Back to Journals » Journal of Blood Medicine » Volume 13
Acquired Hemophilia A: Current Guidance and Experience from Clinical Practice
Received 29 January 2022
Accepted for publication 22 April 2022
Published 11 May 2022 Volume 2022:13 Pages 255—265
DOI https://doi.org/10.2147/JBM.S284804
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Allyson M Pishko,1 Bhavya S Doshi2
1Department of Medicine, Division of Hematology/Oncology, University of Pennsylvania, Philadelphia, PA, USA; 2Department of Pediatrics, Division of Hematology, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
Correspondence: Bhavya S Doshi, Department of Pediatrics, Division of Hematology, Children’s Hospital of Philadelphia, 3501 Civic Center Blvd, Colket Translational Research Building Rm 5024, Philadelphia, PA, 19104, USA, Tel +1 215-590-3437, Fax +1 215-590-3992, Email [email protected]
Abstract: In acquired hemophilia A (AHA), autoantibodies to coagulation factor VIII (FVIII) neutralize FVIII activity leading to a potentially severe bleeding diathesis that carries a high rate of morbidity and mortality. This disorder is rare and occurs mainly in adults over 60 years of age or in the postpartum period. The diagnosis should be suspected in patients with new-onset bleeding without a personal or family history of bleeding and can be confirmed via specific assays for FVIII inhibitors. Treatment involves both hemostatic therapies to decrease bleeding and immune modulation strategies to re-establish immune tolerance to FVIII. There are limited data on treatment for refractory disease, based mostly on small case series. Registry studies have informed consensus guidelines for optimal hemostatic therapies and initial immunosuppressive therapies. Additional studies are needed to evaluate novel hemostatic agents and develop biomarkers to risk-stratify treatment while limiting adverse events.
Keywords: factor VIII, autoantibodies, hemostasis, immune modulation
Introduction
As opposed to alloantibodies that occur in up to 30% of patients with severe congenital hemophilia A (HA), acquired hemophilia A (AHA) is a rare disease that arises from autoantibodies to coagulation factor VIII (FVIII). Of the coagulation factors, autoantibodies to FVIII are the most common with an incidence of approximately 1 to 1.5 per million population per year.1,2 The vast majority of patients are adults with a median age of 64–78 years; however, pediatric cases have been reported3 with an estimated incidence of 0.045 per million per year.4 The disease carries high rates of morbidity and mortality, especially in elderly patients with other comorbidities. However, owing to its rare nature, data that inform consensus guidelines on management and prognostic markers are largely limited to registries and expert opinion.
Presentation and Diagnosis
Although most cases are idiopathic, AHA has been associated with pregnancy, autoimmune disorders such as rheumatoid arthritis or systemic lupus erythematosus, malignancies, and certain medications (Table 1).1,2,5–10 Medications reported to be associated with FVIII antibodies include penicillins, sulfonamides, phenytoin, interferons and fludarabine.11,12 In a systematic review of 42 pediatric cases of acquired coagulation inhibitors, 28 were directed against FVIII and were either associated with infections, medications, and autoimmune disorders or were idiopathic in nature. The median inhibitor titer was 8.5 Bethesda units (BU)/mL (range 1.7–6500). Pediatric patients have a higher response rate than adults, and some have spontaneous resolution (eg, when associated with infection or antibiotic use).3
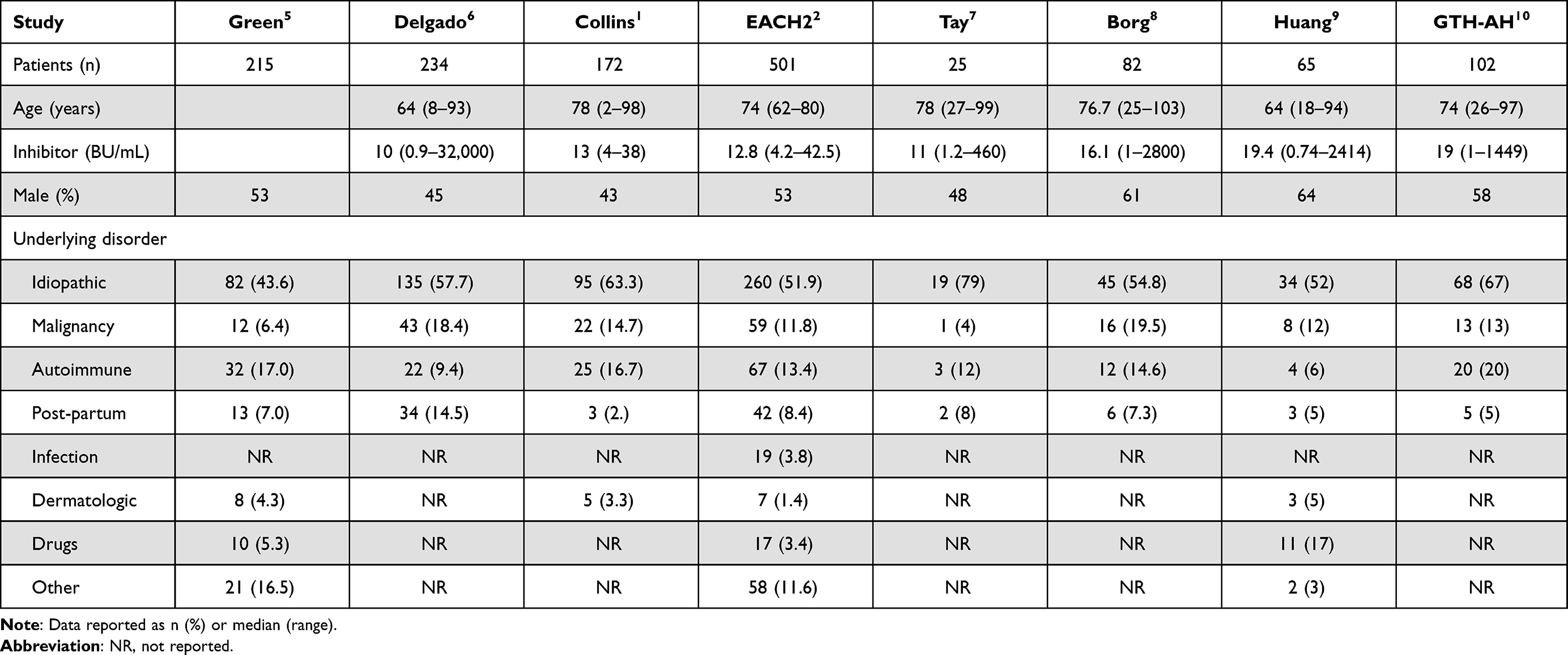 |
Table 1 Demographic and Clinical Characteristics of Patients with Acquired Hemophilia A in Large Registry Studies |
Irrespective of age, these patients typically present with spontaneous bleeding episodes or post-surgical hemorrhage. Mucosal bleeding events, including large hematomas or ecchymoses, severe epistaxis, gastrointestinal bleeding, and gross hematuria, are far more common than joint bleeds as seen in congenital HA. The most common presentation is subcutaneous bleeding (80%) followed by bleeding in muscular (45%), gastrointestinal (21%), genitourinary (9%) and retroperitoneal (9%) spaces.1,6 In post-partum AHA, soft-tissue, muscular, and vaginal bleeding are the common presentations. Transplacental passage of the IgG antibodies may result in bleeding in the neonate.13,14 Some patients may present solely with laboratory abnormalities without bleeding. Bleeding episodes are often serious,2 and the overall mortality rate approaches 20%.1,2 The residual FVIII activity does not always predict the risk of bleeding.
