Back to Journals » OncoTargets and Therapy » Volume 15
Aberrant MAPK Signaling Offers Therapeutic Potential for Treatment of Ovarian Carcinoma
Authors Colic E, Patel PU, Kent OA ![]()
Received 12 June 2022
Accepted for publication 1 November 2022
Published 5 November 2022 Volume 2022:15 Pages 1331—1346
DOI https://doi.org/10.2147/OTT.S361512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Eva Colic *, Preya U Patel *, Oliver A Kent
Department of Pharmacology, adMare BioInnovations, Montreal, Quebec, Canada
*These authors contributed equally to this work
Correspondence: Oliver A Kent, adMare BioInnovations, Centre d’innovation - 7171 Frederick-Banting, Montréal, Quebec, H4S 1Z9, Canada, Tel +1 438 874-6546, Email [email protected]
Abstract: Ovarian cancer remains the most lethal gynecological malignancy worldwide due to lack of effective screening, vague early symptoms, poor description of biomarkers, and absence of effective treatment regimes. Epithelial ovarian carcinoma (EOC) is categorized into five distinct disease subtypes which collectively account for ~90% of ovarian carcinomas. Most women present at advanced stages contributing to a poor overall 5-year survival rate. Standard treatment for EOC is cytoreductive surgery and platinum-based chemotherapy; however, most patients suffer from recurrence and platinum-resistant disease, which highlights an urgent need for targeted therapy. The high frequency of molecular alterations affecting gain-of-function signaling through the RAS mitogen-activated protein kinase (MAPK) pathway in EOC has prompted pre-clinical and clinical efforts toward research into the effectiveness of MAPK pathway inhibition as a second-line treatment. The RAS/MAPK pathway is a highly conserved signal transduction cascade, often disrupted in cancer, that regulates tumorigenic phenotypes including cellular proliferation, survival, migration, apoptosis, and differentiation. Herein, the role of the MAPK pathway in EOC with emphasis on targetability of the pathway is described. Pre-clinical and clinical efforts to target MAPK signaling in EOC have identified several MAPK pathway inhibitors that offer efficacious potential for monotherapy and in combination with other compounds. Thus, inhibition of the RAS/MAPK pathway is emerging as a tractable strategy for treatment of ovarian cancer that may permit development of personalized therapy and improved prognosis for women challenged by this disease.
Keywords: RAS/MAPK, ovarian, serous, clear-cell, mucinous, endometrioid
Introduction
Ovarian cancer is the most lethal gynecological malignancy worldwide. Approximately 90% of malignant ovarian cancers are epithelial ovarian carcinoma (EOC) derived from the epithelial cells lining the ovarian surface, peritoneum, or fallopian tube.1 Due to lack of effective screening methods, poor description of early biomarkers, and vague distinctive symptoms, most women present at advanced stages contributing to a poor overall 5-year survival rate around 30–40%.1–3 Despite an initial response to standard therapy of cytoreductive surgery and platinum-based chemotherapy, most patients suffer disease recurrence following treatment.4,5 Therefore, a personalized regimen or targeted therapy for treating ovarian carcinoma remains an urgent unmet need.
The RAS/MAPK pathway is an essential signaling module that regulates cellular proliferation, cell survival, and apoptosis, all of which are perturbed in cancer.6,7 Somatic mutations leading to constitutive RAS activity are drivers of many lethal cancers including pancreatic, lung, colorectal, melanoma, and certain hematological cancers.6–8 Present, but less commonly described are the pathogenesis and molecular genetic features of a subset of primary EOC with somatic mutations in RAS pathway components.3
The potential for therapeutic targetability of the RAS/MAPK pathway in ovarian cancer has been previously described.9,10 Histological evidence demonstrated punctual mutations are prevalent in EOCs, establishing RAS pathway involvement in some types of ovarian cancers.10,11 Herein, we describe the role of the RAS/MAPK pathway in EOC with emphasis on targetability of the pathway directing novel treatment regimes.
The RAS-MAPK Pathway Signal Transduction Cascade
The RAS mitogen-activated protein kinase (MAPK) pathway is a highly conserved signal transduction cascade that regulates cell growth, survival, migration, apoptosis, and differentiation.6 Activation of the canonical MAPK cascade involves signaling through RAS, RAF, MEK, and extracellular signal-regulated kinase (ERK). Signals received at the cell surface activate receptor tyrosine kinases (RTKs), which are essential to promote activation of RAS and RAF followed by the sequential phosphorylation of MEK and ERK (Figure 1).7 RAS/MAPK signaling is triggered by multiple growth factors, inflammatory cues, and cytokine stimulation, thus modulating diverse cellular and physiological processes.12
 |
Figure 1 RAS signaling engages MAPK and PI3K pathways. Receptor tyrosine kinase (RTK, e.g. HER2, VEGF) activation by mitogenic signaling activates guanine exchange factors (e.g. RasGRP, SOS) that promote exchange of GDP for GTP on Ras. GTPase activating proteins (e.g. RasGAP, p120GAP, NF1) facilitate hydrolysis of GTP to inactivate Ras. Oncogenic mutation (yellow star) traps Ras in a constitutively active state. Activated Ras engages downstream effectors RAF and PI3K to regulate signaling through the MAPK (left) and PI3K (right) pathways affecting pro-tumorigenic phenotypes such as proliferation, survival, and apoptosis. |
RAS is the prototypical member of the Ras family and is encoded by three proto-oncogenes, HRAS, KRAS, and NRAS, all of which are ubiquitously expressed in mammalian cells and interact with the same set of effector proteins.6,8,13,14 RAS is the most frequently mutated gene in human cancer, highlighting an essential role in tumorigenesis.8,14 Unfortunately, direct inhibitors of RAS have eluded researchers for more than three decades, earning RAS the title of “undruggable”. Thus, finding therapeutically tractable targets in RAS effector pathways is critical for the effective treatment of RAS-driven cancers.
RAS proteins are small GTPases that operate as molecular switches to activate signal transduction pathways.14 In the absence of oncogenic mutation, RAS cycles between inactive GDP-bound and active GTP-bound forms. Guanine nucleotide exchange factors (GEFs) promote exchange of GDP for GTP leading to RAS activation.15 RTK activation stimulates GEF activity and promotes GDP exchange on RAS permitting RAS to interact directly with RAF. Intrinsic but weak GTPase activity of RAS is augmented by a class of tumor suppressors called RAS GTPase activating proteins (GAPs), that catalyze the hydrolysis of GTP and thus inactivate RAS.6,15,16 RAS signaling is coupled to the activation of multiple effector pathways including RAF/MEK/ERK and the phosphoinositide 3-kinase (PI3K)/AKT that relay mitogenic signaling to regulate pro-tumorigenic phenotypes (Figure 1).
