Back to Journals » OncoTargets and Therapy » Volume 11
A ten-long non-coding RNA signature for predicting prognosis of patients with cervical cancer
Authors Shen L, Yu H ![]() , Liu M, Wei D, Liu W, Li C, Chang Q
, Liu M, Wei D, Liu W, Li C, Chang Q ![]()
Received 23 May 2018
Accepted for publication 20 August 2018
Published 28 September 2018 Volume 2018:11 Pages 6317—6326
DOI https://doi.org/10.2147/OTT.S175057
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Liang Shen,1 Haochen Yu,2 Ming Liu,1 Deying Wei,1 Wei Liu,1 Changzhong Li,1 Qin Chang2
1Department of Gynecology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong 250021, People’s Republic of China; 2Department of Applied Mathematics, College of Science, China University of Petroleum, Qingdao, Shandong 266580, People’s Republic of China
Purpose: The aim of the present study was to construct a novel long non-coding RNA (lncRNA) signature to predict the prognosis of patients with cervical cancer (CC).
Materials and methods: We downloaded lncRNA expression profiles and clinical characteristics from The Cancer Genome Atlas database and randomly divided them into a training dataset (n=200) and a testing dataset (n=87). Using a Cox-based iterative sure independence screening procedure combined with a resampling technique, a lncRNA signature was calculated from prognostic lncRNAs in the training dataset and was independently verified in the testing and the entire datasets. In addition, multivariate Cox regression and further stratified analyses were performed, taking into consideration the lncRNA signature as well as other clinical characteristics. Finally, we predicted the underlying functional effects of the prognostic lncRNAs by using Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses.
Results: We constructed a promising ten-lncRNA signature that was significantly associated with the prognosis of CC on the basis of a risk score formula. The risk score was used to classify patients into high-risk and low-risk groups with different overall survival in the training dataset, and was confirmed in the testing and entire datasets. Compared with the clinical factors, the ten-lncRNA signature was found to be an independent prognostic indicator and displayed robust prognostic performance. A functional analysis indicated that these ten lncRNAs were enriched in immune response, cell adhesion molecules and nuclear factor kappa B signaling.
Conclusion: Our results demonstrated that this ten-lncRNA signature may serve as a prognostic biomarker for patients with CC.
Keywords: cervical cancer, long non-coding RNAs, survival, prognosis signature
Introduction
Cervical cancer (CC) remains the fourth leading cause of cancer-related mortality among women worldwide.1 Approximately 527,600 cases of CC were diagnosed, resulting in over 265,700 deaths in 2012 globally.2 Despite the fact that the 5-year overall survival (OS) rate for patients with early-stage CC is ~80%, the 5-year OS for stages IIIA, IIIB and IVA disease is 40%, 42% and 22%, respectively.3 Postoperative adjuvant chemoradiotherapy may improve local control, reduce distant metastasis and prolong OS in high-risk patients.4 However, adjuvant chemoradiotherapy may also cause side effects that adversely affect the patients’ quality of life. Hence, prognostic markers able to predict survival in patients with CC may prove valuable for individualized treatment.
Long non-coding RNAs (lncRNAs) are a type of non-coding transcript with a length of more than 200 nucleotides.5 Accumulating evidence indicates that lncRNAs greatly affect gene expression through chromatin modification, transcriptional and post-transcriptional regulation.6,7 Aberrant expression of lncRNAs has been extensively demonstrated in several types of cancer.8,9 LncRNAs have been attracting attention over the past decade, and have prompted a series of studies due to their regulation of multiple CC-related cellular processes, including proliferation, invasion, apoptosis, metastasis and radio-resistance.10–14 Several researchers have focused on detecting prognostic lncRNAs in CC,15–17 and a number of studies have described several prognostic lncRNAs in CC, such as GAS5,18 PANDAR,19 TUG1,20 MEG321 and MALAT1.22 However, the comprehensive strength of a potential lncRNA signature for predicting the prognosis of CC has not been clearly determined. Although a 15-lncRNA signature has been developed to predict the prognosis of patient with CC, that study was only focused on cervical squamous cell carcinoma without including cervical adenocarcinoma, which accounts for ~25% of CC cases.23
In the present study, we screened out the prognostic lncRNAs through investigating lncRNA expression profiles in 287 CC patients from The Cancer Genome Atlas database (TCGA),33 and constructed a ten-lncRNA signature to effectively predict survival in CC.