Clinicians must maintain a high index of suspicion for this diagnosis in adult patients with new onset of bruising or bleeding with a prolonged PTT. Diagnostic delays may result from lack of awareness due to the rarity of AHA and the need for centers to send out the laboratory work-up to a reference laboratory. A retrospective analysis found that delay in diagnosis of greater than one month was associated with extended periods of active bleeding and higher hemostatic factor requirements.15 There was no association with recurrence rate or survival between those patients with and without a diagnostic delay in this series. However, this may be secondary to the small number of patients in the series and potential biases such as lack of reporting of “never-diagnosed” cases.
A diagnostic algorithm based upon consensus guidelines to direct testing is presented in Figure 1 and should be considered in the setting of spontaneous bleeding without prior personal or family history of bleeding.16 Initial testing should include an activated partial thromboplastin time (aPTT) and prothrombin time (PT). AHA patients will have an isolated prolonged aPTT, and initial screening with a 1:1 mixing study with normal pooled plasma does not correct the prolonged clotting time. Interfering substances (eg, heparins) and lupus anticoagulants must be ruled out and FVIII activity assessed. FVIII activity is <1% in approximately 50% of cases and less than 5% in 75% of cases.16 The dilute Russell viper venom test (DRVVT) can be used for lupus anticoagulant testing and is not typically affected by FVIII inhibitors. Chromogenic FVIII activity assays are not sensitive to lupus anticoagulants.17–19 If FVIII is low and other tests are negative, quantification of the inhibitor titer via a Bethesda assay should then be conducted. It should be noted that patients may have concurrent lupus anticoagulants and FVIII inhibitors. Thus, identification of a lupus anticoagulant in a patient with a newly prolonged aPTT and bleeding manifestations does not entirely rule out the presence of an acquired inhibitor to FVIII.20,21
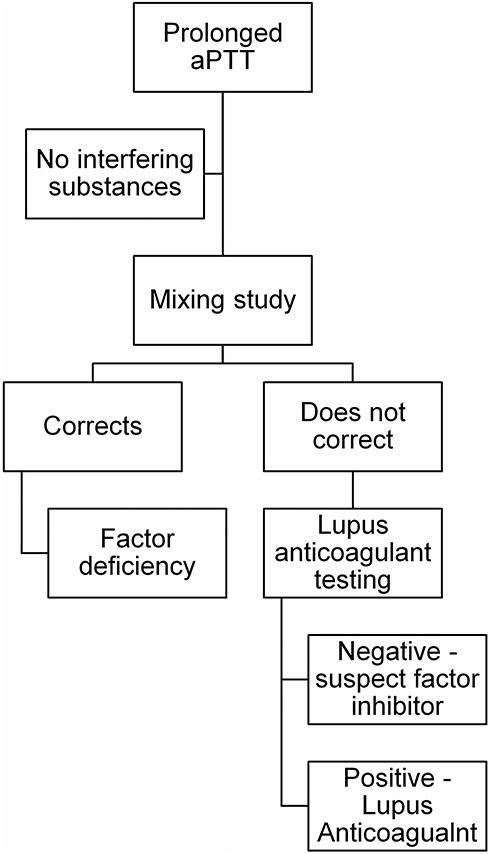 |
Figure 1 Laboratory testing algorithm for suspected acquired hemophilia A. After exclusion of interfering substances as a cause of prolonged aPTT, a two-hour mixing study should be done with normal pooled plasma. For samples where the aPTT corrects, a factor deficiency should be suspected, and specific factor assays conducted. For samples where the aPTT does not correct, testing for lupus anticoagulant should be done and, if negative, specific testing for a FVIII inhibitor should be conducted. Inhibitors can be measured via Bethesda assay ideally with heat inactivation. |
The inhibitor titer is equal to the reciprocal of the plasma dilution that results in 50% inhibition of FVIII in normal plasma after incubation for 2 hours at 37°C.22 Inhibitor titers are measured in Bethesda units (BU) where 1 BU is equal to the amount of antibody that neutralizes 50% FVIII activity. Although quite accurate for type I inhibitors which display linear kinetics, autoantibodies in AHA can display type II kinetics, which have some residual FVIII activity; the Bethesda assay may underestimate the titer in the presence of type II inhibitors.23–25 Sensitivity and specificity of the Bethesda assay is improved by the Nijmegen modification (buffering the normal plasma) and heat inactivation of the patient’s plasma prior to assessment.26,27 Enzyme-linked immunosorbent assays (ELISA) can be used to diagnose FVIII antibodies, but these cannot distinguish neutralizing capacity and are seen to some degree in the normal population (see below). Finally, if recombinant porcine FVIII (rpFVIII) is a therapeutic option, then a Bethesda assay specific to rpFVIII should be considered as it may help guide treatment decisions (see hemostatic therapies section).16
Pathophysiology
The precise trigger for the spontaneous production of neutralizing IgG antibodies to FVIII in patients with AHA is currently unknown. A certain percentage of the normal non-hemophilic population develop non-neutralizing IgG antibodies to FVIII, usually of low affinity and IgG1 subclass.28,29 In congenital HA, longitudinal studies suggest that the alloantibody response matures from a low-affinity predominantly IgG1 antibody to a high-affinity IgG4, which corresponds to FVIII neutralizing activity.30 Whether these non-neutralizing IgG antibodies in non-hemophilic patients are predecessors to or independent of neutralizing autoantibodies in AHA is unclear. Similar IgG subtypes are described in patients with AHA as compared to congenital HA.31 A recent study analyzed affinities and subtypes and noted that the most common subtypes with the highest titers and affinities were IgG1 (88%, KA 5.8 × 1010 M−1) and IgG4 (98%, KA 1.3 × 1010 M−1), though all IgG subclass antibodies and IgA (46%) or IgM (9%) antibodies were detected.32 The IgA antibodies do not correlate with FVIII neutralizing activity but do correlate with outcome (as discussed below).32 The IgG antibodies correlate with inhibitor titers32 and other studies have identified that these antibodies could be proteolytic towards FVIII.24,33 As in congenital HA, limited studies in AHA demonstrate that the IgG antibodies are generally polyclonal and largely bind to the A2 and C2 domains of FVIII.34
As noted in Table 1, 50% or more of AHA patients do not have an underlying disorder that predisposes them to form autoantibodies. Genetic investigations have associated polymorphisms in CTLA4, non-hemophilic F8 gene variants, and human leukocyte antigen (HLA) DRB1*16 and DQB1*0502 with AHA.35–38 Interestingly, these HLA types are noted to be protective towards inhibitors in congenital HA. Further, some studies have delineated FVIII-reactive CD4+ T cells in healthy populations and patients with congenital or acquired HA with inhibitors; albeit the frequency of these cells is lower in the healthy controls.39 Thus, there is a possibility of a trigger leading to a break in tolerance that allows these CD4+ T cells to proliferate and lead to AHA in the right context.
