Back to Journals » OncoTargets and Therapy » Volume 11
Ablation of MCM10 using CRISPR/Cas9 restrains the growth and migration of esophageal squamous cell carcinoma cells through inhibition of Akt signaling
Authors Yan J, Du P, Jia Y, Chang Z, Gan S, Xu X, Wang Y, Qin Y ![]() , Kan Q
, Kan Q
Received 14 November 2017
Accepted for publication 11 April 2018
Published 6 June 2018 Volume 2018:11 Pages 3323—3333
DOI https://doi.org/10.2147/OTT.S157025
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ingrid Espinoza
Jie Yan,1,2 Pan Du,3 Yongxu Jia,2 Zhiwei Chang,2 Silin Gan,4 Xiaohan Xu,5 Yaohe Wang,3 Yanru Qin,2 Quancheng Kan1
1Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China; 2Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China; 3National Center for International Research in Cell and Gene Therapy, Sino-British Research Centre for Molecular Oncology, Zhengzhou University, Zhengzhou, China; 4Department of Hematology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China; 5Department of Anesthesiology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Introduction: Minichromosome maintenance 10 (MCM10) is deregulated in several malignancies including cervical cancer and urothelial carcinoma. However, the expression and biologic role of MCM10 in esophageal squamous cell carcinoma (ESCC) is still unknown.
Methods: In this study, we performed immunohistochemistry and real-time polymerase chain reaction (PCR) analysis to examine the expression of MCM10 in ESCC and adjacent normal esophageal tissues. The associations of MCM10 expression with clinicopathologic parameters of ESCC were analyzed. Ablation of MCM10 through the CRISPR/Cas9 technology was conducted and its impact on ESCC cell growth and migration was investigated.
Results: The mRNA and protein expression levels of MCM10 were significantly greater in ESCC than in normal tissues (P<0.001). The expression of MCM10 was significantly associated with age at diagnosis (P=0.033), but not with gender, differentiation grade, invasion status, or tumor–node–metastasis (TNM) stage. Knockout of MCM10 significantly suppressed the proliferation, colony formation, and migration capacity of EC109 ESCC cells, compared to control cells harboring wild-type MCM10. Mechanistically, MCM10 depletion markedly reduced the phosphorylation of Akt. Overexpression of constitutively active Akt significantly restored the aggressive phenotype of MCM10-null EC109 cells.
Conclusion: In conclusion, these results suggest that MCM10 acts as an oncogene in ESCC through activation of Akt signaling and represents a promising therapeutic target for this malignancy.
Keywords: esophageal cancer, growth, migration, minichromosome maintenance proteins
Introduction
Esophageal cancer is one of the most frequently diagnosed cancers worldwide.1 In Eastern Asian countries including China, esophageal squamous cell carcinoma (ESCC) is the major histologic type of esophageal cancer.2 Despite advances in the prevention and treatment of esophageal cancer, the prognosis of this malignancy is still poor. It is estimated that the overall 5-year survival rate for ESCC is <40%.3,4 Identification of the mechanisms underlying the growth and progression of ESCC is of importance in combating this disease.
Minichromosome maintenance (MCM) proteins are highly conserved in eukaryotes and play an essential role in DNA replication initiation and elongation.5 MCM2–7 proteins are related to each other and form the hexameric complex as a key component of the prereplication complex, contributing to DNA unwinding. MCM1, MCM8, and MCM10 are distinct from MCM2–7 proteins, but also participate in DNA synthesis.6 Despite the lack of enzymatic domains, MCM10 can interact with replication factors including the MCM2–7 complex and thus coordinate DNA replication.7
There is growing evidence for the implication of MCM proteins in cancer progression.8–10 For example, high expression of MCM3 is associated with epithelial–mesenchymal transition and invasion in prostate cancer.8 Likewise, upregulation of MCM7 is linked to aggressive parameters and poor prognosis in pituitary adenoma.9 Blocking MCM2 activity has been reported to enhance DNA damage-induced apoptosis in breast cancer cells.10 A previous study has shown that MCM10 is significantly overexpressed in cervical cancer specimens relative to normal cervical tissues.11 Another study demonstrated that MCM10 overexpression is associated with advanced stage, nodal metastasis, and vascular invasion, and predicts adverse prognosis in urothelial carcinoma.12 However, the expression and biologic role of MCM10 in esophageal cancer is still unclear.
In this study, we examined the expression of MCM10 in 64 pairs of ESCC samples and adjacent normal esophageal tissues and evaluated the relationship between MCM10 expression and clinicopathologic features of ESCC. Moreover, the consequence of deletion of MCM10 through the CRISPR/Cas9 technology in ESCC cells was determined.
Materials and methods
Tissue specimens
We collected 64 pairs of formalin-fixed, paraffin-embedded and 56 pairs of fresh cancerous and normal esophageal tissues from ESCC patients who underwent curative surgery between 2003 and 2004 at Linzhou People’s Hospital (Linzhou, China). No patient received radiotherapy or chemotherapy before operation. All cases were pathologically confirmed as ESCC. Among the patients, there were 28 women and 36 men, with a median age of 60 years (range 40–80 years). Lymph-node metastasis was detected in 38 cases. The study was approved by the Ethical Review Board of Zhengzhou University (Zhengzhou, China). Written informed consent was obtained from each patient for the use of their tissue in research.
Immunohistochemical staining for MCM10
Immunohistochemical analysis of MCM10 was performed following a standard protocol. In brief, tissue sections were deparaffinized, rehydrated, and incubated with 3% H2O2 to eliminate endogenous peroxidase activity. After blocking with 5% normal goat serum, sections were incubated with rabbit anti-MCM10 polyclonal antibody (1:500 dilution; Atlas, Stockholm, Sweden) at 4°C overnight, followed by biotinylated goat anti-rabbit immunoglobulin for 30 min at room temperature. After washing, sections were incubated with 3,3′-diaminobenzidine used as a chromogen and counterstained with hematoxylin. Negative controls were included using nonimmune serum.