Materials and methods
Patient datasets
Clinical information of 287 patients with CC were obtained from TCGA on December 10, 2017. The TCGA CC patients were randomly classified into a 200-sample training dataset and an 87-sample testing dataset. The training samples were used to identify lncRNAs whose expression levels are significantly associated with patients’ survival and to construct a prognostic signature (risk score), while the testing samples were used to verify the efficiency of the constructed signature. The detailed clinical information for CC is listed in Table 1.
 | Table 1 Clinical information of patients with cervical cancer |
LncRNA profile mining
The lncRNAs extracted from TCGA and GENCODE were cross-referenced with Ensembl IDs in order to refine the number of lncRNAs. We then normalized the lncRNA expression profiles by log2 transformation. Finally, the expression profiles of 7,923 lncRNAs were obtained.
Generation of a prognostic lncRNA signature
Cox’s proportional hazards model, which is commonly employed in survival analysis, was used to model the dependence of survival time on lncRNA expression. Since the number of lncRNAs (7,923) was notably higher than the sample size (200), we adopted the iterative sure independence screening (ISIS) procedure for Cox’s model24 to detect the most significant lncRNAs. ISIS starts with ranking covariates by the absolute value of their marginal correlation with the response variable and selecting the top ranked covariates, and then it adjusts the selected covariates according to the regression residual iteratively. This is a very efficient variable selection method in an ultra-high dimensional scenario.24 In this study, the ISIS procedure was implemented using R package “SIS”.25
It needed to be emphasized that only the training dataset was used to screen the survival-associated lncRNAs and to construct the expression-based lncRNA signature. Thus, 200 samples in the entire dataset would be distributed to the training dataset and 87 samples would constitute the hold-out sample which would affect the results of lncRNA selection. How to reduce the effect of this random ‘hold-out’ on the final lncRNA selection is challenging and has been rather overlooked. In the present study, we adopted a sample partitioning strategy inspired by the Jackknife method,26 which was proposed to estimate the properties of an estimator derived from a full sample by systematic partitions of the dataset, whereas what was required here was to find a relatively robust result. Based on similar considerations, we repeatedly (100 times) conducted the Cox-based ISIS procedure described previously and only used a random subset (n=150) of the training data in each repeat. After 100 repeats, we obtained 100 groups of significant lncRNAs. The lncRNAs that appeared in at least one of the groups were listed and then sorted by their frequencies of appearance in the 100 groups. By setting the minimum frequency (count of appearance in the 100 groups ≥3), and the maximum P-value (P<0.1) in Cox’s univariate regression, some candidate prognostic lncRNAs were selected. Then, with the entire training dataset (n=200), the association of the expression level of these lncRNAs with patient survival was further analyzed through a stepwise multivariate Cox regression. Subsequently, prognostic lncRNAs associated with patient survival were obtained.
Finally, we constructed a prognostic lncRNA signature (risk score) by a linear combination of the expression levels of the prognostic lncRNAs with the multivariate Cox regression coefficients estimated previously as the weights:
|
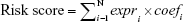
where N stands for the number of selected prognostic lncRNAs, expri stands for the expression level of the ith prognostic lncRNA in each patient, and coefi is the corresponding regression coefficient estimated by the multivariate Cox regression using training data. With these fixed coefi (i=1, ..., N), risk scores could be calculated for all patients. From the form of the Cox proportional hazard model, it is readily observed that patients with a higher risk score are more likely to have poor rates of survival. Using the median risk score in the training dataset as a threshold, the patients were classified into low-risk and high-risk groups. A flow chart was depicted to show the framework of this part (Figure 1).