Management of Acquired Hemophilia A
The initial management of AHA can be broken down into two parts: obtaining and maintaining hemostasis and re-establishing FVIII immune tolerance by eradicating the inhibitor. Given the rarity of this disease, randomized studies of the optimal management of AHA are limited. International consensus AHA guidelines, first published in 2009 and updated in 2020, provide detailed guidance based on expert experience and registry data.16 Below, we provide a brief overview of the clinical management of AHA, highlighting important practical clinical points, and refer the reader to the International AHA guidelines for further details.16
Hemostatic Therapies
Given the rarity of the diagnosis, clinicians should refer patients to an experienced tertiary care center for management.40 Acute bleeding is typically best managed by using a bypassing agent or rpFVIII. Human-derived recombinant factor VIII products and desmopressin (DDAVP) are generally not effective, except in cases of low inhibitor titer (<5 BU), where DDAVP may be considered for minor bleeding episodes.41,42 The initial product depends on institutional experience and provider preference (Table 2). Recombinant factor VIIa (rVIIa) or activated prothrombin complex concentrates (aPCC) are most frequently used. These products require frequent dosing every 2–4 hours for rVIIa or every 8–12 hours for aPCC. Neither rVIIa nor aPCC can be monitored with standard laboratory assays, and thus the frequency and intensity of dosing is based on clinical improvement in bleeding symptoms. A global consensus provides clinical and laboratory markers of appropriate hemostasis and optimal intervals for assessment of bleeding control as pertinent to each type of bleeding that may occur in patients with AHA.40 Generally, experts suggest stabilization of hemoglobin levels, lack of transfusion requirement, and visually or clinically improved signs of bleeding as markers of appropriate hemostasis.40 There is potential for thrombotic risks with rVIIa or aPCC, particularly with repetitive infusions of high doses. Fortunately, these events seem uncommon, with one multicenter European AHA registry (EACH2) reporting thrombotic events in 2.9% (5/174) and 4.8% (3/63) patients with AHA who received rVIIa or aPCC, respectively.43 Case reports support the use of the anti-fibrinolytic agent tranexamic acid either in combination with rVIIa or aPCC.44–46 However, combination therapy may increase thrombotic risk, and clinicians must consider the risks and benefits for each patient.47
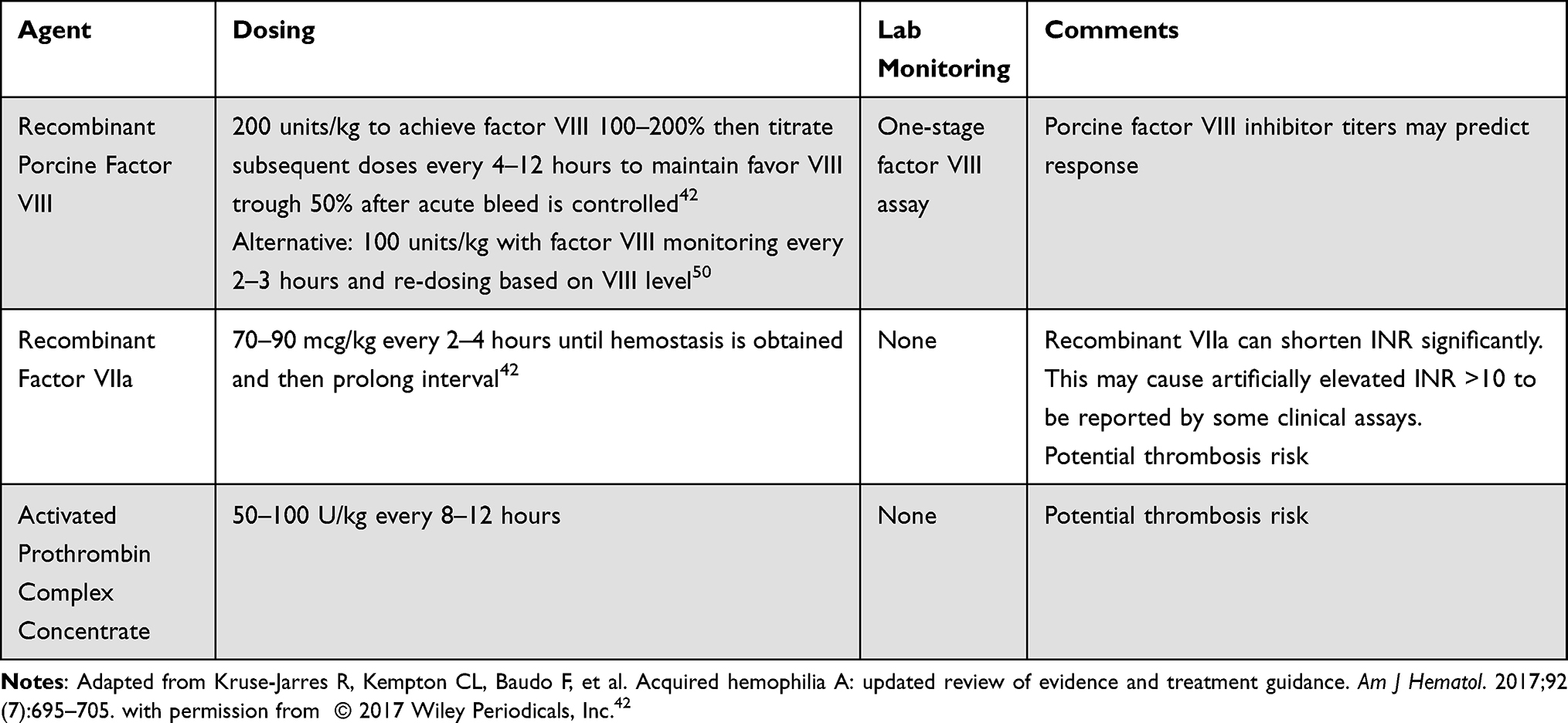 |
Table 2 Hemostatic Therapies for Management of Bleeding in Acquired Hemophilia |
More recent clinical trial data showed rpFVIII had efficacy in AHA.48 rpFVIII is appealing as it can be monitored with one-stage clot-based FVIII assays.16 Inhibitors can be present or develop to rpFVIII, and thus current AHA guidelines suggest porcine inhibitor titers should be measured to determine the likelihood of responsiveness to this agent.16 Anti-rpFVIII titers should also be measured after initiation of therapy as there are report of inhibitors developing which may decrease the efficacy of these products.42 In one study, 5 of the 28 subjects treated with rpFVIII developed de novo anti-rpFVIII inhibitor between 8 and 85 days after first infusion.48 Single institution published small series, unpublished abstract data, as well as a retrospective series report using lower than the approved doses of rpFVIII (100 IU/kg rather than 200 IU/kg) without awaiting rpFVIII titers with good hemostatic efficacy. FVIII activity (FVIII:C) monitoring can guide dosing when rpFVIII is used with goal of obtain FVIII:C levels initially at 100% for severe hemorrhage.16 The optimal dosing and monitoring of rpFVIII require further study.49–51 Martin et al described that their institutional protocol is to measure an FVIII:C 30 minutes after rpFVIII administration and then every 4 hours (immediately prior to next dose). For severe bleeds or bleeding in concerning areas (eg intracranial, neck, retroperitoneal) it is recommended to maintain FVIII:C >80% and >50% for all other bleeds.16 Doses are adjusted to maintain these goals until hemostasis has been obtained.50