The evaluation of immunohistochemical results was done in a blind manner by two independent pathologists. A final staining score was calculated by multiplying the scores for staining intensity (0, negative; 1, weak; 2, intermediate; 3, strong) and percentage of positive cells (0, <25% positive cells; 1, 25%–50% positive cells; 2, 50%–75% positive cells; 3, >75% positive cells). Samples were defined as high MCM10 expression when the final scores were 4–9.
Real-time PCR analysis
Fresh tissue samples were immediately frozen in liquid nitrogen after surgical resection. Total RNA from tissues and cells was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized using the HiScript Q Select RT SuperMix for quantitative PCR and random hexamers from Vazyme Biotech (Nanjing, China). Quantitative real-time PCR was carried out with the SYBR Green PCR Master Mix (Vazyme Biotech). PCR primers are as follows: MCM10 forward, 5′-CACAGAAATGAACAAGAA-3′ and MCM10 reverse, 5′-AATAAGAACAAGGACACA-3′; GAPDH forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′ and GAPDH reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′. GAPDH was used as a loading control. The relative expression of MCM10 was calculated using the 2-ΔΔCt method.13
Reverse transcription PCR (RT-PCR) analysis
Total RNA was extracted and reverse-transcribed to cDNA, as described above. PCR amplification was conducted using the same primers as used in real-time PCR. PCR products were subjected to 1% agarose gel electrophoresis and stained with ethidium bromide.
Cell culture
K520, K510, K410, K30, K180, HKESC1, EC7906, EC18, and EC109 ESCC cells were kindly provided by Professor Srivastava (University of Hong Kong). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific). The usage of the cell lines was approved by the Ethical Review Board of Zhengzhou University.
Plasmid construction
For generation of MCM10-targeting single-guide RNAs (sgRNAs), three pairs of oligonucleotides were designed with the sequences as follows: sgRNA-1 forward, 5′-ACCGGAGGCTGATGATGGAGAAAC-3′ and sgRNA-1 reverse, 5′-AAACGTTTCTCCATCATCAGCCTC-3′; sgRNA-2 forward, 5′-ACCGGAAAATCTGGCCACTCTCTT-3′ and sgRNA-2 reverse, 5′-AAACAAGAGAGTGGCCAGATTTTC-3′; sgRNA-3 forward, 5′-ACCGGGCCACTCTCTTTGGAGATA-3′ and sgRNA-3 reverse, 5′-AAACTATCTCCAAAGAGAGTGGCC-3′. The complementary oligonucleotides were annealed and cloned into the pGL3-U6 expression vector. To evaluate CRISPR-mediated gene knockout efficiency, MCM10 genomic fragments containing sgRNA target sites were inserted into the pmCherry-C1-enhanced green fluorescent protein (EGFP) plasmid. The insertion of MCM10 genomic sequence created a frameshift mutation that led to disruption of the expression of EGFP. When the inserts were removed by sgRNA-guided Cas9 endonuclease, EGFP expression was detected, thus indicating the knockout efficiency. A plasmid expressing human Cas9 was obtained from Addgene. Full-length MCM10 cDNA in pCMV6-Entry vector was obtained from OriGene Technologies (Rockville, MD, USA).
Cell transfection and sorting
EC109 cells (1 × 106 cells/well) were seeded onto six-well plates and transfected with the Cas9 vector, sgRNA-expressing plasmid, and reporter plasmid (0.8 μg for each) with Lipofectamine 3000 (Thermo Fisher Scientific). Forty-eight hours after transfection, transfected cells were observed under a fluorescence microscope and subjected to flow cytometric sorting. Cells showing strong EGFP signals were sorted. Cells without transfection of the sgRNA-expressing plasmid were used as a control. The sorted cells were plated at a density of 1 cell/well onto 96-well plates by limiting dilution. Cell clones were collected and tested for gene mutation or deletion.
In rescue experiments, MCM10-depleted EC109 cells were transfected with a plasmid expressing a constitutively active isoform of Akt or empty vector (Addgene, Cambridge, MA, USA) using Lipofectamine 2000. Twenty-four hours after transfection, cells were tested for proliferation and migration. For inhibitor experiments, K510 ESCC cells were pretreated with LY294002 (25 μM; Sigma, St Louis, MO, USA) or vehicle for 30 min at 37°C before transfection with MCM10-overexpressing plasmid or empty vector.
T7 endonuclease I assay
Genomic fragments containing the sgRNA-1 target site were amplified by PCR with the following primers: forward, 5′-CGTGCTTATTCTCTGTCCTTTCTC-3′ and reverse, 5′-CTGGCCCAAACATTTCATCTACCA-3′. PCR products were purified and mixed with wild-type genomic DNA (in a 1:1 ratio). The mixture was denatured at 100°C for 5 min and annealed at room temperature. After treatment with T7 endonuclease I (New England Biolabs, Ipswich, MA, USA) at 37°C for 2 h, the resulting fragments were subjected to 1% agarose gel electrophoresis and stained with ethidium bromide.
DNA sequencing
PCR fragments containing the sgRNA-1 target site were ligated to the T-simple vector and subjected to DNA sequencing performed by Shanghai Sangon Biotechnology Company (Shanghai, China).
Cell growth assay
Cells were plated in 24-well plates (5 × 103 cells/well) and cultured for 7 days and counted using a hemocytometer. Each experiment with six replicates was repeated three times.
Colony formation assay
EC109 cell clones expressing wild-type and mutant MCM10 were seeded onto six-well plates (1,000 cells/well) and cultured for 3 weeks. Colonies were stained with 1% bromophenol blue and counted. For soft-agar colony formation assay, DMEM containing 0.6% agar and 10% FBS was plated on six-well plates. After solidification, cells (1,000 cells/well) suspended in culture medium containing 0.4% agar and 10% FBS were added on the gel. Cells were incubated for 3 weeks at 37°C. Visible colonies were photographed and counted.
In vitro wound-healing assay
Cells were seeded onto six-well plates (6 × 105 cells/well) and allowed to grow to 90% confluence. The cell monolayer was scratched with a 200-μL pipette tip. To block cell proliferation, mitomycin-C (Sigma; 1 μg/mL) was added in the media. After incubation for 48 h, cells were photographed. Wound healing was quantified by measuring the shortest distance between scratch edges at 0 and 48 h after scratching.