 | Figure 1 Flow chart of the prognostic lncRNA signature generation. |
Statistical analysis
Kaplan-Meier survival curves and two-sided log-rank tests were employed to compare the survival differences between the high-risk and low-risk groups by using the R package ‘survival’. To further investigate whether the lncRNA signature predicts the OS of CC independently of other clinical factors, multivariate Cox regression and stratified analysis were performed. HRs and 95% CIs were computed. The receiver operating characteristic (ROC) curve analysis was performed to assess the predictive power of the lncRNA signature using the R package ‘survivalROC’.27 The statistical analyses were conducted using R packages(version 3.4.3).
Functional enrichment analysis
The Spearman’s rank correlation coefficient between the expression value of each prognostic lncRNA and that of protein-coding genes was computed to infer the potential functional characteristics of the prognostic lncRNAs. Functional enrichment analysis was conducted using DAVID Bioinformatics Resources (version 6.8).28 Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were set on the cut-off criteria of P<0.05 and enrichment score >1.0.
Results
Identification of prognostic lncRNAs associated with OS of patients with CC in the training dataset
The 287 samples were randomly classified into a training dataset (n=200) and a testing dataset (n=87) (Table 1). In the training dataset, we used a Cox-based ISIS procedure as previously described in “Materials and methods” to identify the most significant lncRNAs associated with the OS of CC patients.24 By setting the minimum appearing frequency (count ≥3) in the 100 selected groups, and the maximum P-value (P<0.1) in Cox’s univariate regression, a total of 23 lncRNAs, including three up-regulated (coefficient >0) and 20 down-regulated (coefficient <0) lncRNAs, were identified as candidates. After the stepwise multivariate Cox regression, ten lncRNAs were selected; among those, eight lncRNAs were down-regulated, indicating that their high expression was correlated with better survival, while the remaining two lncRNAs were up-regulated, suggesting that their high expression was correlated with poor survival. The univariate Cox-regression results (P-value and HR) and multivariate Cox-regression results (coefficient) of these ten prognostic lncRNAs using the whole training set are listed in Table 2.
 | Table 2 Detailed information of ten prognostic lncRNAs significantly associated with overall survival in patients with cervical cancer |
Derivation of a ten-lncRNA signature for predicting survival from the training dataset
We constructed a novel lncRNA signature for survival prediction by using the expression value of each lncRNA weighted by their estimated regression coefficients as follows: risk score = (−0.285× expression level of AC005906.2) + (−0.290× expression level of LINC01727) + (−0.255× expression level of AC108868.1) + (−0.547× expression level of AC020978.4) + (0.160× expression level of GNAS.AS1) + (0.570× expression level of AC012306.2) + (−0.351× expression level of AC024270.5) + (−0.410 × expression level of AC122694.1) + (−0.356× expression level of AC015819.2) + (−0.716× expression level of AL590068.1). Risk scores were calculated and ranked for the 200 samples in the training dataset. The median of the risk scores (0.447957) was used as a threshold to stratify the 200 training samples into a low-risk group (n=100) and a high-risk group (n=100). The Kaplan-Meier analysis revealed that survival of patients in the low-risk group was longer than the high-risk group (31.82 vs 17.62 months, respectively; P=1.71E-10; Figure 2A). Moreover, the ROC curve analysis acquired an area under the curve (AUC) of 0.852 at 5 years, indicating their significant prognostic performance (Figure 2B). The distribution of the risk scores, survival status and expression profiles of the prognostic lncRNAs in the training dataset are presented in Figure 2C. It can be seen that patients with high-risk scores have poorer prognosis than patients with low-risk scores. The results of the univariate Cox regression analysis revealed that the ten-lncRNA risk score was significantly associated with patients’ OS in the training dataset (P=1.74E-08, HR =7.149, 95% CI =3.607–14.170; Table 3).