Emicizumab is a bispecific antibody binding coagulation factor IXa and X without requiring FVIII.52,53 Emicizumab is Food and Drug Administration (FDA) approved for patients with moderate-severe congenital HA. The drug is administered subcutaneously and has a long half-life, making it appealing for use in AHA. However, there are still questions regarding thrombotic risk with emicizumab use in AHA as patients with AHA are generally older and have more comorbidities than patients with congenital HA. There were early reports of thrombotic complications and mortality following off-label and compassionate use of emicizumab in AHA patients, but details of additional risk factors in these reports are lacking.54 Subsequently, there is growing experience with the off-label use of the agent, which has been published or presented in abstract form.55 A recent abstract reported a multicenter survey of US hemophilia treatment centers in which twenty-four patients with AHA were treated with emicizumab. The majority of cases obtained bleeding control and tolerated the treatment without complications, but one patient experienced a thrombotic event and two deaths occurred while on emicizumab, which were not attributed to the medication.56 A published case series by Knoebl et al describes 12 patients with AHA and new-onset bleeding treated with emicizumab and “reduced-intensity” immunosuppression.57 Patients were started on emicizumab at 3 mg/kg given weekly via subcutaneous injection for 2–3 weeks followed by 1.5 mg/kg every 3 weeks (with interval prolongation up to 4 weeks) and discontinuation of therapy when FVIII levels were >30%. Of note, aPCC was held or patient switched to rVIIa for 48 hours prior to initiation of emicizumab due to the association of thrombotic microangiopathy in patients with congenital HA treated with emicizumab and concurrent aPCC.58 A rapid clinical improvement in bleeding was seen in this study at a median of 3 days (range 2–15 days). It should also be noted that as emicizumab immediately and completely corrects the aPTT, FVIII levels need to be measured by bovine chromogenic (not clot-based) assays in patients on this therapy. We currently recommend the use of emicizumab in AHA only in the clinical trial setting until more data are available regarding its safety and efficacy in AHA.
Patients with AHA are not protected from thrombotic events and may be at increased risk of thrombosis during treatment with bypassing agents. The incidence of thromboembolic events in patients receiving bypassing agents varies by study with some registries not reporting any thromboembolic events due to bypassing agents2,43 and others reporting an incidence of 2–10%.8,10,42 Current International AHA guidelines recommend initiating venous thromboembolism (VTE) prophylaxis once FVIII:C returns to normal if the patient remains hospitalized or has another indication for VTE prophylaxis.16 As AHA often occurs in the elderly where anticoagulation is often used for primary stroke prevention for atrial fibrillation and/or treatment and prevention of thrombosis, it is essential to restart appropriate anticoagulation in patients with a pre-existing indication once FVIII:C levels return to normal.16
Inhibitor Eradication
Obtaining hemostasis is the immediate priority after which strategies to eliminate the inhibitor should be promptly employed. Current guidelines recommend immunosuppressive therapy in most patients diagnosed with AHA.16 Many of the patients with AHA who develop inhibitors to FVIII are elderly, and thus clinicians must carefully consider the toxicities of therapy when choosing a regimen.