Commonly observed in cancer, gain-of-function mutations trap RAS in a constitutively activated, GTP-bound state potentiating MAPK signaling. Mutations in codons 12, 13, or 61 of RAS disrupt GAP-mediated GTP hydrolysis, allowing these mutants to accumulate in a persistently GTP-bound state.8,14 For RAS-driven cancers, the lack of RAS mutant specific therapeutics underscores an urgent need to identify therapeutic strategies targeting RAS effectors, which may demonstrate selectivity and improved outcomes over conventional chemotherapy.10,17
The Dichotomy of Ovarian Carcinoma: Type I and Type II Cancer
EOC are a heterogeneous group of cancers that account for ~90% of all ovarian cancers.1,18,19 Although EOC share a common site of tumor growth in the ovary, evidence suggests EOC encompasses five distinct diseases, with differing clinical presentation, genetic background, response to chemotherapy, prognosis, and sites of origin.19–21 The major histologically distinct subtypes include serous high- and low-grade, clear cell, endometrioid, and mucinous carcinomas, each characterized by specific genetic backgrounds (Figure 2A). Importantly, the molecular alterations that define subtypes of EOC offer the potential for targeted therapies including those subtypes that may respond to RAS/MAPK inhibition.20
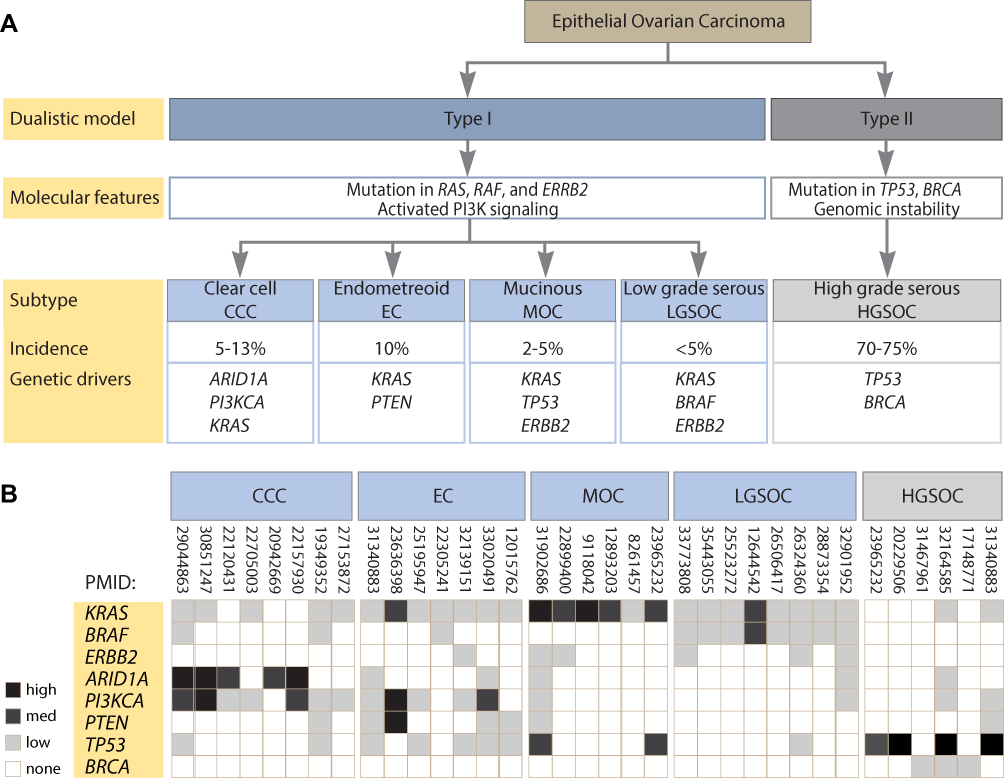 |
Figure 2 Analysis of genetic aberrations in epithelial ovarian carcinoma. (A) Classification of epithelial ovarian cancers using the type I and type II dualistic approach into five distinct subtypes based on genetic abnormalities and histopathology. (B) Heat map presentation of frequency of mutation for each of the indicated genes in the five subtypes of epithelial ovarian cancer. Percentage of mutation frequency reported in the indicated references was converted to high (>80%), medium (40–80%), and low (<40%) expression value. None may refer to zero or not reported. References indicated as PubMed reference number (PMID). |
Studies suggest a dualistic model of ovarian carcinogenesis that correlates with the clinical, histopathological, genetic, and molecular features of the disease. In this model, ovarian tumors are divided into two groups designated as type I and type II.22 Based on molecular and cell of origin studies, type II tumors can be classed together; however, type I tumors are not homogeneous, even within distinct histological subtypes arguing for a refinement of the classification.23,24 Type I cancers include low-grade serous ovarian carcinoma (LGSOC), clear cell, endometrioid, and mucinous carcinomas, whereas type II cancers contain high-grade serous ovarian carcinoma (HGSOC), undifferentiated carcinoma and malignant mixed mesodermal carcinosarcoma.1 Although HGSOC and LGSOC best fit the description of the dualistic model, endometrioid, mucinous, and clear cell type I tumors include an uncertain origin, heterogeneous mutational spectrum, and clinical behavior, thus complicating classification.24
Type I tumors are often diagnosed at an early stage, are slow growing, generally confined to the ovary at diagnosis, and develop from well-established precursor lesions termed “borderline” tumors.25 Often resisting conventional chemotherapy, type I tumors are genetically stable with wild-type TP53 and BRCA1/BRCA2. Type I tumors are frequently characterized by mutations affecting the RAS/MAPK pathway including KRAS, BRAF, and ERBB2 (HER2/neu), as well as having aberrant activation of the PI3K/AKT pathway, likely associated with increased RAS activity and mutation or loss of PTEN.9,25 In contrast, type II tumors are rapidly growing highly aggressive neoplasms for which well-defined morphological precursor lesions have not been described.22 Type II tumors have a high level of genomic instability, are nearly ubiquitous for mutations in TP53, and nearly half harbor BRCA1/BRCA2 mutations.22,25,26 Although type II tumors typically present with wild-type RAS genes, type I tumors frequently harbor gain-of-function RAS mutations critical to delineate predictive therapeutic decisions.22,25,27
Pathogenic Features of EOC Subtypes: Targeting the RAS/MAPK Pathway
Evidence from mutational analysis of EOC tumors, cell lines, and micro-dissected tumors have established the mutational frequency of RAS/MAPK genes associated with the pathogenesis of EOC (Figure 2B). Analysis of the clinical features, frequency of mutations in genes affecting RAS pathway activity, and the potential for targeting the MAPK in studies supported by pre-clinical and clinical research are described below for each EOC subtype (Figure 3).
 |
Figure 3 Treatment strategies for epithelial ovarian carcinoma. (A–E) Treatment regimes described in the text for subtypes of epithelial ovarian cancer in (A) high-grade serous HGSOC, (B) clear cell carcinoma CCC, (C) endometrioid EC, (D) mucinous MOC, and (E) low-grade serous LGSOC with the compounds listed. Compounds in clinical trials are shown with the associated clinical trial numbers from the ClinicalTrials.gov database if applicable. Not available (n/a) refers to compounds involved in studies without clinical support. Molecular targets of each compound affecting RAS/MAPK pathway (blue), receptors (green), and other targets (yellow) are shown. |
High-Grade Serous Carcinoma (HGSOC)
HGSOC is the most common histological subtype of EOC representing ~70% of cases, presenting at an advanced stage, and accounting for the most deaths from ovarian cancer.9 HGSOC is typically driven by copy number abnormalities and genomic instability.9 Evidence suggests that HGSOC tumors originate in fallopian tubal secretory epithelial cells (FTSEC) and serous tubal intraepithelial carcinoma (STIC) has been established as the precursor lesion for HGSOC in animal models, leading to progression of advanced stage disease.26,28
TP53 mutations occur in 96% of HGSOC presenting more frequently in advanced ovarian carcinomas and mutations in BRCA1/BRCA2 are also frequently observed.26 Early TP53 mutation and BRCA loss cause deficiencies in DNA repair pathways which trigger chromosomal instability and widespread somatic copy number changes.21 Genetic mutations driving HGSOC oncogenesis are supported by a genetically engineered mouse model demonstrating Pax8, a marker of the fallopian secretory cells, which drove Cre-mediated inactivation of Brca1, Brca2, Tp53, and Pten, transformed fallopian tube epithelial cells which led to the development of STIC lesions.28
In some HGSOC tumors, oncogenesis appears to be driven by amplification of genes that activate the RAS/MAPK and PI3K/AKT pathways.9 One study has extensively characterized KRAS and BRAF mutations in 264 ovarian neoplasms and identified KRAS mutations in up to 12% of HGSOC tumors.29 Although RAS mutation rates are low in HGSOC, high transcript levels of RAF1, ERK1, and ERK2 have been significantly correlated with poor platinum-free survival in patients with advanced stage disease.30 Analysis of differential gene expression in 408 HGSOC tumors from the cancer genome atlas demonstrated a high intratumor MAPK-activated gene set was prognostic for poor outcome and decreased survival.31 The study demonstrated that selumetinib (a MEK inhibitor) reversed expression of a subset of MAPK-activated genes in vitro and in xenografts, suggesting the gene signature was predictive of a MEK inhibitor response.31
For HGSOC, cytoreductive surgery and combination chemotherapy with platinum compounds and taxanes is the first-line treatment,32 but due to high incidence of resistant disease, secondary treatments must be explored (Figure 3A). Although chemotherapy with carboplatin and paclitaxel achieves complete response, platinum resistance occurs at an alarming rate, with disease recurrence observed in 75% of HGSOC patients, thus contributing to an abysmal 5-year survival rate.9 Molecular characterization of platinum-resistant HGSOC has identified activation of pro-proliferative signaling pathways, including the RAS/MAPK in disease resistance.30 In resistant tumors, crosstalk drives numerous downstream targets of RAS/MAPK signaling including activation of ERK regulated transcription factors leading to upregulation of pro-tumorigenic gene signatures. In cisplatin-resistant HGSOC cell lines, resistance was attributed to activation of mutant p53 by MEK phosphorylation, arguing that MEK inhibition may be a therapeutic option for refractory HGSOC.33
Vascular endothelial growth factor (VEGF) is a membrane receptor that regulates a complex signaling cascade involving multiple kinases including the RAS/MAPK pathway to stimulate angiogenesis. Anti-angiogenesis agents including those that target VEGF are being investigated as second-line therapy for recurrent HGSOC, since high VEGF expression has been observed in serous carcinomas and most of these tumors are dependent on VEGF for progression.34 Regorafenib, a multikinase inhibitor that blocks signaling through VEGF receptors and the MAPK pathway,35 is currently in a phase II clinical trial for anti-tumor activity against persistent ovarian carcinoma in patients harboring tumors that are predominantly HGSOC. Unfortunately, no significant difference has been noted in progression-free survival (PFS), objective response rates (ORR), or overall survival (OS) between regorafenib or tamoxifen treatment groups (Table 1: NCT02584465). A study in phase III has examined pazopanib (a VEGF inhibitor) in patients with advanced ovarian cancer. Although pazopanib prolonged PFS to 17.9 months compared with 12.3 months in placebo, there was no associated improvement in median OS (Table 1: NCT00866697).