 | Figure 2 Association between the ten-lncRNA expression signature and overall survival of patients with CC in the training dataset. |
 | Table 3 Univariate and multivariate Cox regression analyses in each dataset |
Further validation of the ten-lncRNA signature for survival prediction in the testing and the entire datasets
To validate our findings, risk scores were calculated for each of the 87 patients in the testing dataset, dividing them into a high-risk group (n=56) and a low-risk group (n=31), according to the previously mentioned risk score model and the threshold derived from the training dataset. In accordance with the findings in the training dataset, patients with high-risk scores had markedly worse OS compared with those with low-risk scores (P=1.06E-02, median 20.63 vs 38.97 months, respectively; Figure 3A). As shown in Figure 3C, the ROC curve analysis yielded an AUC of 0.743 at 5 years in the testing dataset. The HR of the high-risk vs low-risk group for OS was 3.820 (P=1.68E-02, 95% CI =1.273–11.460; Table 3), suggesting that the association of the ten-lncRNA risk score and OS was also significant. Similar findings were observed in the entire dataset, which consisted of 156 high-risk patients with a median OS of 18.5 months and 131 low-risk patients with a median OS of 32.87 months (P=1.13E-10; Figure 3B). The ROC analysis for the ten-lncRNA achieved an AUC of 0.837 (Figure 3D). As shown in Table 3, the ten-lncRNA signature was found to be significantly correlated with patients’ survival in the entire dataset (P=5.54E-09, HR =5.448, 95% CI =3.081–9.633).
 | Figure 3 The ten-lncRNA related risk score predicts overall survival of patients with CC in the testing dataset and the entire dataset. |
Independence of the prognostic power of the ten-lncRNA signature from other clinical factors
As shown in Table 3, the results demonstrated that the risk score obtained from the ten-lncRNA signature maintained a significant association with OS, with the other three clinical factors serving as covariates in each dataset. However, we also found that stage was significantly associated with OS in all three datasets. Therefore, stratification analysis was required to determine the prognostic power of the ten-lncRNA signature for the CC stage; the entire dataset was stratified into an early-stage group (I and II, n=223) and a late-stage group (III and IV, n=64). Moreover, the ten-lncRNA signature subdivided CC patients into high-risk and low-risk subgroups in each stage. This analysis demonstrated that patients in the high-risk subgroups had significantly shorter OS than those in the low-risk subgroup for both early-stage (P=1.31E-09) and late-stage disease (P=1.25E-03) (Figure 4).
 | Figure 4 Survival prediction, stratified by stage, of the lncRNA signature in patients. |
Functional roles of the ten prognostic lncRNAs
In order to understand the functional implication of the ten prognostic lncRNAs in the development of CC, we carried out a functional enrichment analysis to elucidate their roles. We identified 347 protein-coding genes that were significantly correlated with at least one of the ten prognostic lncRNAs (Spearman |R| >0.4). GO and KEGG pathway enrichment analyses were performed with these genes to identify their associated KEGG pathways and GO annotations. GO analysis consisted of three domains, including biological process, molecular function and cellular component. It was demonstrated that these genes were mainly associated with immune response in biological process, receptor activity and binding in molecular function, and plasma membrane in cellular component (Figure 5A–C). These genes were also significantly enriched for cell adhesion molecules (CAMs) and nuclear factor kappa B (NF-κB) signaling (Figure 5D).
 | Figure 5 Gene Ontology and pathway analysis of protein-coding genes correlated with prognostic lncRNAs in the signature. |
Discussion
In this study, we identified ten lncRNAs that were significantly associated with OS in CC patients. The ten-lncRNA risk score signature demonstrated superior ability to divide CC patients into high-risk and low-risk groups with significantly different OS in each dataset. Further studies indicated that the ten-lncRNA signature is an independent predictor of OS with other clinical factors, including age, stage and histology, taken into account simultaneously. Therefore, we demonstrated that this ten-lncRNA signature is a promising prognostic biomarker in the progression of CC.