First-Line Immunosuppressant
The recommended first line of therapy is corticosteroids, typically 1 mg/kg prednisone (Table 3). Traditionally, a second agent (either rituximab or cyclophosphamide) was added in patients who failed to respond to steroids alone. However, more recent observational studies suggest higher complete remission (CR) rates when combination therapy is utilized (35.2% CR with steroid monotherapy vs 67.7–83.3% with combination therapy).59 Current International AHA guidelines now suggest adding rituximab or cyclophosphamide to first-line therapy in patients with inhibitor titer >20 BU.16 The choice of cyclophosphamide versus rituximab depends on the comorbidities of the patient as well as provider and institutional experience. Elimination of the inhibitor is achieved with immunosuppression at a median of 5–6 weeks.42 While CR rates appear higher with combination therapy, a significant proportion of mortality is related to infection due to immunosuppression (outlined further in prognosis section below). Coupled with the fact that there are a percentage of patients that will have spontaneous remissions (in one series up to 35% achieved remission by 1 year) without immunosuppression, a careful patient-centered risk versus benefit analysis must be employed for each case.5
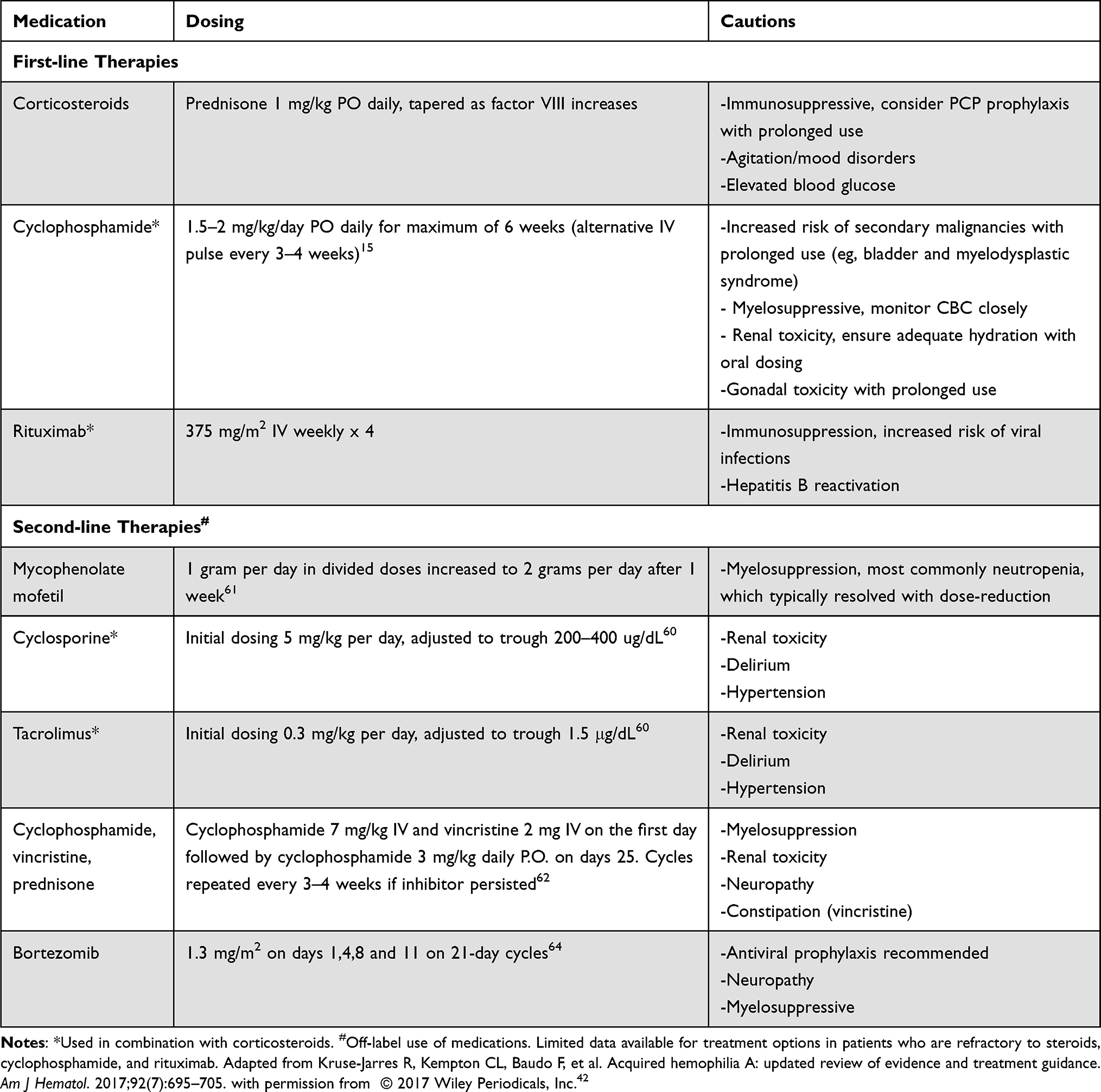 |
Table 3 Immunosuppressant Therapies for Acquired Hemophilia A |
Second-Line Immunosuppressant
A second-line therapy should be considered if there is no response after 3–5 weeks of first-line therapy.42 International AHA guidelines suggest first using whichever agent (cyclophosphamide or rituximab) that was not used in the first-line setting.16 Unfortunately, there are very limited data about additional options for patients refractory to steroids, rituximab, and cyclophosphamide. An overview of some studied regimens is outlined in Table 3. Pardos-Gea et al performed a prospective study of 11 patients with AHA treated with calcineurin inhibitors (tacrolimus or cyclosporine) and corticosteroids in the first-line setting. They reported responses in 10 out of the 11 patients with one patient experiencing a major side effect (hypertensive posterior progressive encephalopathy).60 If calcineurin inhibitors are used, clinicians must monitor closely for renal toxicity and hypertensive complications, particularly in elderly patients with AHA.60 Small series also suggest that mycophenolate mofetil has efficacy in AHA, although there were lower rates of initial response than prednisone and rituximab. Mycophenolate mofetil maintained remission in two patients with AHA who previously did not obtain CR with first-line therapy.61 Combining multiple immunosuppressants and inclusion of vincristine has also been reported in a case series of 6 patients with AHA, with 5 patients obtaining CR after 1–7 cycles of therapy.62 Case reports also describe responses to plasma-cell directed therapies, such as bortezomib, in patients with refractory AHA.63,64
Immune tolerance induction (ITI) protocols with high-dose FVIII (as used in congenital HA) have been trialed in AHA,65 but their efficacy in addition to standard immunosuppressive therapy is unclear. Current guidelines do not recommend ITI unless a patient has severe bleeding failing first-line hemostatic therapies.16
Prognosis and Response
Table 4 outlines the complete response and relapse rates as well as mortality in patients with AHA from selected registry studies. Approximately 60–80% of patients will respond to first-line immunosuppressive therapy (corticosteroids with or without either rituximab or cyclophosphamide). Relapses are not uncommon during corticosteroid withdrawal, occurring in 15% of patients in a Dutch registry.59 After complete cessation of immunosuppressive therapy, an estimated 25% of patients will experience a relapse with a median time to relapse of 14.7 weeks (IQR 2.9–66.6 weeks).59 Given the relapse rate, clinicians should counsel patients during weaning of immunosuppression to monitor for increased bruising or other signs of bleeding. The optimal frequency of monitoring PTT and/or FVIII levels following CR is not well defined. One of the author’s practices is to monitor complete blood count and PTT and FVIII levels every 2 weeks while weaning prednisone and immediately following immunosuppression withdrawal and then monthly for at least the next 3-6 months.