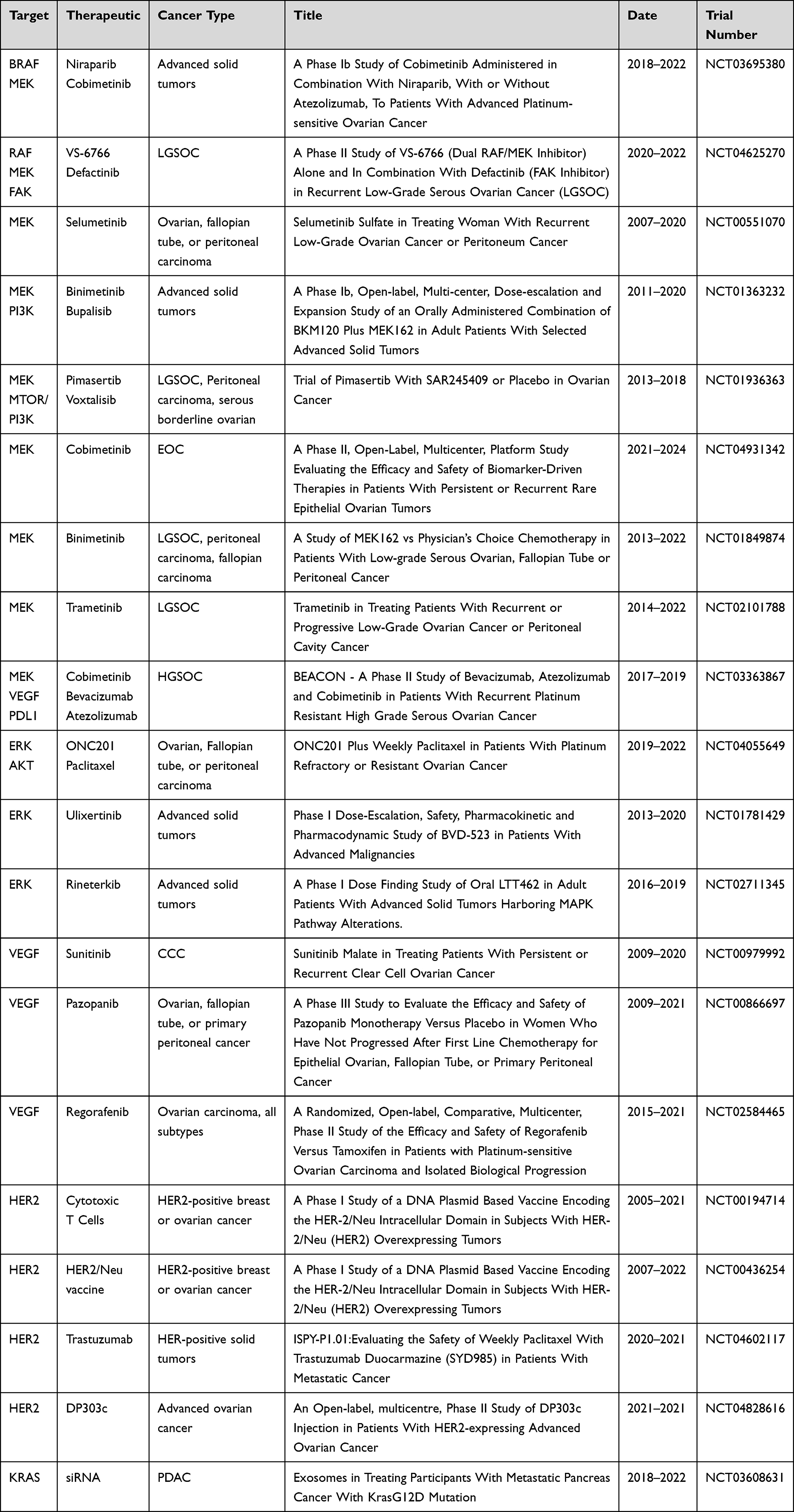 |
Table 1 Summary of Clinical Trials Targeting MAPK Signaling in Ovarian Carcinoma |
Additional evidence for VEGF inhibition as a treatment for HGSOC comes from a clinical trial that has tested cobimetinib (a MEK inhibitor) in combination with bevacizumab (a VEGF inhibitor) and PD-L1 inhibition. The PD-L1 pathway regulates adaptive immune resistance often exerted by tumor cells in response to anti-tumor activity.36 MEK inhibition with cobimetinib in combination with a monoclonal antibody inhibiting PD-L1 (atezolizumab) has previously been used to treat patients with solid tumors and shown to have potential synergistic activity in metastatic colon cancer.37 Referred to as the ABC study, the triple combination of atezolizumab, bevacizumab, and cobimetinib is being explored in a phase II clinical trial for treatment of recurrent platinum-resistant HGSOC (Table 1: NCT03363867). Cobimetinib is FDA-approved for use in combination with a BRAF inhibitor vemurafenib for the treatment of advanced melanomas harboring BRAF (V600E/K) mutation, suggesting additional combination therapies could be explored for ovarian cancer.
Clear Cell Carcinoma (CCC)
CCC is the second most common type of epithelial ovarian cancer and accounts for approximately 5–13% of EOC.38–40 CCC has been reported to arise from endometriosis that has implanted on the ovaries or in the peritoneal cavity and undergone malignant transformation. Research suggests that endometriosis may be the precursor of CCC and is thought to develop from retrograde menstruation.22 CCC frequently presents at an early stage with a high propensity for recurrence after primary chemotherapy.40 Patients with early-stage disease who underwent platinum-based therapy corroborate a high recurrence rate and decreased survival.41 Since CCC display aggressive clinical phenotypes which rarely respond to platinum-based chemotherapy, prognosis is reported as worse than in HGSOC.41,42
The majority of CCC express wild type TP53 and rarely harbor germline mutations in BRCA1/BRCA2. Consequently, CCC show low levels of chromosomal instability with fewer copy number variants.43,44 Collectively, the most common molecular aberrations involve ARID1A, the PI3K/AKT pathway, and RTK signaling through the RAS/MAPK pathway.44 Somatic inactivating mutation of ARID1A is reported to occur in up to 50% of CCC cases.21,22 The ARID1A gene encodes a protein involved in chromatin remodeling which functions as a tumor suppressor in ovarian clear cell carcinogenesis.45 In addition, approximately 50% of CCC harbor inactivating PTEN mutations and an activating mutation of PIK3CA, the catalytic subunit of PI3K, in up to 40% of cases.21,22 KRAS mutations have been identified in 15–30% of CCC with mutations specifically located in codon 12 but not in codon 13.46,47
Currently, the preferred treatment for CCC is a complete resection of the tumor which can be complicated in advanced stage disease.9 Further, drug resistance contributes to the poor prognosis of patients with CCC. An in vitro study revealed that low clear cell proliferation may account for chemoresistance to cisplatin via mechanisms that include decreased drug accumulation, increased drug detoxification, and increased DNA repair activity.42 Additional treatment options are thus being investigated (Figure 3B).