To the best of our knowledge, none of the ten prognostic lncRNAs have been reported in literature to date. Therefore, we screened out protein-coding genes that are intensively correlated with the ten lncRNAs (Spearman |R| >0.4) in the entire database. We conducted an integrated analysis to predict the potential biological roles of the ten lncRNAs through those correlating genes in CC. The results demonstrated that the lncRNA may exert their effects through several known GO annotations and KEGG pathways. The biological processes of the genes were mainly associated with immune suppression. Infection by human papillomavirus (HPV), a causative factor of CC, induces a cellular immune response with regulatory T cells and maintains local immune suppression in HPV-associated CC.29 These genes were also significantly enriched in CAMs and the NF-κB signaling pathway. Recent studies have found that CAMs were associated with adhesion or signaling status of tumor cells, promoting acquisition of a more invasive phenotype. It is reported that L1CAM may be a helpful prognostic marker to predict locoregional recurrences in CC.30 The NF-κB signaling pathway has been found to play a critical role in the pathogenesis and progression of CC.31 It was demonstrated that MAFIP may also act as a suppressor in CC by restraining the activation of the NF-κB pathway.32
There were several limitations to this study. First, metastasis was not included, as this information was not available for predicting survival. Second, data from TCGA were built on the RNA-Seq technique, and more experimental methods were needed to confirm the results. Third, the exact mechanisms of action of the ten lncRNAs in CC remain to be fully elucidated; further experiments should be designed to explore these mechanisms in future.
Conclusion
In the present study we identified a ten-lncRNA signature that may prove to be a critical prognostic tool for patients with CC. These lncRNAs modulate genes associated with immune response, CAMs and NF-κB signaling, which has previously been associated with CC tumorigenesis. Ultimately, we expect this lncRNA signature to be helpful for predicting prognosis and uncovering the mechanisms underlying CC development.
Acknowledgments
This study was supported in part by the Natural Science Foundation of Shandong Province (grant nos 2R2013HM060 and ZR2015AL014) and the Fundamental Research Funds for the Central Universities (grant no 15CX02064A).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Quinn MA, Benedet JL, Odicino F, et al. Carcinoma of the cervix uteri. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;95 Suppl 1:S43–S103. | ||
Chen K, Ge JJ, Yan SX, Ke SB. Microarray gene expression profiling for identifying different responses to radiotherapy and chemoradiotherapy in patients with cervical cancer. Eur J Gynaecol Oncol. 2017;38(1):106–112. | ||
Kong D, Wang Y. Knockdown of lncRNA HULC inhibits proliferation, migration, invasion, and promotes apoptosis by sponging miR-122 in osteosarcoma. J Cell Biochem. 2018;119(1):1050–1061. | ||
Liang WC, Ren JL, Wong CW, et al. LncRNA-NEF antagonized epithelial to mesenchymal transition and cancer metastasis via cis-regulating FOXA2 and inactivating Wnt/β-catenin signaling. Oncogene. 2018;37(11):1445–1456. | ||
Zhang Y, Lun L, Li H, et al. The value of lncRNA NEAT1 as a prognostic factor for survival of cancer outcome: a meta-analysis. Sci Rep. 2017;7(1):13080. | ||
Bian Z, Jin L, Zhang J, et al. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci Rep. 2016;6:23892. | ||
Gooding AJ, Zhang B, Jahanbani FK, et al. The lncRNA BORG drives breast cancer metastasis and disease recurrence. Sci Rep. 2017;7(1):12698. | ||
Yan Q, Tian Y, Hao F. Downregulation of lncRNA UCA1 inhibits proliferation and invasion of cervical cancer cells through miR-206 expression. Oncol Res. Epub 2018 Mar 9. | ||
