Back to Journals » OncoTargets and Therapy » Volume 11
A small compound spindlactone A sensitizes human endometrial cancer cells to TRAIL-induced apoptosis via the inhibition of NAD(P)H dehydrogenase quinone 1
Received 16 February 2018
Accepted for publication 12 May 2018
Published 21 June 2018 Volume 2018:11 Pages 3609—3617
DOI https://doi.org/10.2147/OTT.S165723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Xiang-Zhai Zhao,1 Xiao-Hua Wu2,3
1Department of Gynecology and Obstetrics, The Third Hospital of Hebei Medical University, Hebei 050051, People’s Republic of China; 2Department of Gynecology and Obstetrics, Hebei Medical University, Hebei 050000, People’s Republic of China; 3Department of Gynecology and Obstetrics, Shijiazhuang Obstetrics and Gynecology Hospital, Hebei Medical University, Hebei 050000, People’s Republic of China
Introduction: Spindlactone A (SPL-A) is a novel small molecule inhibitor of TACC3 that selectively inhibits the nucleation of centrosome microtubules and induces mitotic arrest in ovarian cancer cells. SPL-A is derived from dicoumarol which inhibits the activity of NAD(P)H dehydrogenase quinone oxidoreductase 1 (NQO1). This study aimed to investigate the mechanism by which SPL-A enhances TRAIL-induced apoptosis in endometrial carcinoma cells.
Materials and methods: Endometrial carcinoma cells were treated with SPL-A and/or TRAIL, and the apoptosis and protein expression in the treated cells were examined.
Results: Combined treatment with SPL-A and TRAIL significantly induced apoptosis in various human endometrial carcinoma cells, but not in normal human endometrial stromal cells and endometrial epithelial cells. Notably, both NQO1 inhibitor ES936 and NQO1 siRNA enhanced TRAIL-induced apoptosis of endometrial carcinoma cells. Furthermore, SPL-A downregulated the expression of c-FLIP, Bcl-2, Bcl-xl, and Mcl-1, while increasing p53 expression.
Conclusion: In particular, luciferase assay showed that SPL-A inhibited Bcl-2 promoter activity, and p53 inhibitor PFT-α could reverse the effect of SPL-A on Bcl-2 expression. Moreover, Bcl-2 overexpression inhibited the apoptosis induced by SPL-A and TRAIL. Taken together, our results suggest that SPL-A sensitizes endometrial cancer cells to TRAIL-induced apoptosis via the regulation of apoptosis-related proteins and the inhibition of NQO1 activity.
Keywords: dicoumarol derivative, SPL-A, endometrial carcinoma, TRAIL, Bcl-2, apoptosis, NQO1
Corrigendum for this paper has been published
Introduction
Endometrial carcinoma is the fourth most common type of uterine cancer among women. Although most endometrial carcinomas are diagnosed in an early stage and have favorable prognosis, women diagnosed with advanced and recurrent endometrial carcinoma have poor prognosis.1 At present, the initiation and progression of endometrial carcinoma remain poorly understood. Thus, there is a great need to investigate molecular mechanism of endometrial carcinoma and develop novel targeted therapies against endometrial carcinomas.2
TRAIL could promote apoptosis in various cancer cells but not in normal cells.3 However, TRAIL resistance occurs in many carcinomas, including endometrial carcinoma. TRAIL resistance is proposed to be due to the action of decoy receptors such as mutation in DR4 (TRAIL-RI) or DR5 (TRAIL-RII), and the dysfunction of DISC components, such as FADD, caspase-8 or 10, and c-FLIP.4,5 Overexpression of c-FLIP, Bcl-2, or survivin or the inactivation of Bax, Bak, or Bid could block TRAIL-induced apoptosis and contribute to TRAIL resistance in numerous cancers.6
Spindlactone A (SPL-A) is a novel small molecule inhibitor of TACC3 that selectively inhibits the nucleation of centrosome microtubules and induces mitotic arrest in ovarian cancer cells.7 SPL-A is derived from dicoumarol, which is widely applied as an anticoagulant to inhibit vitamin K-dependent blood coagulation.8,9 Furthermore, dicoumarol and its derivative SPL-A can compete with NAD(P)H for binding to NAD(P)H quinone oxidoreductase 1 (NQO1), leading to the inhibition of NQO1 enzymatic activity.10 Dicoumarol has been shown to enhance proapoptotic effect of chemotherapy agents in various carcinoma cells.11–13 Nevertheless, the efficacy of SPL-A on TRAIL-induced apoptosis remains unclear. In this study, we aimed to evaluate the efficacy of SPL-A to enhance TRAIL-mediated apoptosis in human endometrial cancer cells.