CCC has been reported to express actionable levels of ERBB2 pointing to a potential role for HER2 inhibitors in treatment.3 In ongoing clinical trials, HER2 positive ovarian cancer patients are being treated with trastuzumab, a monoclonal antibody against HER2 (Table 1: NCT00194714) and in other ongoing studies, antibody-drug conjugates targeting HER2 are being investigated in advanced solid tumors (Table 1: NCT04602117 and NCT04828616).
A case study reported advanced chemo-resistant CCC that was also resistant to PI3K/AKT pathway inhibitors despite harboring a PIK3CA mutation.48 The tumor responded to treatment with combination therapy of sunitinib, a small-molecule with activity against multiple RTKs and sorafenib (a RAF-kinase inhibitor) which suggested that blocking MAPK signaling in these tumors could be a targetable option.48 A phase II clinical study in patients with recurrent CCC demonstrated sunitinib monotherapy had minimal activity in persistent or recurrent CCC with 2 of 35 (6.7%) patients showing a complete or partial response (Table 1: NCT00979992). Combination treatments should be explored in this subtype.
Endometroid Carcinoma (EC)
EC is tied with clear cell as the second-most common EOC subtype, accounting for approximately 10% of cases.39 EC typically presents as a unilateral mass confined to the ovary, which is difficult to distinguish from HGSOC, especially in patients with solid tumors.38 Overall survival is a promising 10 years when tumors primarily display endometrioid histology.49
EC is frequently associated with endometrioid borderline tumors and like CCC, has been suggested to arise from endometriosis that has implanted on the ovaries or in the peritoneal cavity and undergone malignant transformation.49,50 Endometriosis is a benign disease that is considered a precancerous lesion with potential for metastasis to the colon or lung.49,51 Endometriosis can affect up to 15% of women and those with a history of endometriosis are at increased risk of ovarian cancer.51
KRAS is mutated in 46% of ovarian endometrioid cases, with KRAS (G12V) mutant allele most frequently detected.52 The PI3K/AKT pathway has been implicated in development of chemotherapy resistance in EC.53 EC typically presents with somatic mutations in ARID1A, CTNNB1, PI3KCA, and PTEN.1,21,38 Mutation of ARID1A and CTNNB1 occurs in 30% of cases.21 The CTNNB1 gene encodes for beta-catenin, an essential component of the Wnt signaling pathway which regulates cell growth and proliferation. Mutations in CTNNB1 are also linked to colorectal cancer, medulloblastoma, and ovarian cancer.
Analysis of underlying genetic changes demonstrated mutations in PTEN (32%), TP53 (17%), CTNNB1 (13%), and KRAS (13%) in 53 sporadic EC tumors.54 Molecular stratification of 112 EC cases using whole exome sequencing provided robust assessment of the mutational landscape of EC detecting mutation in CTNNB1 (43%), PIK3CA (43%), ARID1A (36%), PTEN (29%), KRAS (26%), TP53 (26%), and SOX8 (19%) correlating with previous results.55 Upon examination of the frequency of these mutations in clinically distinct EC subtypes, cases with TP53 mutation demonstrated greater genomic complexity concurrent with decreased survival, and cases with CTNNB1 mutation, which was mutually exclusive with TP53 mutation, demonstrated low genomic complexity and better clinical outcome. Moreover, high frequency of mutations in the RAS/MAPK and PI3K/AKT pathways emerged suggesting therapeutic potential.
PTEN mutations are common in EC but less common in other subtypes of ovarian cancer. PTEN is a tumor suppressor frequently found to be mutated or deleted in a wide range of human cancers which drives dysregulated signaling through the PI3K/AKT pathway.56 Conditional inactivation of Pten in mice resulted in formation of adenocarcinomas morphologically like human EC with 100% penetrance, short latency, and rapid progression toward metastatic disease.57 The murine model supports the idea that inactivation of PTEN is an early event in EC tumorigenesis. Genetic mouse models of endometriosis and EC have been described based on the expression of oncogenic Kras or conditional Pten deletion within the ovarian surface epithelium. Both models gave rise to preneoplastic ovarian lesions suggesting endometriosis may be the cause of malignant transformation leading to ovarian cancer.58 The combination of Kras mutation and conditional Pten deletion led to the induction of invasive metastatic endometrioid ovarian adenocarcinomas with complete penetrance.58
Treatment for EC is typically cytoreduction therapy followed by chemotherapy which may improve survival and decrease recurrence; however, following first-line platinum therapy many patients relapse with resistant disease.59,60 Although genetic models have clearly demonstrated oncogenic KRAS induces endometriosis and may lead to the development of invasive EC, treatment options for EC targeting RAS effectors such as RAF and MEK are lagging (Figure 3C). Currently, cobimetinib, a MEK inhibitor described above for use in HGSOC, is in an ongoing phase I clinical trial evaluating combination with a poly ADP-ribose polymerase (PARP) inhibitor in patients with high grade serous or EC (Table 1: NCT03695380).
Targeting the MAPK pathway has been examined in patient-derived xenograft (PDX) models from platinum-relapsing endometrioid tumors, chosen based on the presence of activated RAS, mutant TP53, lack of PTEN, and activation of the PI3K pathway.60 The study employed a combination regimen containing binimetinib (a MEK inhibitor), bevacizumab (a VEGF inhibitor), and paclitaxel as second-line therapy. The triple combination induced long-lasting response without increased toxicity better than dual combinations, demonstrating MEK inhibition can augment anti-tumor activity of the other compounds.60
Mucinous Ovarian Cancer (MOC)
MOC is a rare histological subtype representing less than 5% of all EOC.22 Mucinous tumors have a multicystic structure with more than 90% of cells filled with conspicuous amounts of mucin. Mucinous ovarian tumors are postulated to arise from benign lesions, which transform into mucinous borderline tumors, and then into invasive cancer. MOCs morphologically resemble adenocarcinomas of the pancreas and gastrointestinal tract, complicating the differentiation of primary ovarian tumors from metastatic disease.61 Tumor progression of mucinous carcinoma shows benign-appearing, borderline, non-invasive, and invasive components.21 Patient outcomes and response to conventional chemotherapy are often poor.38
The most common genetic defects in mucinous tumors affect the RAS/MAPK pathway. More than 75% of mucinous ovarian tumors harbor activating KRAS mutations occurring predominantly at codon 12.27,29,38,62 Other frequently mutated genes include BRAF, TP53, PTEN, PI3KCA, and CDKN2A.38 ERBB2 amplification is relatively common in MOC and is observed in 20–40% of cases.63,64 ERBB2 has been linked to enhanced survival of MOC patients and may be a potential biomarker.65 In some tumors, mutation of KRAS and amplification of ERBB2 have been observed in the same cell populations, with consistent KRAS allelic frequency in both ERBB2 amplified and non-amplified regions, suggesting KRAS mutation occurred in advance of the amplification event.21,63,66
TP53 mutations are common and occur concomitantly with KRAS mutations in MOC in 36% of cases.67 Ovarian cells sourced from Pten/Kras (G12D) mice develop serous EOC with 100% penetrance.68 When the Pten/Kras (G12D) mutant mouse strain was crossed with mutant Tp53(R172H) heterozygous mice, resulting WT/Tp53(R172H) mice presented with mucinous cystadenocarcinomas at 12 weeks of age, recapitulating human mucinous ovarian tumors.68 Although tumors were derived from Pten/Kras (G12D) and promoted EOC, differential effects on disease features and progression depended on the presence or absence of the wild-type TP53 allele. This model provides genetic evidence that mutant TP53 promotes EOC differentiation and metastasis.
MOC is frequently resistant to conventional platinum-based chemotherapy with low response rates and is unlikely to respond to PARP inhibition commonly used to treat other EOC cases.9,64 Due to the high frequency of activating KRAS mutations, the RAS/MAPK pathway has been suggested as a therapeutic target.2 However, perhaps due to the rarity of MOC, targeted therapies against downstream RAS pathway components have not been explored clinically (Figure 3D). However, some research suggests targeting HER2 is a viable option for ERBB2 amplified advanced or recurrent MOC.63,66 In one study, three patients with MOC displaying ERRB2 amplification received trastuzumab in combination with conventional chemotherapy and one patient showed a positive response.69 In addition, a current phase I trial is addressing the safety of a plasmid-based vaccine directed against the ICD of HER2 in combination with an immunostimulatory agent, in patients with HER2 amplified advanced breast or ovarian cancers to stimulate HER2-specific immune responses (Table 1: NCT00436254).