Western blot analysis
Cell lysates were prepared in lysis buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP40, 0.5% deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) containing 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 mmol/L phenylmethylsulfonyl fluoride (Sigma). Protein concentration was measured using the Protein Assay kit (Bio-Rad, Hercules, CA, USA). Equal amounts of protein samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated with anti-Akt (#9272, Cell Signaling Technology, Danvers, MA, USA; 1:500 dilution), anti-phospho-Akt (#9271, Cell signaling; 1:300 dilution), and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:2,000 dilution). Horseradish peroxidase-conjugated immunoglobulin G (Santa Cruz Biotechnology; 1:5,000 dilution) was used as a secondary antibody. Signals were visualized by enhanced chemiluminescence (Amersham Biosciences, Buckinghamshire, UK).
Statistical analysis
Comparison of quantitative data was determined by the Student’s t-test, unless otherwise specified. The relationship between MCM10 expression and clinicopathologic factors was analyzed using the chi-square test. P<0.05 was considered statistically significant.
Results
MCM10 is upregulated in ESCC
Immunohistochemical staining for MCM10 was performed in 64 pairs of formalin-fixed, paraffin-embedded ESCC samples and adjacent normal esophageal tissues. It was found that MCM10 was predominantly detected in the cytoplasm of tumor cells (Figure 1A). Low and high expression of MCM10 was detected in 27% (17/64) and 73% (47/64) of the ESCC specimens and 70% (45/64) and 30% (19/64) of normal esophageal tissues, respectively (Figure 1B). There was a significant difference in MCM10 protein levels between ESCC and adjacent normal tissues (P<0.0001; Figure 1B). We also examined the mRNA expression of MCM10 in 56 pairs of fresh ESCC samples and corresponding normal tissues by real-time PCR analysis. The results showed that the amounts of MCM10 transcripts were significantly greater in ESCC than in normal tissues (P<0.001; Figure 1C). Compared to the other ESCC cell lines tested, K520, K510, and K410 cells showed lower levels of MCM10 mRNA (Figure 1D).
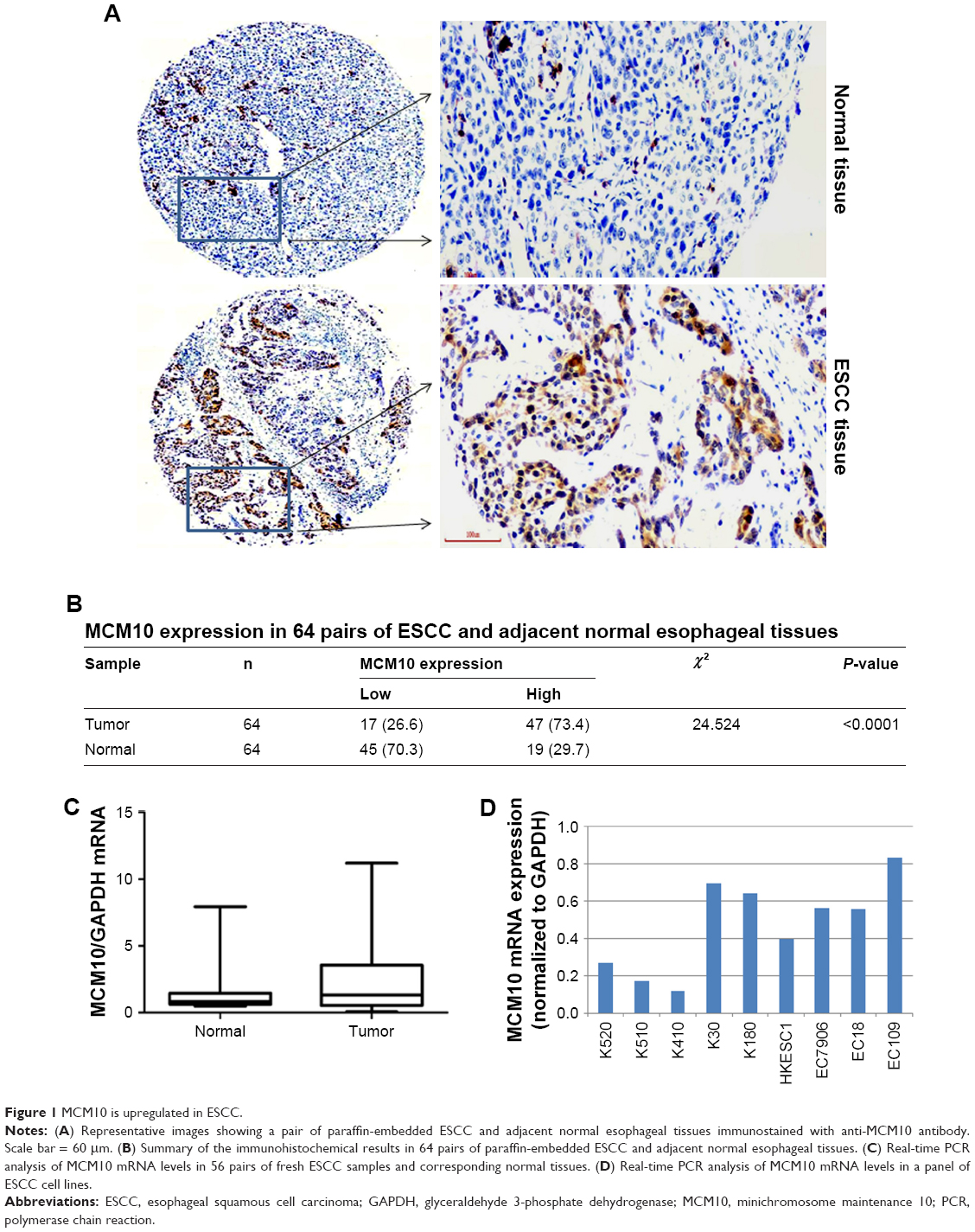 | Figure 1 MCM10 is upregulated in ESCC. |
Relationship between MCM10 expression and clinicopathologic features of ESCC
Next, we assessed the correlations between MCM10 expression and clinicopathologic features of ESCC. As summarized in Table 1, the expression of MCM10 was significantly associated with age at diagnosis (P=0.033). However, no significant difference was detected between MCM10 levels and other parameters including gender, differentiation grade, invasion status, and TNM stage.