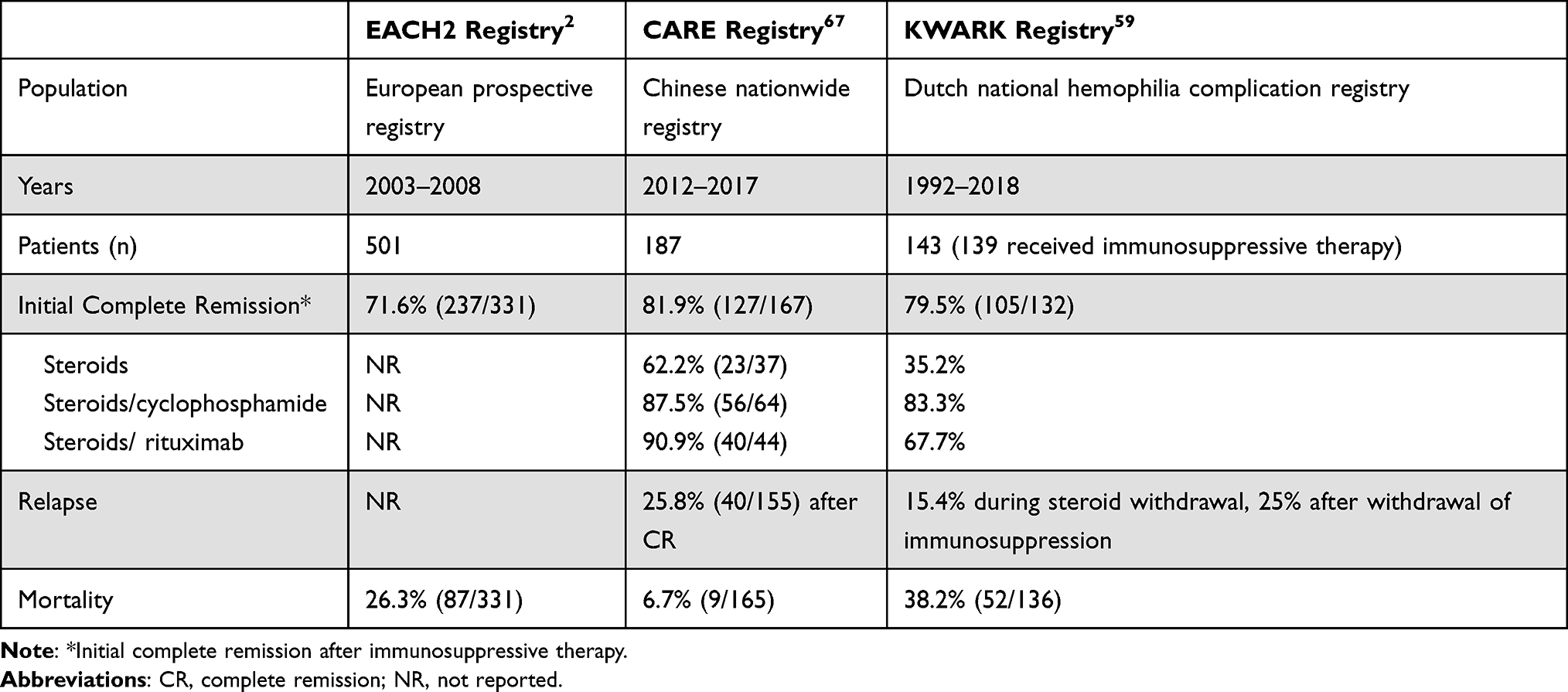 |
Table 4 Initial Complete Remission, Relapse, and Mortality Rates in Selected Registry Studies of Acquired Hemophilia A |
Registry studies have assessed the influence of various clinical and laboratory characteristics on outcome of AHA therapy. Spontaneous remission is noted, especially in pediatric patients with AHA.3 In the prospective GTH registry study, which assessed a standardized approach of steroids alone (first line), steroids with cyclophosphamide (second line) or rituximab (refractory or relapse), predictors of response included presence of high-titer (>1:80) anti-FVIII IgA antibodies but not IgG antibodies.32 In multivariable analysis, clinical predictors of poor response rates and overall survival in this study included baseline FVIII activity <1% and WHO-PS scores >2.10 A recent conference presentation displayed differential cytokine production from FVIII-stimulated peripheral blood mononuclear cells of 11 AHA patients when comparing baseline samples of healthy controls and relapsed patients but not between CR and relapsed patients.66 However, these are preliminary findings and further studies need to be conducted to verify their predictive relevance in AHA. Additional prospective studies and biorepositories are needed to guide biomarkers of tolerance and overall response in AHA.
AHA has significant mortality, ranging from 6.7% to 38% in registry data.2,59 Data from multiple registries support that infection is the leading cause of mortality in this disease, likely due to the need for immunosuppression.2,8,10,59 For example, in the Dutch Cohort, overall mortality was 38.2% (52/132 patients) with the cause of death identified as infection in 19.2% (10/52 patients), malignancy in 13.5% (7/52) and fatal bleeding in 7.7% (4/52 patients).59 Some studies have reported rare fatal thrombotic events in patients with AHA receiving hemostatic agents (see above).10
Conclusions
Although rare, AHA carries a high risk of morbidity and mortality and requires prompt treatment, ideally by a specialized center well versed in treating these patients. Immediate attention to control bleeding should be followed closely by therapy to re-establish FVIII immune tolerance. Initial treatment choice should involve corticosteroids with or without additional immunosuppressants based upon the risk profile of the patients. Although treatment should be initiated quickly, it can pose safety risks necessitating close monitoring of patients. Most patients will respond to initial immunosuppressive therapy with corticosteroids alone or in combination with cyclophosphamide or rituximab. Vigilance is required during discontinuation of immunosuppression to allow early identification of relapse. Additional prospective studies are necessary to determine the optimal management and identify biomarkers of response that can inform therapeutic decisions.