Low-Grade Serous Carcinoma (LGSOC)
LGSOC is the rarest subtype of EOC accounting for less than 5% of all EOC cases.39,70 LGSOC is typically diagnosed in younger women and presents as a distinct pathologic and clinical entity characterized by less aggressive behavior compared with HGSOC, a more indolent growth pattern, and poor response to systemic therapy compared with HGSOC.2,9,26 LGSOC may arise from serous tumors with low malignant potential called borderline serous carcinomas or de novo from the ovary or peritoneum.9,71 Advanced LGSOC is associated with poor long-term prognosis.72
Although LGSOC tumors evolve slowly, evidence suggests limited benefit of cytotoxic therapy due to high resistance of LGSOC to platinum-based neoadjuvant chemotherapy.71 Surgical cytoreduction is associated with improved PFS and OS in women with advanced-stage disease.73 However, high risk of recurrence and cancer-related death is a major concern. Because LGSOC is relatively chemo-resistant, the need for targeted therapies is an urgent demand.
LGSOC appears to be driven by RAS/MAPK pathway activation with wild type TP53 on a background of a relatively normal karyotype.9 Most low-grade tumors typically harbor KRAS mutation in 30–50% of cases or BRAF mutation in 15–40% of cases.21,27,29,74–76 Somatic mutation of KRAS in codons 12 and 13 and BRAF mutation in codon 599 are most common.77 Activating NRAS (Q61R) mutation has been detected in 4% of LGSOC tumors, indicating that NRAS plays a limited role in this subtype.78 Copy number aberrations have been observed with loss of 9p and homozygous deletions of the CDKN2A/2B locus.79
Somatic ovarian cells derived from Tp53 deficient mice can be transformed by oncogenic Kras demonstrating the ovarian surface epithelium (OSE) is the precursor tissue for ovarian carcinomas.80 In a mouse model of LGSOC, expression of Kras (G12D) in OSE cells with loss of Pten induced low-grade ovarian serous papillary adenocarcinomas at an early age with 100% penetrance.81 Transformation of immortalized human ovarian serous cystadenoma cells by oncogenic KRAS (G12V) and PIK3CA have been shown to drive development of tumors that histologically resemble LGSOC.82 Analysis of oncogenic KRAS gene signature datasets from human ovarian tumors and the OSE ovarian mouse model documented significant overlap, consistent with an essential role of RAS in transformation.
In contrast to HGSOC, LGSOC shows a low response rate to cytotoxic chemotherapy.83 Like other type I cancers, LGSOC responds poorly to platinum-based chemotherapy due to the high frequency of RAS/RAF mutations suggesting patients with LGSOC may derive clinical benefit from MAPK-targeted therapies.9,26 Many studies confirm that LGSOC can be defined by somatic mutations in RAS which points to novel treatment paradigms.84 Recurrent or persistent LGSOC following prior platinum-based chemotherapy is the subject of ongoing evaluation with BRAF and MEK inhibitors in clinical trials (Figure 3E).
Response to BRAF inhibitors has been reported in case studies of LGSOC. Partial and durable responses following BRAF inhibition with dabrafenib or vemurafenib have been observed in patients with BRAF (V600E) mutant LGSOC.74,85,86 Additional studies have demonstrated partial responses to BRAF inhibitors when combined with MEK inhibitors. Patients with LGSOC harbouring a BRAF (V600E) mutation developed sustained clinical response with combination treatment of dabrafenib and trametinib, an allosteric MEK1/2 inhibitor.87 The combination of both drugs mitigated tumor-acquired resistance and decreased the incidence of secondary malignancies. However, research has not established if combination therapy with BRAF and MEK inhibitors is superior to monotherapy in BRAF mutated LGSOC.87 Trametinib monotherapy is emerging as a promising treatment option for LGSOC with efficacy over chemotherapy.84 PDX models of LGSOC that harbored KRAS (G12V) mutation showed effective sensitivity to trametinib.88
A phase II/III clinical trial on patients with LGSOC harboring activating KRAS, NRAS, or BRAF mutations demonstrated trametinib significantly improved PFS and ORR compared with standard chemotherapy in those with recurrent or persistent disease (Table 1: NCT02101788). The promising results from this trial have led to the inclusion of trametinib as a therapeutic option for LGSOC and provide the first strong evidence that LGSOC should be treated differently than HGSOC based on aberrant MAPK pathway activation.
Several other studies evaluating different MEK inhibitors in clinical trials for LGSOC have been conducted with varying degrees of success. In a phase II clinical trial, 15% of patients with recurrent LGSOC responded to selumetinib monotherapy which was four times that of cytotoxic chemotherapy.89 Similarly, a single-arm phase II study in 52 patients with recurrent LGSOC demonstrated selumetinib was well-tolerated and showed a modest ORR with one complete response and seven partial responses. Notably, 35 patients (65%) achieved stable disease (Table 1: NCT00551070).
Binimetinib, a MEK inhibitor approved for treatment of BRAF (V600E) mutant tumors in patients with unresectable or metastatic melanoma, was explored for use in patients with recurrent or persistent LGSOC (Table 1: NCT01849874). Binimetinib treatment did not improve PFS at the time of preliminary analysis, leading to early closure of the phase II study. However, post-hoc analysis revealed that patients with KRAS mutations were 3.4 times more likely to respond to binimetinib than patients without KRAS mutations.90 Currently, binimetinib is included in the National Cancer Network compendium for treatment of recurrent LGSOC.
Binimetinib in combination with bulparlisib (a PI3K inhibitor) was examined in a clinical trial conducted in patients with advanced solid tumors including RAS/BRAF mutant ovarian carcinomas (Table 1: NCT01363232). Although the dual inhibition showed promising activity with 12% patients with RAS/BRAF mutant ovarian cancer showing a partial response, the continuous dosing regimen led to intolerable toxicity.91 Consequently, alternative dosing strategies when combining therapies must be explored to achieve maximum benefit.
Pimasertib, an allosteric MEK inhibitor, has been shown to possess efficacy in combination with voxtalisib (a PI3K inhibitor) in 65 patients with unresectable LGSOC, peritoneal carcinoma, or serous borderline ovarian tumors.92 Although monotherapy resulted in slightly better PFS and ORR than combination therapy, encouragingly, stable disease was achieved in 50% of patients in the combination group and 40% of patients in the pimasertib monotherapy group (Table 1: NCT01936363). The study was discontinued early because of the lack of ORR suggesting further research is needed to study the utility of combined MEK and PI3K inhibition.
A dual RAF/MEK covalent inhibitor called VS-6766 is currently in a phase II clinical trial to compare the efficacy of monotherapy to combination with defactinib, a focal adhesion kinase (FAK) inhibitor, in recurrent LGSOC (Table 1: NCT04625270). The most recent update of the clinical study reported the combination elicited an ORR of 70% in patients with KRAS-mutant LGSOC and 44% in those with KRAS wild-type. Based on the promising results, the FDA has granted a breakthrough therapy designation to the combination, irrespective of KRAS mutational status.
Perspective and Future Directions
Promising pre-clinical and clinical studies have demonstrated the potential for targeting the RAS/MAPK in EOC subtypes. Combination therapy with novel agents targeting RAF and MEK may benefit patients with type I ovarian cancers and should be investigated. Patients that received combination therapy with RAF and MEK inhibitors derived promising early clinical results demanding future research into new combinations for ovarian cancer. Although combination therapy has demonstrated benefits, most patients develop resistance to RAF and MEK inhibitors concomitant with disease progression in less than a year.93
Unfortunately, in response to MAPK pathway inhibition, compensatory mechanisms are activated that cause resistance by activating feedback loops in tumor cells.94 A clinical study treating metastatic melanoma harboring BRAF mutation found combined BRAF and MEK inhibition resulted in significant reduction of disease progression. The results suggested that 25% decreased risk of resistance could be achieved by inhibition of the MAPK pathway at two nodes rather than one.95 In addition, several clinical trials have investigated the efficacy of ERK inhibitors in treating advanced staged malignancy which displays resistance to targeting upstream nodes of the MAPK pathway (Table 1: NCT04055649, NCT01781429, and NCT02711345). Future studies will establish the clinical efficacy of pharmacologic inhibition of ERK in combination with other MAPK inhibitors to promote increased tumor sensitivity.