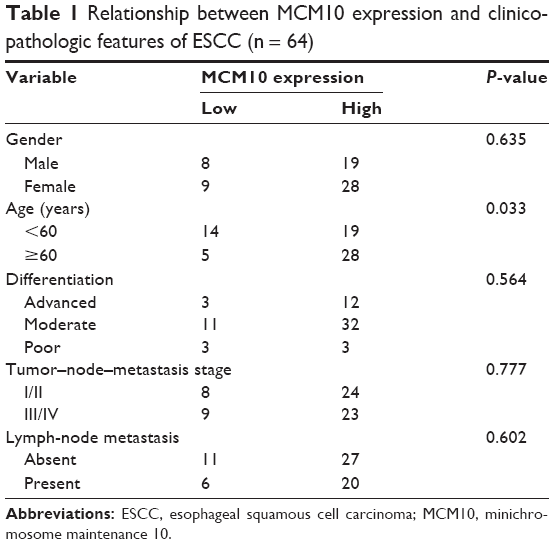 | Table 1 Relationship between MCM10 expression and clinicopathologic features of ESCC (n = 64) |
Deletion of MCM10 through CRISPR/Cas9 technology
EC109 cells were transfected with the Cas9, sgRNAs, and reporter constructs, and after incubation for 48 h, the reporter expression was examined. The expression of EGFP was found in some reporter-transfected clones, indicating the removal of the MCM10 sgRNA target site in the reporter by sgRNA-guided Cas9 (Figure 2A). Moreover, delivery of sgRNA-1 yielded the highest knockout efficiency, compared to the other two sgRNAs (data not shown). Therefore, in the following experiments, we used sgRNA-1 to eliminate MCM10 expression. Flow cytometric sorting showed that 18% of sgRNA-1-transfected cells emitted both red and green fluorescence (data not shown). Flow cytometry sorted cells were expanded to form single-cell clones. PCR amplicons were subjected to the mismatch-sensitive T7 endonuclease assay (Figure 2B). It was found that out of the 96 cell clones tested, 72 had mutations induced by sgRNA-guided Cas9. Sequencing of the PCR products demonstrated that 30 of the 33 samples (90.9%) had deletion mutations, two (6%) frameshift mutations, and one wild-type. Additionally, RT-PCR and Western blot analyses confirmed the ablation of MCM10 in the Clone 5 subline (Figure 2C).
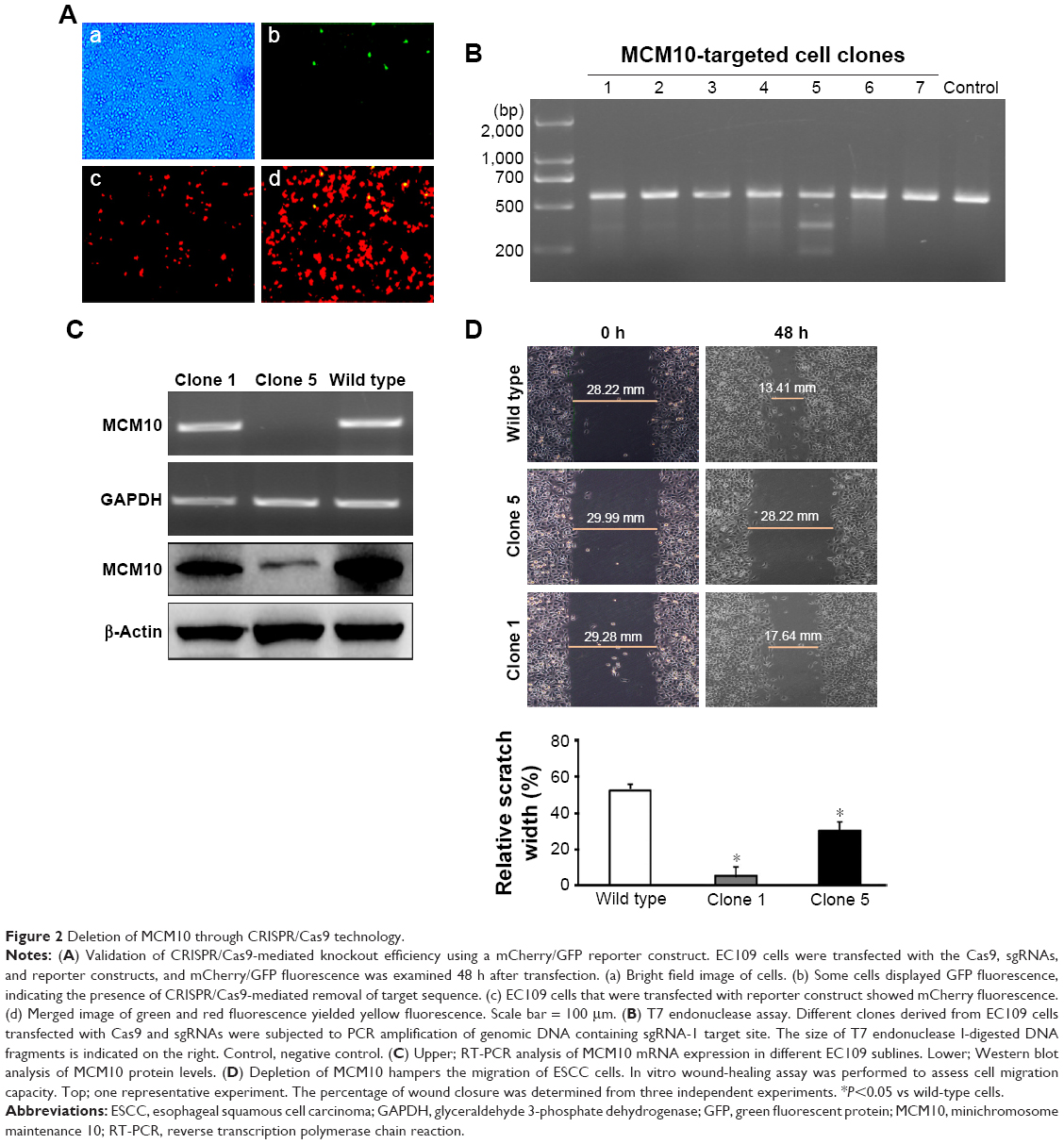 | Figure 2 Deletion of MCM10 through CRISPR/Cas9 technology. |
Depletion of MCM10 suppresses the migration of ESCC cells
We also checked the effect of MCM10 knockout on the migration of ESCC cells. The percentage of wound closure was markedly reduced in the Clone 5 subline compared to that in the Clone 1 subline (Figure 2D). Moreover, the Clone 1 subline exhibited a reduced migration capacity relative to wild-type EC109 cells (Figure 2D). These observations suggest an important role for MCM10 in the regulation of ESCC cell migration.