Materials and methods
Cell culture and reagents
Ishikawa, HEC-1A, and RL-952, and human endometrial epithelial cells were purchased from ATCC (Manassas, VA, USA), and cultured in Dulbecco’s Modified Eagle’s Medium/F12 supplemented with 10% fetal bovine serum (Life Technologies Gibico, Grand Island, NE, USA), 20 mM HEPES buffer, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine at 37°C in a 5% CO2 humidified incubator. SPL-A was prepared as described previously.7 Recombinant human TRAIL was purchased from Sigma-Aldrich (St Louis, MO, USA). 6-Hydroxy-2,5,7,8-tetramethychroman-2-carboxylic acid (Trolox) and N-acetyl-L-cysteine were obtained from Beyotime Biotechnology (Suzhou, People’s Republic of China). Other reagents such as propidium iodide and radio immunoprecipitation assay (RIPA) lysis buffer were purchased from Sigma-Aldrich.
Western blot analysis
The cells were washed twice with PBS at 4°C and lysed on ice in RIPA lysis buffer containing 50 mM tris(hydroxymethyl)aminomethane (Tris)–HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 0.25% Na-deoxycholate, 1 mM Na3VO4, and 1 mM NaF and 2 mM phenylmethylsulfonyl fluoride, 2 mM EDTA, 10 μg/mL pepstatin, and 10 μg/mL leupeptin. The lysates were centrifuged at 12,000 rpm for 20 min at 4°C, and then the supernatants were collected for bicinchoninic acid protein assay. Total proteins (30 μg) were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membrane. The membranes were incubated with primary antibodies for cleaved PARP, Bcl-2, Mcl-1, Bcl-xl, c-FLIP, CIAP1, XIAP, p53, NQO1, and β-actin, which were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), at 4°C overnight, washed, and then incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) at 37°C for 1 h. The membranes were washed and detected using enhanced chemiluminescence detection system according to the manufacturer’s protocols.
Flow cytometry analysis
The cells were washed twice with PBS at 4°C, centrifuged at 1,500 rpm for 5 min at 4°C, and the precipitated cells were resuspended in 500 μL of PBS with 85% ethanol and fixed at −20°C overnight. Next, the cells were washed with PBS twice and resuspended in 500 μL PBS with RNase (0.05 g/L), incubated for 30 min at 37°C. The DNA was then stained by propidium iodide (25 μg/mL) for 30 min in the dark at room temperature. Apoptotic cells were subsequently analyzed by flow cytometer FC500 (Beckman Coulter, Brea, CA, USA).
Caspase activity assay
Cell lysates were prepared and incubated in 96-well plates in reaction buffer (100 μL) containing NaCl (137 mM), NP-40 (1%), Tris–HCl (20 mM pH 7.5), glycerol (10%), and Asp–Glu–Val–Asp–chromophore p-nitroanilide (5 μM) (caspase substrate) for 2 h at 37°C. After that, the absorbance at 405 nm was detected to measure caspase activity.
DNA fragmentation assay
Apoptosis of the cells was detected by the analysis of DNA fragments released to the cytoplasm. Briefly, the cells were centrifuged at 1,500 rpm for 10 min, and the pellet was lysed for 30 min. The lysates were centrifuged at 1,500 rpm for 10 min, and the supernatants containing cytoplasmic DNA fragments were harvested and analyzed by using Cellular DNA Fragmentation enzyme linked immunosorbent assay kit (Sigma-Aldrich) following the manufacturer’s protocols.