While standard chemotherapy with carboplatin and paclitaxel achieves an improved clinical response, resistance to chemotherapy is a major impediment in the management of ovarian cancer.2 In the clinic it has been observed that patients with type I ovarian cancers harboring gain-of-function MAPK activation respond poorly to platinum-based chemotherapy.26 Early studies have demonstrated the combination of EGFR, BRAF, or MEK inhibitors with standard chemotherapy agents significantly improves clinical efficacy and delays drug resistance in platinum refractory cancer.26 The combined treatment of trametinib, an allosteric MEK1/2 inhibitor and selumetinib have identified KRAS mutational status, EGFR and PKC-alpha protein expression as predictive biomarkers that distinguish MEK inhibitor sensitive and MEK resistant LGSOC cell lines.96 The data suggest that combination therapy of MEK inhibition with EGFR inhibitors may represent a promising new therapy for MEK-resistant LGSOC.
Clinical studies have demonstrated the efficacy of both trametinib and selumetinib monotherapy with promise in LGSOC over cytotoxic chemotherapy. Trametinib may be more potent compared with selumetinib with efficacy correlated to degree of inhibition of ERK phosphorylation.96 Mechanistically, trametinib shows equal potency for targeting MEK1 and MEK2 and preferentially binds unphosphorylated MEK preventing RAF-dependent MEK activation.97 In contrast, selumetinib does not block MEK binding and phosphorylation by RAF. With demonstrated occurrence of gain-of-function MAPK mutation in multiple subtypes of EOC, future clinical studies should address the efficacy of MEK inhibition as a general treatment option.
PARP inhibitors have valuable roles as maintenance therapy in EOC; however, like platinum chemotherapy, most patients acquire resistance.97 Clinical trials in EOC testing combinations of MAPK pathway inhibitors in combination with PARP inhibitors have presented with limited success. Promisingly, an in vitro study demonstrated the combined treatment with talazoparib (PARP inhibitor) and trametinib (MEK inhibitor) evoked synergistic cytotoxic effects in ovarian cancer cell lines. Further, MEK inhibition by trametinib was capable of desensitizing talazoparib-resistant cells.98 Another study which combined olaparib (PARP inhibitor) with the pimasertib (MEK inhibitor) demonstrated that pimasertib treatment enhanced PARP inhibition sensitivity in BRCA2-proficient ovarian cancer cell lines.99 These results suggest the utility of MAPK pathway inhibition in combination with PARP inhibitors may offer unexplored therapeutic potential.
Novel approaches to target RAS-driven cancers, including ovarian carcinomas, must be developed and take into consideration isoform and codon-specific mutations in RAS genes observed in tumors. Currently, enhanced efforts are being applied to develop specific inhibitors of all RAS isoforms and mutant RAS alleles that will eventually provide an approach for all RAS-driven cancers including EOC.8 Due to lack of RAS mutant-specific inhibitors, other strategies, such as RNA interference (RNAi) using small interfering RNAs (siRNA) or antisense oligonucleotides (ASO) to target mutant RAS mRNA are emerging to inhibit RAS-driven cancer.100 Research efforts are currently focused on modifications and delivery platforms to improve activity, stability, and biosafety.101 An ongoing phase I trial is attempting to establish preliminary efficacy of exosomes loaded with KRAS (G12D) siRNA for treating metastatic pancreatic cancer (Table 1: NCT03608631). If successful, siRNA therapies may eventually be extended to treat ovarian cancer with known RAS mutations.
Currently, there is a critical need to identify biomarkers as effective predictors for ovarian cancer diagnosis and prognosis.20 Biomarkers will increase the capacity to predict therapeutic efficacy and response to treatment, and potentially could enable improvements in early diagnosis and survival of patients with ovarian cancer.102 A phase II clinical study has been initiated to evaluate biomarker-driven therapies in patients with persistent or recurrent EOC (Table 1: NCT04931342). The study will evaluate the efficacy and safety profile of multiple biomarker-selected treatments with one experimental arm designed to treat patients with cobimetinib whose tumors harbor activating mutations in KRAS, NRAS, BRAF, and NF1. The study has the potential to detect biomarkers that predict response of cancers to RAS/MAPK targeted therapies.
Conclusion
In summary, the RAS/MAPK pathway has been implicated in ovarian cancer cell survival, tumorigenesis, invasiveness, angiogenesis, and platinum resistance.10,103,104 The numerous studies discussed herein suggest oncogenic mutations that promote gain-of-function MAPK signaling are prognostic markers and promising therapeutic targets in all subtypes of ovarian cancer. Indeed, targeting the RAS/MAPK pathway in the clinical setting is an emerging tractable strategy for personalized therapy and improved prognosis for woman with ovarian cancer.
Clinical Data Collection
The search strategy for identifying clinical studies included keywords chosen to perform a systematic search of ClinicalTrials.gov database (https://www.clinicaltrials.gov/). Keywords and terms for finding clinical studies included ovarian cancer, type I, type II epithelial ovarian cancer, high and low grade serous ovarian carcinoma, clear cell carcinoma, mucinous carcinoma, endometrioid carcinoma, MAPK, RAS, RAF, MEK, ERK, HER2, ERBB2, and receptor tyrosine kinase.
Acknowledgments
We would like to thank the adMare co-op student journal club for critically reading our manuscript and offering excellent suggestions for improvement.
Disclosure
The authors report no conflict of interest in this work.
References
1. Maru Y, Hippo Y. Current status of patient-derived ovarian cancer models. Cells. 2019;8(5):505. doi:10.3390/cells8050505
2. Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013;19(5):961–968. doi:10.1158/1078-0432.CCR-12-2243
3. Lapke N, Chen CH, Chang TC, et al. Genetic alterations and their therapeutic implications in epithelial ovarian cancer. BMC Cancer. 2021;21(1):499. doi:10.1186/s12885-021-08233-5
4. Bast RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nature Publishing Group. 2009. doi:10.1038/nrc2644
5. Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133(3):624–631. doi:10.1016/j.ygyno.2014.02.038
6. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. doi:10.1038/nrm2438
7. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4:11. doi:10.1101/cshperspect.a011254
8. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–552. doi:10.1038/s41573-020-0068-6