MCM10 is required for the growth of ESCC cells
Next, we assessed the impact of MCM10 deficiency on the growth of EC109 cells. We found that the Clone 5 subline had a significantly lower proliferation capacity than the Clone 1 subline (P<0.05; Figure 3A). Colony formation assay confirmed that the Clone 5 subline formed significantly fewer colonies than the Clone 1 subline (P<0.05; Figure 3B). To investigate the role of MCM10 in the anchorage-independent growth of ESCC cells, soft-agar colony formation assay was performed. As depicted in Figure 3C, the number of colonies formed by the Clone 5 subline was significantly lower than that by the Clone 1 subline (P<0.05). These results indicate that MCM10 plays an essential role in the growth of ESCC cells.
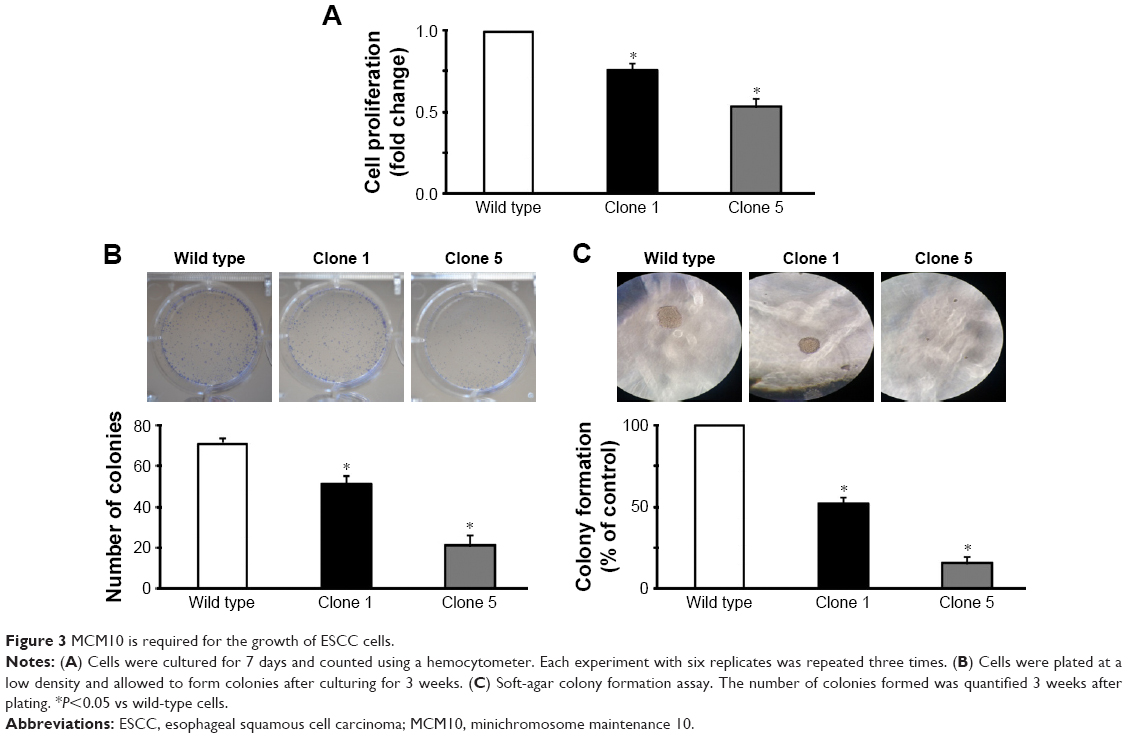 | Figure 3 MCM10 is required for the growth of ESCC cells. |
MCM10 overexpression enhances the growth and migration of ESCC cells
Next, we explored the effect of ectopic expression of MCM10 on the aggressive phenotype of ESCC cells. It was found that overexpression of MCM10 (Figure 4A) significantly promoted the growth (Figure 4B) and migration (Figure 4C) of both K510 and K520 ESCC cells.