Reverse-transcription PCR
Total RNA was extracted from the cells using TRIZOL reagent (Life Technologies, Gaithersburg, MD, USA). cDNA was synthesized from RNA using reverse transcriptase. The cDNA was amplified by PCR on Perkin-Elmer 9600 PCR machine (Applied Biosystems, Foster, CA, USA) using specific primers: Bcl-2 (sense) 5′-GTCCTCAGCCCTCGCTCT-3′ and (antisense) 5′-CACCTAATTGGGCTCCATCT-3′; β-actin (sense) 5′-GGCATCGTCACCAACTGGGAC-3′; and (antisense) 5′-CGATTTCCCGCTCGGCCGTGG-3′. The amplification conditions were as follows: 95°C for 5 min followed by 20 cycles (β-actin) or 26 cycles (Bcl-2) of 95°C for 30 s, 57°C for 30 s, 72°C for 1 min, and final extension at 72°C for 10 min. Results are shown as the ratio of the mean threshold cycle (Ct) values of experimental groups to that of the control group after normalization to β-actin.
Luciferase assay
Ishikawa cells were grown to 70%–80% confluence. Bcl-2 promoter luciferase construct was transfected into Ishikawa cells using Lipofectamine™ 2000 (Thermo Fisher Scientific, Waltham, MA, USA). After 48 h, cells were collected, and cell lysates were analyzed using Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) following the manufacturer’s instructions.
Transfection
Ishikawa cells were transfected with pEGFP-N1-Bcl-2 plasmid or pEGFP-N1 as control using Lipofectamine™ 2000 according to the manufacturer’s instructions. Ishikawa cells were transfected with control or NQO1 siRNAs (Santa Cruz Biotechnology) using transfection reagent according to the manufacturer’s protocols.
Reactive oxygen species (ROS) measurement
Ishikawa cells were seeded into 6-well plates at 1×105 cells/well. Cells were treated with SPL-A, then incubated with H2DCF-DA (10 μM) probe at 37°C for 30 min, and then observed using a fluorescence microscope (Leica, Wetzlar, Germany).
NQO1 activity assay
Ishikawa cells were seeded into 96-well plates. Cell were treated with SPL-A, and then lysed with 2 mM EDTA (50 μL) with 0.8% digitonin at room temperature for 10 min. The lysates were incubated with 3-(4,5-dmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide as the substrate and measured at 620 nm in a microplate reader (Model 550, Bio-Rad, Hercules, CA, USA).
Statistical analysis
All data were analyzed with a 1-way analysis of variance and Student–Newman–Keuls test using the SPSS 16.0 (SPSS Inc., Chicago, IL, USA).
Results
SPL-A sensitizes human endometrial cancer cells to TRAIL-induced apoptosis
Because SPL-A has demonstrated anticancer effect on various types of carcinoma cells, we examined whether SPL-A could sensitize human endometrial cancer cells resistant to TRAIL-induced apoptosis. Our results showed that cotreatment with SPL-A and TRAIL increased the percentage of sub-G1 cell population (apoptotic cells) in Ishikawa cells (Figure 1A). In contrast, SPL-A and TRAIL did not affect the percentage of sub-G1 population in normal human endometrial stromal cells and human endometrial epithelial cells (Figure 1B). In addition, combined treatment with SPL-A and TRAIL increased PARP cleavage in Ishikawa cells (Figure 1C). Moreover, combined treatment with SPL-A and TRAIL enhanced the activity of caspases (Figure 1D) and increased DNA fragmentation in Ishikawa cells (Figure 1E).