9. Romero I, Bast RC. Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153(4):1593–1602. doi:10.1210/en.2011-2123
10. Guan LY, Lu Y. New developments in molecular targeted therapy of ovarian cancer. Discov Med. 2018;26(144):219–229.
11. López-Reig R, López-Guerrero JA. The hallmarks of ovarian cancer: proliferation and cell growth. EJC Suppl. 2020;15:27–37. doi:10.1016/j.ejcsup.2019.12.001
12. Zhao H, Wu L, Yan G, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):263. doi:10.1038/s41392-021-00658-5
13. Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2(3):344–358. doi:10.1177/1947601911411084
14. Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779–827. doi:10.1146/annurev.bi.56.070187.004023
15. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10(12):842–857. doi:10.1038/nrc2960
16. Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul. 2014;55:1–14. doi:10.1016/j.jbior.2014.04.002
17. Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9(3):167–181. doi:10.1038/nrc2583
18. House CD, Hernandez L, Annunziata CM. Recent technological advances in using mouse models to study ovarian cancer. Front Oncol. 2014;4:26. doi:10.3389/fonc.2014.00026
19. Gilks CB, Ionescu DN, Kalloger SE, et al. Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Hum Pathol. 2008;39(8):1239–1251. doi:10.1016/j.humpath.2008.01.003
20. Köbel M, Kalloger SE, Boyd N, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5(12):e232. doi:10.1371/journal.pmed.0050232
21. Prat J, D’Angelo E, Espinosa I. Ovarian carcinomas: at least five different diseases with distinct histological features and molecular genetics. Hum Pathol. 2018;80:11–27. doi:10.1016/j.humpath.2018.06.018
22. Kurman RJ, Shih IM. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008;27(2):151–160. doi:10.1097/PGP.0b013e318161e4f5
23. Salazar C, Campbell IG, Gorringe KL, When I. “Type I” ovarian cancer not “type i”? Indications of an out-dated dichotomy. Front Oncol. 2018;8:654. doi:10.3389/fonc.2018.00654
24. Devouassoux-Shisheboran M, Genestie C, Ray-Coquard I. La classification histo-moléculaire des cancers épithéliaux ovariens en 2 types est-elle pertinente? [Dualistic classification of epithelial ovarian cancer: is it clinically relevant?]. Bull Cancer. 2016;103(3):252–258. French. doi:10.1016/j.bulcan.2015.12.005
25. Vang R, Shih IM, Kurman RJ. Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009;16(5):267–282. doi:10.1097/PAP.0b013e3181b4fffa
26. Emmanuel C, Chiew YE, George J, et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin Cancer Res. 2014;20(24):6618–6630. doi:10.1158/1078-0432.CCR-14-1292
27. Vereczkey I, Serester O, Dobos J, et al. Molecular characterization of 103 ovarian serous and mucinous tumors. Pathol Oncol Res. 2011;17(3):551–559. doi:10.1007/s12253-010-9345-8
28. Perets R, Wyant GA, Muto KW, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24(6):751–765. doi:10.1016/j.ccr.2013.10.013
29. Sieben NLG, Macropoulos P, Roemen GMJM, et al. In ovarian neoplasms, BRAF, but not KRAS, mutations are restricted to low-grade serous tumours. J Pathol. 2004;202(3):336–340. doi:10.1002/path.1521
30. Ghoneum A, Almousa S, Warren B, et al. Exploring the clinical value of tumor microenvironment in platinum-resistant ovarian cancer. Semin Cancer Biol. 2021;77:83–98. doi:10.1016/j.semcancer.2020.12.024
31. Hew KE, Miller PC, El-Ashry D, et al. MAPK activation predicts poor outcome and the MEK inhibitor, selumetinib, reverses antiestrogen resistance in ER-positive high-grade serous ovarian cancer. Clin Cancer Res. 2016;22(4):935–947. doi:10.1158/1078-0432.CCR-15-0534
32. Marchetti C, Muzii L, Romito A, Benedetti Panici P. First-line treatment of women with advanced ovarian cancer: focus on bevacizumab. Onco Targets Ther. 2019;12:1095–1103. doi:10.2147/OTT.S155425
33. Bhatt M, Ivan C, Xie X, Siddik ZH. Drug-dependent functionalization of wild-type and mutant p53 in cisplatin-resistant human ovarian tumor cells. Oncotarget. 2017;8(7):10905–10918. doi:10.18632/oncotarget.14228
34. Mukherjee S, Pal M, Mukhopadhyay S, et al. VEGF expression to support targeted therapy in ovarian surface epithelial neoplasms. J Clin Diagn Res. 2017;11(4):EC43–EC46. doi:10.7860/JCDR/2017/24670.9737
35. Subramonian D, Phanhthilath N, Rinehardt H, et al. Regorafenib is effective against neuroblastoma in vitro and in vivo and inhibits the RAS/MAPK, PI3K/Akt/mTOR and Fos/Jun pathways. Br J Cancer. 2020;123(4):568–579. doi:10.1038/s41416-020-0905-8
36. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi:10.1073/pnas.192461099
37. Hellmann MD, Kim TW, Lee CB, et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. 2019;30(7):1134–1142. doi:10.1093/annonc/mdz113
38. Sadłecki P, Walentowicz-Sadłecka M, Grabiec M. Molecular diagnosis in type I epithelial ovarian cancer. Ginekol Pol. 2017;88(12):692–697. doi:10.5603/GP.a2017.0123
39. Shih IM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164(5):1511–1518. doi:10.1016/s0002-9440(10)63708-x
40. Behbakht K, Randall TC, Benjamin I, Morgan MA, King S, Rubin SC. Clinical characteristics of clear cell carcinoma of the ovary. Gynecol Oncol. 1998;70(2):255–258. doi:10.1006/gyno.1998.5071
41. Sugiyama T, Kamura T, Kigawa J, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000;88(11):2584–2589. doi:10.1002/1097-0142(20000601)88:11<2584::AID-CNCR22>3.0.CO;2-5
42. Itamochi H, Kigawa J, Terakawa N. Mechanisms of chemoresistance and poor prognosis in ovarian clear cell carcinoma. Cancer Sci. 2008;99(4):653–658. doi:10.1111/j.1349-7006.2008.00747.x
43. Willner J, Wurz K, Allison KH, et al. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum Pathol. 2007;38(4):607–613. doi:10.1016/j.humpath.2006.10.007
44. de Pauw A, Naert E, van de Vijver K, Philippe T, Vandecasteele K, Denys H. A clearer view on ovarian clear cell carcinoma. Acta Clin Belg. 2021;1–13. doi:10.1080/17843286.2021.1964051
45. Jones S, Wang TL, Shih IM, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–231. doi:10.1126/science.1196333
46. Auner V, Kriegshäuser G, Tong D, et al. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC Cancer. 2009;9:111. doi:10.1186/1471-2407-9-111
47. Zannoni GF, Improta G, Chiarello G, et al. Mutational status of KRAS, NRAS, and BRAF in primary clear cell ovarian carcinoma. Virchows Arch. 2014;465(2):193–198. doi:10.1007/s00428-014-1599-1
48. Rahman M, Nakayama K, Ishibashi T, et al. A case of stage III C ovarian clear cell carcinoma: the role for predictive biomarkers and targeted therapies. Int J Mol Sci. 2013;14(3):6067–6073. doi:10.3390/ijms14036067
49. Swiersz LM. Role of endometriosis in cancer and tumor development. Ann N Y Acad Sci. 2002;955:281–285. doi:10.1111/j.1749-6632.2002.tb02788.x
50. Soliman PT, Broaddus RR, Schmeler KM, et al. Women with synchronous primary cancers of the endometrium and ovary: do they have Lynch syndrome? J Clin Oncol. 2005;23(36):9344–9350. doi:10.1200/JCO.2005.03.5915
51. Yachida N, Yoshihara K, Yamaguchi M, Suda K, Tamura R, Enomoto T. How does endometriosis lead to ovarian cancer? The molecular mechanism of endometriosis-associated ovarian cancer development. Cancers. 2021;13(6). doi:10.3390/cancers13061439
52. Yachida N, Yoshihara K, Suda K, et al. Biological significance of KRAS mutant allele expression in ovarian endometriosis. Cancer Sci. 2021;112(5):2020–2032. doi:10.1111/cas.14871
53. Rinne N, Christie EL, Ardasheva A, et al. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021;4(3):573–595. doi:10.20517/cdr.2021.05
54. Koul A, Willén R, Bendahl PO, Nilbert M, Borg A. Distinct sets of gene alterations in endometrial carcinoma implicate alternate modes of tumorigenesis. Cancer. 2002;94(9):2369–2379. doi:10.1002/cncr.10498
55. Hollis RL, Thomson JP, Stanley B, et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat Commun. 2020;11(1):4995. doi:10.1038/s41467-020-18819-5
56. Ince FA, Shariev A, Dixon K. PTEN as a target in melanoma. J Clin Pathol. 2022;75(9):581–584. doi:10.1136/jclinpath-2021-208008
57. Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11(4):321–333. doi:10.1016/j.ccr.2007.02.016
58. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11(1):63–70. doi:10.1038/nm1173
59. Barlin JN, Puri I, Bristow RE. Cytoreductive surgery for advanced or recurrent endometrial cancer: a meta-analysis. Gynecol Oncol. 2010;118(1):14–18. doi:10.1016/j.ygyno.2010.04.005
60. Ricci F, Guffanti F, Damia G, Broggini M. Combination of paclitaxel, bevacizumab and MEK162 in second line treatment in platinum-relapsing patient derived ovarian cancer xenografts. Mol Cancer. 2017;16(1):97. doi:10.1186/s12943-017-0662-3
61. Seidman JD, Kurman RJ, Ronnett BM. Primary and metastatic mucinous adenocarcinomas in the ovaries: incidence in routine practice with a new approach to improve intraoperative diagnosis. Am J Surg Pathol. 2003;27(7):985–993. doi:10.1097/00000478-200307000-00014
62. Sato N, Saga Y, Mizukami H, et al. Cetuximab inhibits the growth of mucinous ovarian carcinoma tumor cells lacking KRAS gene mutations. Oncol Rep. 2012;27(5):1336–1340. doi:10.3892/or.2012.1626
63. Anglesio MS, Kommoss S, Tolcher MC, et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol. 2013;229(1):111–120. doi:10.1002/path.4088
64. Gorringe KL, Cheasley D, Wakefield MJ, et al. Therapeutic options for mucinous ovarian carcinoma. Gynecol Oncol. 2020;156(3):552–560. doi:10.1016/j.ygyno.2019.12.015
65. Kim SK, Cho NH. HER2-positive mucinous adenocarcinomas of the ovary have an expansile invasive pattern associated with a favorable prognosis. Int J Clin Exp Pathol. 2014;7(7):4222–4230.