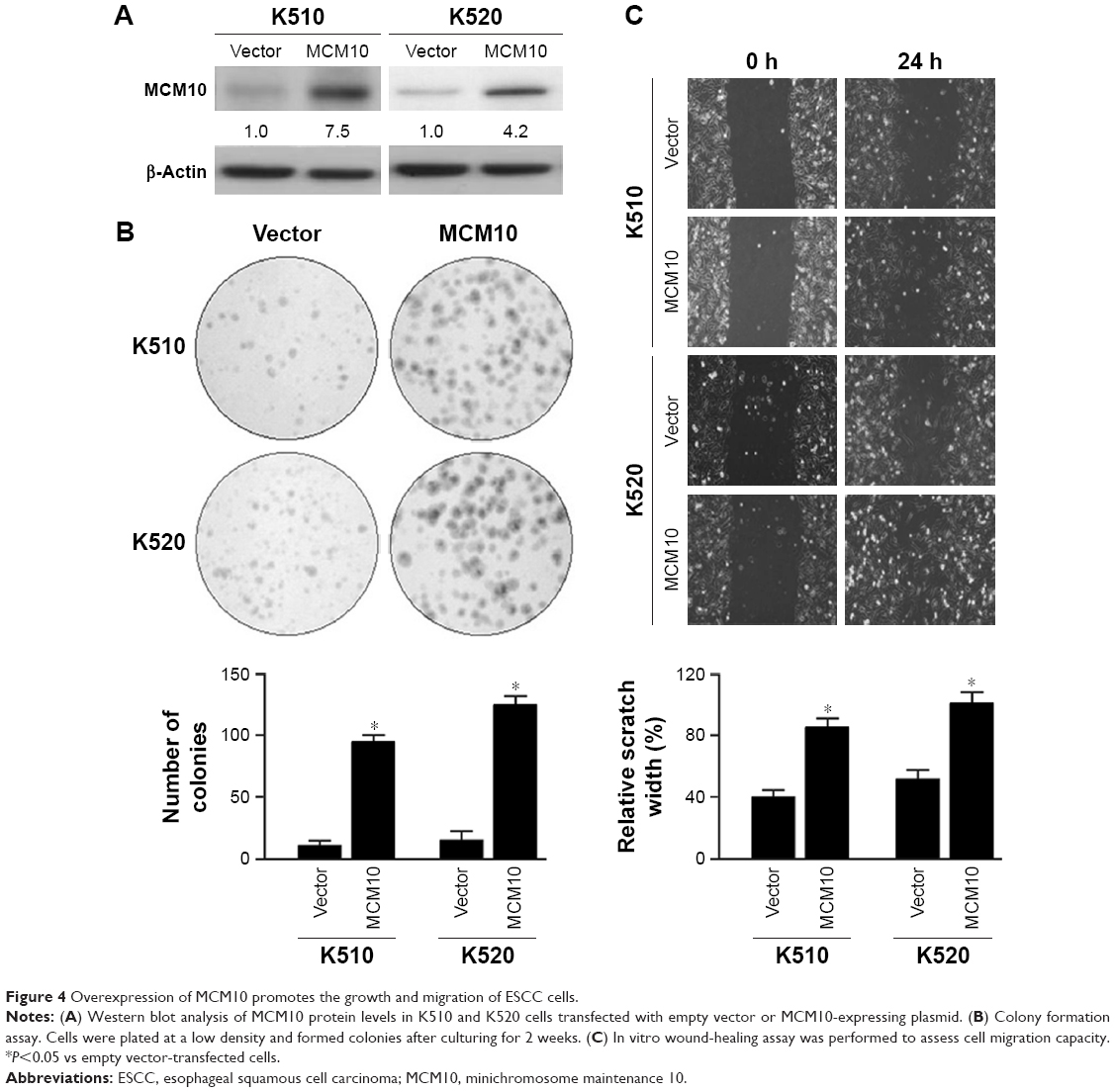 | Figure 4 Overexpression of MCM10 promotes the growth and migration of ESCC cells. |
MCM10 loss-induced suppression of growth and migration involves inhibition of Akt signaling
Western blot analysis revealed that MCM10 depletion led to a marked inhibition of Akt phosphorylation on Ser473 in EC109 cells (Figure 5A). However, the phosphorylation levels of nuclear factor kappa B (NF-κB), extracellular signal-regulated kinase (ERK), and p38 protein were not altered by MCM10 depletion (data not shown). To confirm the role of Akt signaling in the action of MCM10, we performed rescue experiments in the Clone 5 subline using constitutively active Akt (Figure 5B). Of note, overexpression of constitutively active Akt restored cell growth (Figure 5C) and migration (Figure 5D) in MCM10-deficient cells. In contrast, ectopic expression of active Akt had no significant impact on the proliferation of wild-type EC109 cells. In addition, the migration was moderately increased by overexpression of active Akt in wild-type EC109 cells.
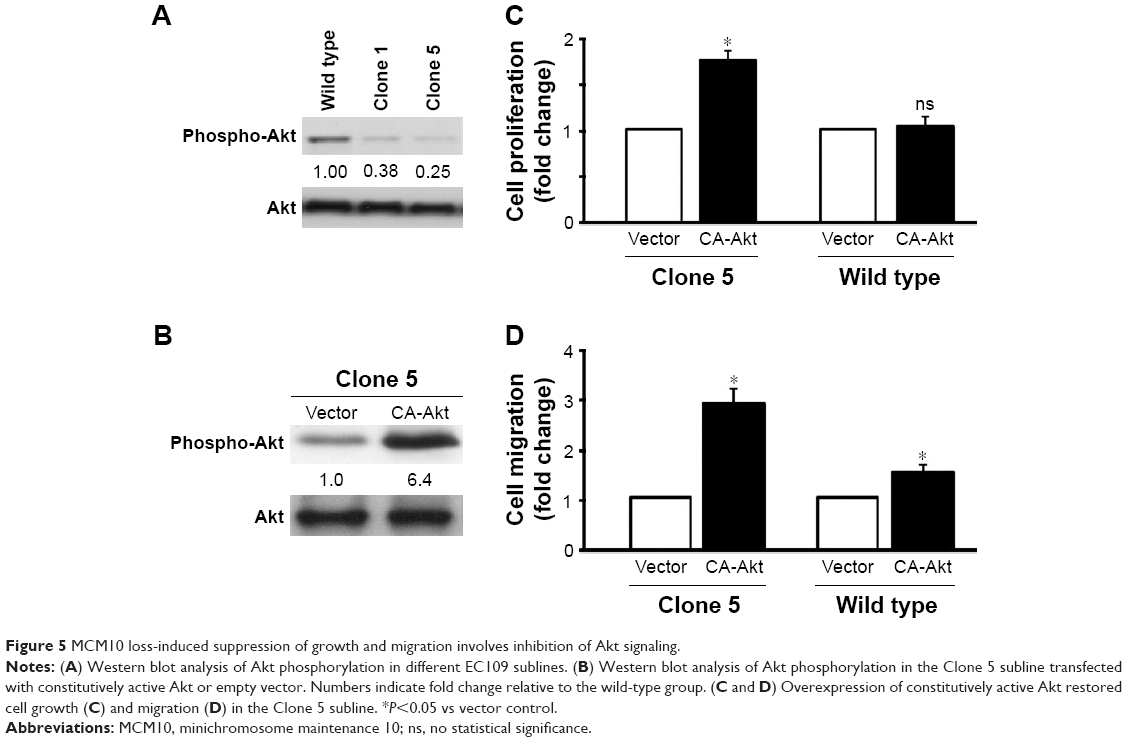 | Figure 5 MCM10 loss-induced suppression of growth and migration involves inhibition of Akt signaling. |
Inhibition of Akt signaling blocks MCM10-induced cell proliferation and migration
Western blot analysis indicated that overexpression of MCM10 promoted Akt phosphorylation in K520 cells transfected with MCM10-expressing plasmid (Figure 6A). We pretreated K520 cells with LY294002 or vehicle before transfection with empty vector or MCM10-expressing plasmid. Inhibition of Akt phosphorylation can restrain cell growth (Figure 6B) and cell migration capacity (Figure 6C) in MCM10-overexpressed cells.