Disclosure
Dr Allyson M Pishko reports grants from Hemostasis and Thrombosis Research Society, grants from Sanofi Genzyme, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870–1877. doi:10.1182/blood-2006-06-029850
2. Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
3. Franchini M, Zaffanello M, Lippi G. Acquired hemophilia in pediatrics: a systematic review. Pediatr Blood Cancer. 2010;55(4):606–611. doi:10.1002/pbc.22657
4. Charlebois J, Rivard GE, St-Louis J. Management of acquired hemophilia A: review of current evidence. Transfus Apher Sci. 2018;57(6):717–720. doi:10.1016/j.transci.2018.10.011
5. Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb Haemost. 1981;45(3):200–203. doi:10.1055/s-0038-1650169
6. Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003;121(1):21–35. doi:10.1046/j.1365-2141.2003.04162.x
7. Tay L, Duncan E, Singhal D, et al. Twelve years of experience of acquired hemophilia A: trials and tribulations in South Australia. Semin Thromb Hemost. 2009;35(8):769–777. doi:10.1055/s-0029-1245109
8. Borg JY, Guillet B, Le Cam-duchez V, Goudemand J, Levesque H, Group SS. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hemophilie Acquise) registry. Haemophilia. 2013;19(4):564–570. doi:10.1111/hae.12138
9. Huang SY, Tsay W, Lin SY, Hsu SC, Hung MH, Shen MC. A study of 65 patients with acquired hemophilia A in Taiwan. J Formos Med Assoc. 2015;114(4):321–327. doi:10.1016/j.jfma.2013.01.006
10. Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH-AH 01/2010 study. Blood. 2015;125(7):1091–1097. doi:10.1182/blood-2014-07-587089
11. Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005;80(1):55–63. doi:10.1002/ajh.20390
12. Sborov DW, Rodgers GM. Acquired hemophilia a: a current review of autoantibody disease. Clin Adv Hematol Oncol. 2012;10(1):19–27.
13. Gunel Karaburun IE, Kayki G, Aytac SS, Celik HT, Gumruk F, Yigit S. Transplacental hemophilia A and prophylactic treatment with intravenous immunoglobulin and recombinant factor VIIa in the newborn period: a case report. Blood Coagul Fibrinolysis. 2021;32(2):151–154. doi:10.1097/MBC.0000000000000978
14. Lulla RR, Allen GA, Zakarija A, Green D. Transplacental transfer of postpartum inhibitors to factor VIII. Haemophilia. 2010;16(1):14–17. doi:10.1111/j.1365-2516.2009.02049.x
15. Pardos-Gea J, Fernandez-Diaz N, Parra R, Cortina V, Altisent C. Diagnostic delay in acquired haemophilia: analysis of causes and consequences in a 20-year Spanish cohort. Haemophilia. 2018;24(3):e163–e166. doi:10.1111/hae.13499
16. Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020;105(7):1791–1801. doi:10.3324/haematol.2019.230771
17. de Maistre E, Wahl D, Perret-Guillaume C, et al. A chromogenic assay allows reliable measurement of factor VIII levels in the presence of strong lupus anticoagulants. Thromb Haemost. 1998;79(1):237–238. doi:10.1055/s-0037-1614254
18. Chandler WL, Ferrell C, Lee J, Tun T, Kha H. Comparison of three methods for measuring factor VIII levels in plasma. Am J Clin Pathol. 2003;120(1):34–39. doi:10.1309/C8T8-YNB4-G3W4-5PRF
19. Blanco AN, Cardozo MA, Candela M, Santarelli MT, Perez Bianco R, Lazzari MA. Anti-factor VIII inhibitors and lupus anticoagulants in haemophilia A patients. Thromb Haemost. 1997;77(4):656–659. doi:10.1055/s-0038-1656029
20. Taher A, Abiad R, Uthman I. Coexistence of lupus anticoagulant and acquired haemophilia in a patient with monoclonal gammopathy of unknown significance. Lupus. 2003;12(11):854–856. doi:10.1191/0961203303lu463cr
21. Seethala S, Collins NP
22. Kasper CK. Measurement of factor VIII inhibitors. Prog Clin Biol Res. 1984;150:87–98.
23. Gawryl MS, Hoyer LW. Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood. 1982;60(5):1103–1109. doi:10.1182/blood.V60.5.1103.1103
24. Mahendra A, Padiolleau-Lefevre S, Kaveri SV, Lacroix-Desmazes S. Do proteolytic antibodies complete the panoply of the autoimmune response in acquired haemophilia A? Br J Haematol. 2012;156(1):3–12. doi:10.1111/j.1365-2141.2011.08890.x
25. Hoyer LW, Scandella D. Factor VIII inhibitors: structure and function in autoantibody and hemophilia A patients. Semin Hematol. 1994;31(2 Suppl 4):1–5.
26. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73(2):247–251. doi:10.1055/s-0038-1653759
27. Boylan B, Miller CH. Effects of pre-analytical heat treatment in factor VIII (FVIII) inhibitor assays on FVIII antibody levels. Haemophilia. 2018;24(3):487–491. doi:10.1111/hae.13435
28. Hofbauer CJ, Whelan SF, Hirschler M, et al. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. 2015;125(7):1180–1188. doi:10.1182/blood-2014-09-598268
29. Whelan SF, Hofbauer CJ, Horling FM, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039–1048. doi:10.1182/blood-2012-07-444877
30. Reipert BM, Gangadharan B, Hofbauer CJ, et al. The prospective Hemophilia Inhibitor PUP Study reveals distinct antibody signatures prior to FVIII inhibitor development. Blood Adv. 2020;4(22):5785–5796. doi:10.1182/bloodadvances.2020002731
31. Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1- and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost. 2002;88(4):568–575. doi:10.1055/s-0037-1613257
32. Tiede A, Hofbauer CJ, Werwitzke S, et al. Anti-factor VIII IgA as a potential marker of poor prognosis in acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood. 2016;127(19):2289–2297. doi:10.1182/blood-2015-09-672774
33. Wootla B, Dasgupta S, Dimitrov JD, et al. Factor VIII hydrolysis mediated by anti-factor VIII autoantibodies in acquired hemophilia. J Immunol. 2008;180(11):7714–7720. doi:10.4049/jimmunol.180.11.7714
34. Scandella DH, Nakai H, Felch M, et al. In hemophilia A and autoantibody inhibitor patients: the factor VIII A2 domain and light chain are most immunogenic. Thromb Res. 2001;101(5):377–385. doi:10.1016/s0049-3848(00)00418-7
35. Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia. 2010;16(Suppl 3):41–45. doi:10.1111/j.1365-2516.2010.02259.x
36. Pavlova A, Diaz-Lacava A, Zeitler H, et al. Increased frequency of the CTLA-4 49 A/G polymorphism in patients with acquired haemophilia A compared to healthy controls. Haemophilia. 2008;14(2):355–360. doi:10.1111/j.1365-2516.2007.01618.x