66. Mackenzie R, Kommoss S, Winterhoff BJ, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015;15:415. doi:10.1186/s12885-015-1421-8
67. Rechsteiner M, Zimmermann AK, Wild PJ, et al. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp Mol Pathol. 2013;95(2):235–241. doi:10.1016/j.yexmp.2013.08.004
68. Ren YA, Mullany LK, Liu Z, Herron AJ, Wong KK, Richards JS. Mutant p53 promotes epithelial ovarian cancer by regulating tumor differentiation, metastasis, and responsiveness to steroid hormones. Cancer Res. 2016;76(8):2206–2218. doi:10.1158/0008-5472.CAN-15-1046
69. McAlpine JN, Wiegand KC, Vang R, et al. HER2 overexpression and amplification is present in a subset of ovarian mucinous carcinomas and can be targeted with trastuzumab therapy. BMC Cancer. 2009;9:433. doi:10.1186/1471-2407-9-433
70. Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23(1):41–44. doi:10.1097/01.pgp.0000101080.35393.16
71. Shvartsman HS, Sun CC, Bodurka DC, et al. Comparison of the clinical behavior of newly diagnosed stages II-IV low-grade serous carcinoma of the ovary with that of serous ovarian tumors of low malignant potential that recur as low-grade serous carcinoma. Gynecol Oncol. 2007;105(3):625–629. doi:10.1016/j.ygyno.2007.01.030
72. Wohlmuth C, Djedovic V, Kjaer SK, et al. CA-125 levels are predictive of survival in low-grade serous ovarian cancer-a multicenter analysis. Cancers. 2022;14:8. doi:10.3390/cancers14081954
73. Nickles Fader A, Java J, Ueda S, et al. Survival in women with grade 1 serous ovarian carcinoma. Obstet Gynecol. 2013;122(2 Pt 1):225–232. doi:10.1097/AOG.0b013e31829ce7ec
74. Moujaber T, Etemadmoghadam D, Kennedy CJ, et al. BRAF mutations in low-grade serous ovarian cancer and response to BRAF inhibition. JCO Precis Oncol. 2018;2:1–14. doi:10.1200/PO.17.00221
75. Tone AA, McConechy MK, Yang W, et al. Intratumoral heterogeneity in a minority of ovarian low-grade serous carcinomas. BMC Cancer. 2014;14:982. doi:10.1186/1471-2407-14-982
76. Zhang X, Devins K, Ko EM, et al. Mutational spectrum in clinically aggressive low-grade serous carcinoma/serous borderline tumors of the ovary-Clinical significance of BRCA2 gene variants in genomically stable tumors. Gynecol Oncol. 2021;161(3):762–768. doi:10.1016/j.ygyno.2021.03.019
77. Singer G, Oldt R
78. Xing D, Suryo Rahmanto Y, Zeppernick F, et al. Mutation of NRAS is a rare genetic event in ovarian low-grade serous carcinoma. Hum Pathol. 2017;68:87–91. doi:10.1016/j.humpath.2017.08.021
79. Hunter SM, Anglesio MS, Ryland GL, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6(35):37663–37677. doi:10.18632/oncotarget.5438
80. Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1(1):53–62. doi:10.1016/s1535-6108(01
81. Mullany LK, Fan HY, Liu Z, et al. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene. 2011;30(32):3522–3536. doi:10.1038/onc.2011.70
82. Dey P, Nakayama K, Razia S, et al. Development of Low-grade serous ovarian carcinoma from benign ovarian serous cystadenoma cells. Cancers. 2022;14:6. doi:10.3390/cancers14061506
83. Gershenson DM, Sun CC, Bodurka D, et al. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant. Gynecol Oncol. 2009;114(1):48–52. doi:10.1016/j.ygyno.2009.03.001
84. Moujaber T, Balleine RL, Gao B, Madsen I, Harnett PR, DeFazio A. New therapeutic opportunities for women with low-grade serous ovarian cancer. Endocr Relat Cancer. 2021;29(1):R1–R16. doi:10.1530/ERC-21-0191
85. Combe P, Chauvenet L, Lefrère-Belda MA, et al. Sustained response to vemurafenib in a low grade serous ovarian cancer with a BRAF V600E mutation. Invest New Drugs. 2015;33(6):1267–1270. doi:10.1007/s10637-015-0297-4
86. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373(8):726–736. doi:10.1056/NEJMoa1502309
87. Lima B, Abreu MH, Sousa S, Bartosch C, Pereira D. Impressive and durable clinical responses obtained with dabrafenib and trametinib in low-grade serous ovarian cancer harbouring a BRAF V600E mutation. Gynecol Oncol Rep. 2022;40:100942. doi:10.1016/j.gore.2022.100942
88. de Thaye E, van de Vijver K, van der Meulen J, et al. Establishment and characterization of a cell line and patient-derived xenograft (PDX) from peritoneal metastasis of low-grade serous ovarian carcinoma. Sci Rep. 2020;10(1):6688. doi:10.1038/s41598-020-63738-6
89. Farley J, Brady WE, Vathipadiekal V, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, Phase 2 study. Lancet Oncol. 2013;14(2):134–140. doi:10.1016/S1470-2045(12)70572-7
90. Grisham RN, Vergote I, Banerjee SN, et al. Molecular results and potential biomarkers identified from MILO/ENGOT-ov11 Phase 3 study of binimetinib versus physicians choice of chemotherapy (PCC) in recurrent low-grade serous ovarian cancer (LGSOC). J Clin Oncol. 2021;39(15_suppl):5519. doi:10.1200/JCO.2021.39.15_suppl.5519
91. Bardia A, Gounder M, Rodon J, et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist. 2020;25(1):e160–e169. doi:10.1634/theoncologist.2019-0297
92. Arend RC, Davis AM, Chimiczewski P, et al. EMR 20006-012: a phase II randomized double-blind placebo controlled trial comparing the combination of pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) to pimasertib alone in patients with previously treated unresectable borderline or low grade. Gynecol Oncol. 2020;156(2):301–307. doi:10.1016/j.ygyno.2019.12.002
93. Germann UA, Furey BF, Markland W, et al. Targeting the MAPK signaling pathway in cancer: promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther. 2017;16(11):2351–2363. doi:10.1158/1535-7163.MCT-17-0456
94. Ali ES, Akter S, Ramproshad S, et al. Targeting Ras-ERK cascade by bioactive natural products for potential treatment of cancer: an updated overview. Cancer Cell Int. 2022;22(1):246. doi:10.1186/s12935-022-02666-z
95. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–1888. doi:10.1056/NEJMoa1406037
96. Fernandez ML, Dawson A, Hoenisch J, et al. Markers of MEK inhibitor resistance in low-grade serous ovarian cancer: EGFR is a potential therapeutic target. Cancer Cell Int. 2019;19:10. doi:10.1186/s12935-019-0725-1
97. McMullen M, Karakasis K, Madariaga A, Oza AM. Overcoming platinum and PARP-inhibitor resistance in ovarian cancer. Cancers. 2020;12:6. doi:10.3390/cancers12061607
98. Sun C, Fang Y, Yin J, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9:392. doi:10.1126/scitranslmed.aal5148
99. Vena F, Jia R, Esfandiari A, et al. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget. 2018;9(14):11592–11603. doi:10.18632/oncotarget.24294
100. Hattab D, Gazzali AM, Bakhtiar A. Clinical advances of siRNA-based nanotherapeutics for cancer treatment. Pharmaceutics. 2021;13(7):1009. doi:10.3390/pharmaceutics13071009
101. Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020;5(1):101. doi:10.1038/s41392-020-0207-x
102. Atallah GA, Abd Aziz NH, Teik CK, Shafiee MN, Kampan NC. New predictive biomarkers for ovarian cancer. Diagnostics. 2021;11(3):465. doi:10.3390/diagnostics11030465
103. Hsu CY, Bristow R, Cha MS, et al. Characterization of active mitogen-activated protein kinase in ovarian serous carcinomas. Clin Cancer Res. 2004;10(19):6432–6436. doi:10.1158/1078-0432.CCR-04-0893
104. Pohl G, Ho CL, Kurman RJ, Bristow R, Wang TL, Shih IM. Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Res. 2005;65(5):1994–2000. doi:10.1158/0008-5472.CAN-04-3625
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.