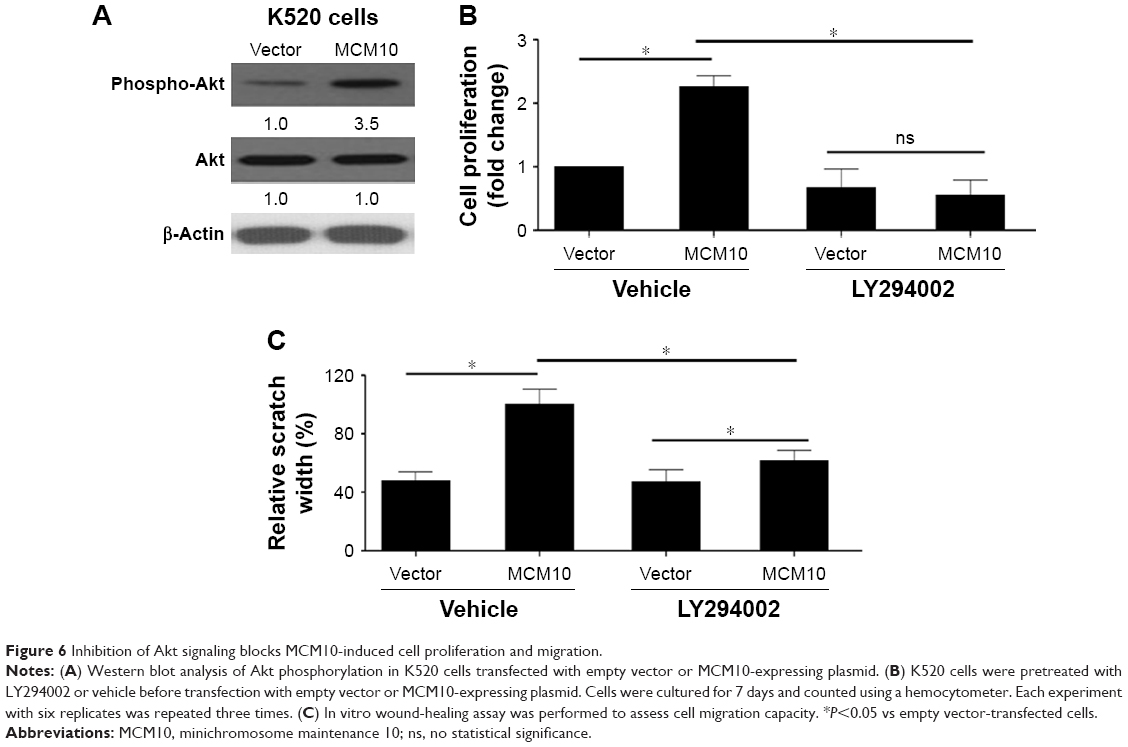 | Figure 6 Inhibition of Akt signaling blocks MCM10-induced cell proliferation and migration. |
Discussion
In this study, we showed that MCM10 was upregulated at both protein and mRNA levels in ESCC relative to adjacent normal esophageal tissues. Consistent with our findings, MCM10 overexpression is detected in cervical cancer and urothelial carcinoma.11,12 A previous study has reported that MCM10 is frequently mutated in early gastric cancer.14 These results suggest a possible role for MCM10 in carcinogenesis. It has been documented that MCM10 overexpression correlates with advanced stage, nodal metastasis, and vascular invasion in urothelial carcinoma. However, our data showed that there was no significant relationship between MCM10 expression and aggressive parameters of ESCC. The conflicting findings may be explained by different cancer types. Further studies are needed to explore the clinical significance of MCM10 overexpression in ESCC.
The emerging genome-editing technology CRISPR/Cas9 is based on RNA-guided nuclease Cas9 and guided RNA, which enables rapid, specific, and efficient modification of target genes in many cell types.15,16 Zhai et al employed the CRISPR/Cas9 system to inactivate PLCE1 gene in esophageal cancer cells.17 Another study has documented the knockout of p53 gene in esophageal adenocarcinoma cells through the CRISPR/Cas9 approach.18 In this study, we also successfully inactivated MCM10 in EC109 cells through the CRISPR/Cas9 technology. Of note, knockout of MCM10 significantly impaired the growth and colony formation of EC109 cells. Moreover, MCM10 knockout suppressed the anchorage-independent growth of EC109 cells on soft agar. In addition, the migration capacity was reduced in MCM10-null EC109 cells relative to wild-type equivalents. These results collectively indicate that MCM10 is required for the maintenance of the aggressive phenotype in ESCC cells.