Shan D, Shang Y, Hu T. Long noncoding RNA BLACAT1 promotes cell proliferation and invasion in human cervical cancer. Oncol Lett. 2018;15(3):3490–3495. | ||
Zhang M, Song Y, Zhai F. ARFHPV E7 oncogene, lncRNA HOTAIR, miR-331-3p and its target, NRP2, form a negative feedback loop to regulate the apoptosis in the tumorigenesis in HPV positive cervical cancer. J Cell Biochem. 2018;119(6):4397–4407. | ||
Li Q, Feng Y, Chao X, et al. HOTAIR contributes to cell proliferation and metastasis of cervical cancer via targetting miR-23b/MAPK1 axis. Biosci Rep. 2018;38(1):BSR20171563. | ||
Han D, Wang J, Cheng G. LncRNA NEAT1 enhances the radio-resistance of cervical cancer via miR-193b-3p/CCND1 axis. Oncotarget. 2018;9(2):2395–2409. | ||
Li Y, Wan YP, Bai Y. Correlation between long strand non-coding RNA GASS expression and prognosis of cervical cancer patients. Eur Rev Med Pharmacol Sci. 2018;22(4):943–949. | ||
Wang L, Zhu H. Long non-coding nuclear paraspeckle assembly transcript 1 acts as prognosis biomarker and increases cell growth and invasion in cervical cancer by sequestering microRNA-101. Mol Med Rep. 2018;17(2):2771–2777. | ||
Yang JP, Yang XJ, Xiao L, Wang Y. Long noncoding RNA PVT1 as a novel serum biomarker for detection of cervical cancer. Eur Rev Med Pharmacol Sci. 2016;20(19):3980–3986. | ||
Cao S, Liu W, Li F, Zhao W, Qin C. Decreased expression of lncRNA GAS5 predicts a poor prognosis in cervical cancer. Int J Clin Exp Pathol. 2014;7(10):6776–6783. | ||
Huang HW, Xie H, Ma X, Zhao F, Gao Y. Upregulation of LncRNA PANDAR predicts poor prognosis and promotes cell proliferation in cervical cancer. Eur Rev Med Pharmacol Sci. 2017;21(20):4529–4535. | ||
Hu Y, Sun X, Mao C, et al. Upregulation of long noncoding RNA TUG1 promotes cervical cancer cell proliferation and migration. Cancer Med. 2017;6(2):471–482. | ||
Zhang J, Lin Z, Gao Y, Yao T. Downregulation of long noncoding RNA MEG3 is associated with poor prognosis and promoter hypermethylation in cervical cancer. J Exp Clin Cancer Res. 2017;36(1):5. | ||
Yang L, Bai HS, Deng Y, Fan L. High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur Rev Med Pharmacol Sci. 2015;19(17):3187–3193. | ||
Mao X, Qin X, Li L, et al. A 15-long non-coding RNA signature to improve prognosis prediction of cervical squamous cell carcinoma. Gynecol Oncol. 2018;149(1):181–187. | ||
Fan J, Feng Y, Wu Y. High-dimensional variable selection for Cox’s proportional hazards model. Statistics. 2010;6:70–86. | ||
Saldana DF, Feng Y. SIS: An R package for sure independence screening in ultrahigh-dimensional statistical models. J Stat Softw. 2018;83(2). | ||
Gentle JE. Elements of computational statistics. Publications of the American Statistical Association. NY: Springer-Verlag New York, Inc; 2002. | ||
Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–344. | ||
Huang Daw, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
van Hede D, Langers I, Delvenne P, Jacobs N. Origin and immunoescape of uterine cervical cancer. Presse Med. 2014;43(12 Pt 2):e413–e421. | ||
Schrevel M, Corver WE, Vegter ME, et al. L1 cell adhesion molecule (L1CAM) is a strong predictor for locoregional recurrences in cervical cancer. Oncotarget. 2017;8(50):87568–87581. | ||
Tilborghs S, Corthouts J, Verhoeven Y, et al. The role of nuclear factor-kappa B signaling in human cervical cancer. Crit Rev Oncol Hematol. 2017;120:141–150. | ||
Li Y, Yu Y, Zhang Y, et al. MAFIP is a tumor suppressor in cervical cancer that inhibits activation of the nuclear factor-kappa B pathway. Cancer Sci. 2011;102(11):2043–2050. | ||
National Cancer Institute [home page on the Internet]. The Cancer Genome Atlas. Available from: https://cancergenome.nih.gov/. Accessed September 17, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.