, van Dijk W, Beckers E, et al. Treatment of acquired hemophilia A, a balancing act: results from a 27-year Dutch cohort study. Am J Hematol. 2021;96(1):51–59. doi:10.1002/ajh.26009
20. Holstein K, Liu X, Smith A, et al. Bleeding and response to hemostatic therapy in acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood. 2020;136(3):279–287. doi:10.1182/blood.2019003639
21. Mahendra A, Padiolleau-Lefevre S, Kaveri SV, et al. Do proteolytic antibodies complete the panoply of the autoimmune response in acquired haemophilia A? Br J Haematol. 2012;156(1):3–12. doi:10.1111/j.1365-2141.2011.08890.x
22. Collins P, Baudo F, Knoebl P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;120(1):47–55. doi:10.1182/blood-2012-02-409185
23. Leebeek FWG. New developments in diagnosis and management of acquired hemophilia and acquired von Willebrand Syndrome. Hemasphere. 2021;5(6):e586. doi:10.1097/HS9.0000000000000586
24. Hay CR. Acquired haemophilia. Bailliere’s Clin Haematol. 1998;11(2):287–303. doi:10.1016/S0950-3536(98)80049-8
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.