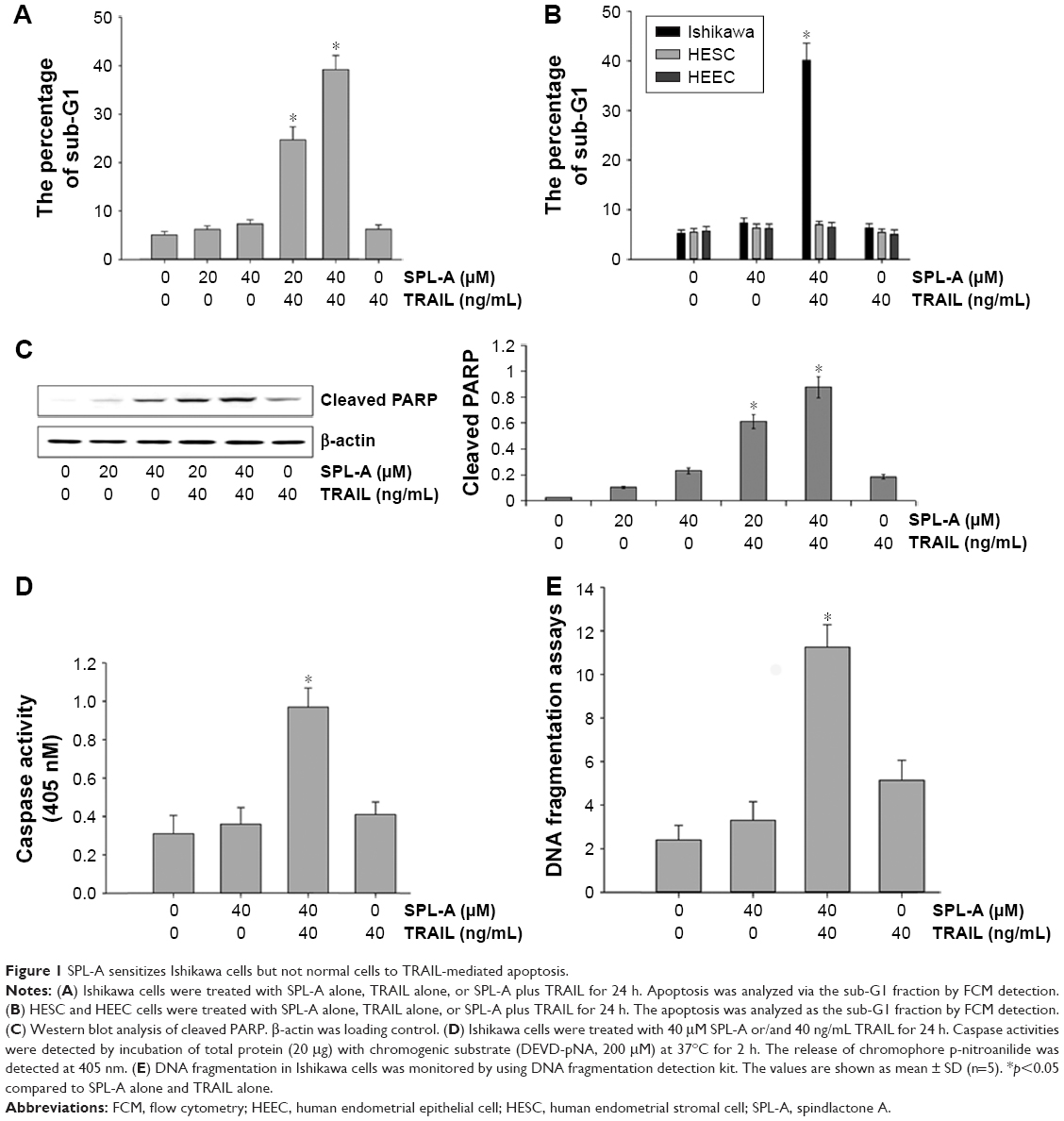 | Figure 1 SPL-A sensitizes Ishikawa cells but not normal cells to TRAIL-mediated apoptosis. |
We further detected whether SPL-A plus TRAIL affected other types of endometrial cancer cells, including HEC-1A and RL-952 cells. Results showed that combined treatment with SPL-A plus TRAIL effectively increased TRAIL-mediated apoptosis and PARP cleavage in HEC-1A and RL-952 cells (Figure 2A and B). Western blot analysis showed that SPL-A reduced the levels of c-FLIP, Bcl-2, Bcl-xL, and Mcl-1, but not cIAP1 and XIAP, in a dose-dependent manner in Ishikawa cells (Figure 2C). These data indicate that downregulation of the expression of c-FLIP, Bcl-2, Bcl-xL, and Mcl-1 may sensitize Ishikawa cells to TRAIL-mediated apoptosis by SPL-A.