MCM10 is implicated in MCM2–7 remodeling and cell-cycle progression and its functional deletion causes S phase defects,19 which may explain the reduced growth in MCM10-null EC109 cells. In addition to regulation of cell-cycle progression, MCM proteins have the capacity to modulate cell migration and invasion. It has been documented that overexpression of MCM2, MCM3, and MCM7 facilitates cell migration and invasion in medulloblastoma cells.20 Another study showed that MCM7 knockdown hinders the migration of KYSE510 and EC9706 cells in vitro.21 In this study, we provide evidence for the importance of MCM10 in the migration of ESCC cells. Therefore, the interaction between MCM10 and MCM2–7 proteins may be critical for the growth and progression of ESCC.
To get more insight into the action of MCM10, we examined the signaling pathways involved. We noted that MCM10 null suppressed the phosphorylation of Akt, but spared NF-κB, ERK, and p38 in EC109 cells, suggesting the involvement of Akt signaling in the activity of MCM10. Rescue experiments provided direct evidence that overexpression of constitutively active Akt accelerated the growth and migration of MCM10-null EC109 cells. Activation of the Akt pathway is implicated in the growth and migration of ESCC cells induced by matrix metalloproteinase-1.22 Another study demonstrated that overexpression of inhibitor of differentiation or DNA binding promotes growth and invasion of esophageal cancer cells through the Akt signaling pathway.23 These studies, combined with our data, point toward the importance of Akt signaling in the action of MCM10 in ESCC. However, further studies are needed to clarify the mechanism by which MCM10 regulates the activation of Akt signaling.
A major limitation of this study is that the role of MCM10 was tested in only one ESCC cell line. Ongoing studies are designed to address the effect of targeted ablation of MCM10 on tumor growth and metastasis in mouse models inoculated with multiple ESCC cell lines.
In conclusion, our data demonstrate that MCM10 is upregulated in ESCC and contributes to the aggressive phenotype of ESCC cells. Activation of Akt signaling is involved in the oncogenic activity of MCM10 in ESCC. Targeting MCM10 may provide a potential therapeutic benefit in the treatment of ESCC.
Data sharing statement
All the data obtained from the present study are available from the corresponding author under reasonable request.
Acknowledgments
This work was funded under the Youth Fund of First Affiliated Hospital of Zhengzhou University. The authors are grateful to all study participants.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Zhang P, Li XM, Zhao XK, et al. Novel genetic locus at MHC region for esophageal squamous cell carcinoma in Chinese populations. PLoS One. 2017;12(5):e0177494. | ||
van Hagen P, Hulshof MC, van Lanschot JJ, et al; CROSS Group. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366(22):2074–2084. | ||
Best LM, Mughal M, Gurusamy KS. Non-surgical versus surgical treatment for oesophageal cancer. Cochrane Database Syst Rev. 2016;3: CD011498. | ||
Ganaie SS, Zou W, Xu P, Deng X, Kleiboeker S, Qiu J. Phosphorylated STAT5 directly facilitates parvovirus B19 DNA replication in human erythroid progenitors through interaction with the MCM complex. PLoS Pathog. 2017;13(5):e1006370. | ||
Maiorano D, Lutzmann M, Méchali M. MCM proteins and DNA replication. Curr Opin Cell Biol. 2006;18(2):130–136. | ||
Lõoke M, Maloney MF, Bell SP. MCM10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 2017;31(3):291–305. | ||
Stewart PA, Khamis ZI, Zhau HE, et al. Upregulation of minichromosome maintenance complex component 3 during epithelial-to-mesenchymal transition in human prostate cancer. Oncotarget. 2017;8(24):39209–39217. | ||
Coli A, Asa SL, Fadda G, et al. Minichromosome maintenance protein 7 as prognostic marker of tumor aggressiveness in pituitary adenoma patients. Eur J Endocrinol. 2016;174(3):307–314. | ||
Abe S, Yamamoto K, Kurata M, et al. Targeting MCM2 function as a novel strategy for the treatment of highly malignant breast tumors. Oncotarget. 2015;6(33):34892–34909. | ||
Das M, Prasad SB, Yadav SS, et al. Over expression of minichromosome maintenance genes is clinically correlated to cervical carcinogenesis. PLoS One. 2013;8(7):e69607. | ||
Li WM, Huang CN, Ke HL, et al. MCM10 overexpression implicates adverse prognosis in urothelial carcinoma. Oncotarget. 2016;7(47): 77777–77792. | ||
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. | ||
Kang G, Hwang WC, Do IG, et al. Exome sequencing identifies early gastric carcinoma as an early stage of advanced gastric cancer. PLoS One. 2013;8(12):e82770. | ||
Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I. CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids. 2017;7:211–222. | ||
Zhu P, Wu F, Mosenson J, Zhang H, He TC, Wu WS. CRISPR/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol Ther Nucleic Acids. 2017;7:31–41. | ||
Zhai S, Liu C, Zhang L, et al. PLCE1 promotes esophageal cancer cell progression by maintaining the transcriptional activity of snail. Neoplasia. 2017;19(3):154–164. | ||
Liu DS, Read M, Cullinane C, et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut. 2015;64(10):1506–1516. | ||
Quan Y, Xia Y, Liu L, et al. Cell-cycle-regulated interaction between MCM10 and double hexameric Mcm2-7 is required for helicase splitting and activation during S Phase. Cell Rep. 2015;13(11): 2576–2586. | ||
Lau KM, Chan QK, Pang JC, et al. Minichromosome maintenance proteins 2, 3 and 7 in medulloblastoma: overexpression and involvement in regulation of cell migration and invasion. Oncogene. 2010;29(40): 5475–5489. | ||
Qiu YT, Wang WJ, Zhang B, Mei LL, Shi ZZ. MCM7 amplification and overexpression promote cell proliferation, colony formation and migration in esophageal squamous cell carcinoma by activating the AKT1/mTOR signaling pathway. Oncol Rep. 2017;37(6):3590–3596. | ||
Liu M, Hu Y, Zhang MF, et al. MMP1 promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2016; 377(1):97–104. | ||
Li B, Tsao SW, Li YY, et al. Id-1 promotes tumorigenicity and meta-stasis of human esophageal cancer cells through activation of PI3K/AKT signaling pathway. Int J Cancer. 2009;125(11):2576–2585. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.