37. Pavlova A, Zeitler H, Scharrer I, Brackmann HH, Oldenburg J. HLA genotype in patients with acquired haemophilia A. Haemophilia. 2010;16(102):107–112. doi:10.1111/j.1365-2516.2008.01976.x
38. Tiede A, Eisert R, Czwalinna A, Miesbach W, Scharrer I, Ganser A. Acquired haemophilia caused by non-haemophilic factor VIII gene variants. Ann Hematol. 2010;89(6):607–612. doi:10.1007/s00277-009-0887-3
39. Reding MT, Wu H, Krampf M, et al. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. Thromb Haemost. 2000;84(4):643–652. doi:10.1055/s-0037-1614081
40. Tiede A, Giangrande P, Teitel J, et al. Clinical evaluation of bleeds and response to haemostatic treatment in patients with acquired haemophilia: a global expert consensus statement. Haemophilia. 2019;25(6):969–978. doi:10.1111/hae.13844
41. Franchini M, Lippi G. Acquired factor VIII inhibitors. Blood. 2008;112(2):250–255. doi:10.1182/blood-2008-03-143586
42. Kruse-Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92(7):695–705. doi:10.1002/ajh.24777
43. Baudo F, Collins P, Huth-Kuhne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;120(1):39–46. doi:10.1182/blood-2012-02-408930
44. Fisher C, Mo A, Warrillow S, Smith C, Jones D. Utility of thromboelastography in managing acquired Factor VIII inhibitor associated massive haemorrhage. Anaesth Intensive Care. 2013;41(6):799–803. doi:10.1177/0310057X1304100617
45. Holmstrom M, Tran HT, Holme PA. Combined treatment with APCC (FEIBA(R)) and tranexamic acid in patients with haemophilia A with inhibitors and in patients with acquired haemophilia A–a two-centre experience. Haemophilia. 2012;18(4):544–549. doi:10.1111/j.1365-2516.2012.02748.x
46. Takedani H, Shima M, Horikoshi Y, et al. Ten-year experience of recombinant activated factor VII use in surgical patients with congenital haemophilia with inhibitors or acquired haemophilia in Japan. Haemophilia. 2015;21(3):374–379. doi:10.1111/hae.12611
47. Taparia M, Cordingley FT, Leahy MF. Pulmonary embolism associated with tranexamic acid in severe acquired haemophilia. Eur J Haematol. 2002;68(5):307–309. doi:10.1034/j.1600-0609.2002.01607.x
48. Kruse-Jarres R, St-Louis J, Greist A, et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015;21(2):162–170. doi:10.1111/hae.12627
49. Ellsworth P, Chen S-L, Wang C, Key NS, Ma A. Rituximab monotherapy is effective for inhibitor eradication with concomitant porcine factor viii followed by emicizumab for bleed control in acquired hemophilia A. Blood. 2021;138:348. doi:10.1182/blood-2021-154207
50. Martin K, Kasthuri R, Mooberry MJ, Chen SL, Key NS, Ma AD. Lower doses of recombinant porcine factor VIII maintain excellent haemostatic efficacy. Haemophilia. 2016;22(6):e549–e551. doi:10.1111/hae.13038
51. Tarantino MD, Cuker A, Hardesty B, Roberts JC, Sholzberg M. Recombinant porcine sequence factor VIII (rpFVIII) for acquired haemophilia A: practical clinical experience of its use in seven patients. Haemophilia. 2017;23(1):25–32. doi:10.1111/hae.13040
52. Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody ACE910 prevents joint bleeds in a long-term primate model of acquired hemophilia A. Blood. 2014;124(20):3165–3171. doi:10.1182/blood-2014-07-585737
53. Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. doi:10.1038/nm.2942
54. Makris M, Iorio A, Lenting PJ. Emicizumab and thrombosis: the story so far. J Thromb Haemost. 2019;17(8):1269–1272. doi:10.1111/jth.14556
55. Dane KE, Lindsley JP, Streiff MB, Moliterno AR, Khalid MK, Shanbhag S. Successful use of emicizumab in a patient with refractory acquired hemophilia A and acute coronary syndrome requiring percutaneous coronary intervention. Res Pract Thromb Haemost. 2019;3(3):420–423. doi:10.1002/rth2.12201
56. Poston JN, Al-Banaa K, von Drygalski A, et al. Emicizumab for the treatment of acquired hemophilia A: a multicenter US case series. Blood. 2021;138:496. doi:10.1182/blood-2021-148040
57. Knoebl P, Thaler J, Jilma P, Quehenberger P, Gleixner K, Sperr WR. Emicizumab for the treatment of acquired hemophilia A. Blood. 2021;137(3):410–419. doi:10.1182/blood.2020006315
58. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
59. Schep SJ, van Dijk WEM, Beckers EAM, et al. Treatment of acquired hemophilia A, a balancing act: results from a 27-year Dutch cohort study. Am J Hematol. 2021;96(1):51–59. doi:10.1002/ajh.26009
60. Pardos-Gea J, Altisent C, Parra R, Vilardell-Tarres M, Ordi-Ros J. Acquired haemophilia A. First line treatment with calcineurin inhibitors and steroid pulses: a 10-year follow-up study. Haemophilia. 2012;18(5):789–793. doi:10.1111/j.1365-2516.2012.02772.x
61. Obaji S, Rayment R, Collins PW. Mycophenolate mofetil as adjunctive therapy in acquired haemophilia A. Haemophilia. 2019;25(1):e59–e65. doi:10.1111/hae.13658
62. Lian EC, Villar MJ, Noy LI, Ruiz-Dayao Z. Acquired factor VIII inhibitor treated with cyclophosphamide, vincristine, and prednisone. Am J Hematol. 2002;69(4):294–295. doi:10.1002/ajh.10070
63. McFadyen JD, Tran H, Kaplan ZS. Factor VIII inhibitor eradication with bortezomib in acquired haemophilia A. Br J Haematol. 2017;178(6):986–987. doi:10.1111/bjh.14185
64. Ratnasingam S, Walker PA, Tran H, et al. Bortezomib-based antibody depletion for refractory autoimmune hematological diseases. Blood Adv. 2016;1(1):31–35. doi:10.1182/bloodadvances.2016001412
65. Zeitler H, Ulrich-Merzenich G, Hess L, et al. Treatment of acquired hemophilia by the Bonn-Malmo Protocol: documentation of an in vivo immunomodulating concept. Blood. 2005;105(6):2287–2293. doi:10.1182/blood-2004-05-1811
66. Frade-Guanaes JO, Racanelli AP, Siqueira LH, et al. Acquired hemophilia A relapse is related to Th2 response, and increased expression of B-Cell Activating Factor (BAFF). Blood. 2021;138:497. doi:10.1182/blood-2021-152589
67. Sun B, Xue F, Feng Y, et al. Outcome of CARE: a 6-year national registry of acquired haemophilia A in China. Br J Haematol. 2019;187(5):653–665. doi:10.1111/bjh.16128
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.