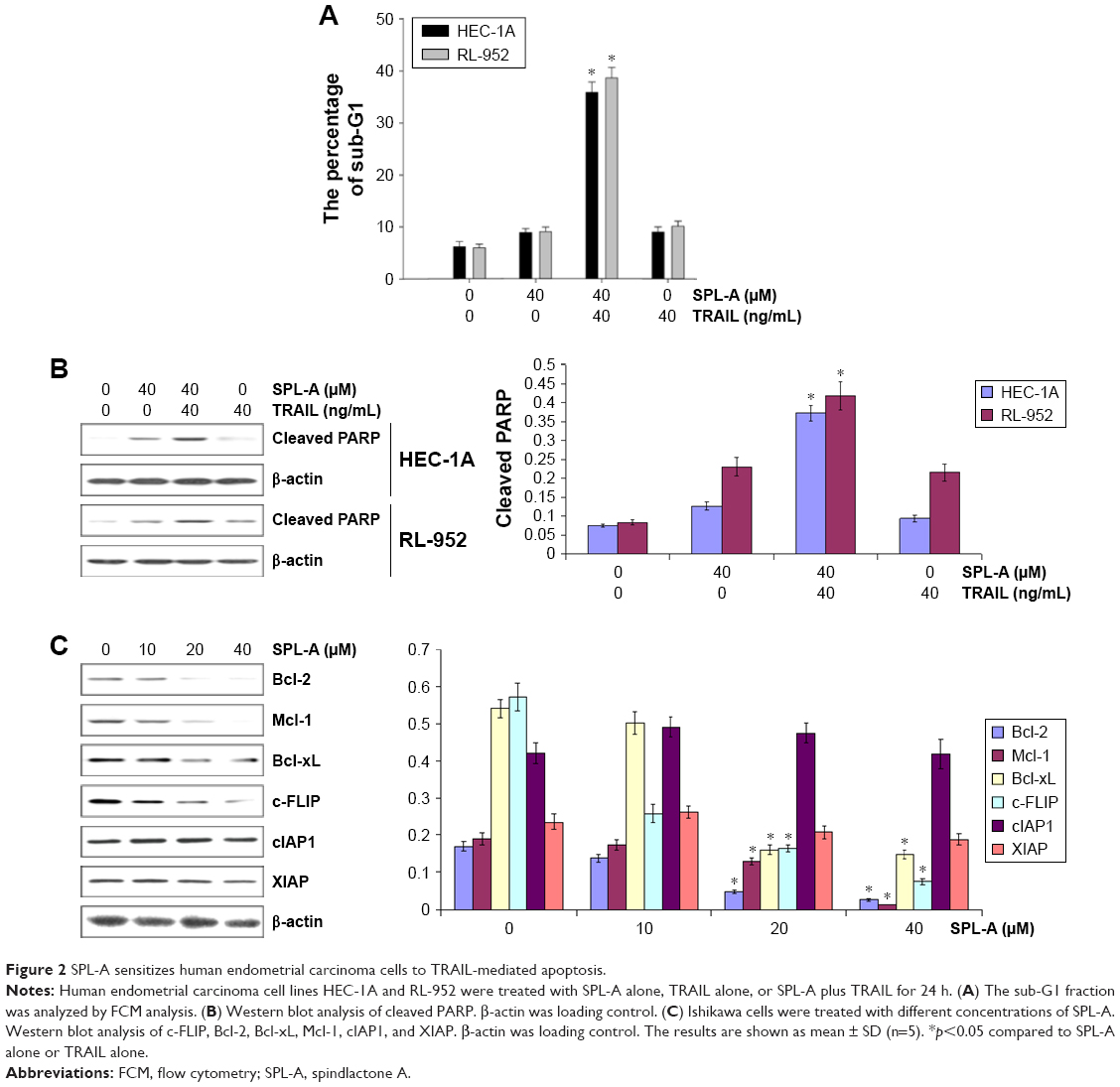 | Figure 2 SPL-A sensitizes human endometrial carcinoma cells to TRAIL-mediated apoptosis. |
SPL-A upregulates p53 expression and downregulates Bcl-2 expression
To further understand how SPL-A sensitizes endometrial cancer cells to apoptosis, we focused on Bcl-2. SPL-A decreased Bcl-2 mRNA expression in a dose-dependent manner and reduced Bcl-2 mRNA expression in a time-dependent manner (Figure 3A). Furthermore, luciferase assay showed that SPL-A inhibited Bcl-2 promoter activity in a dose-dependent manner (Figure 3B).
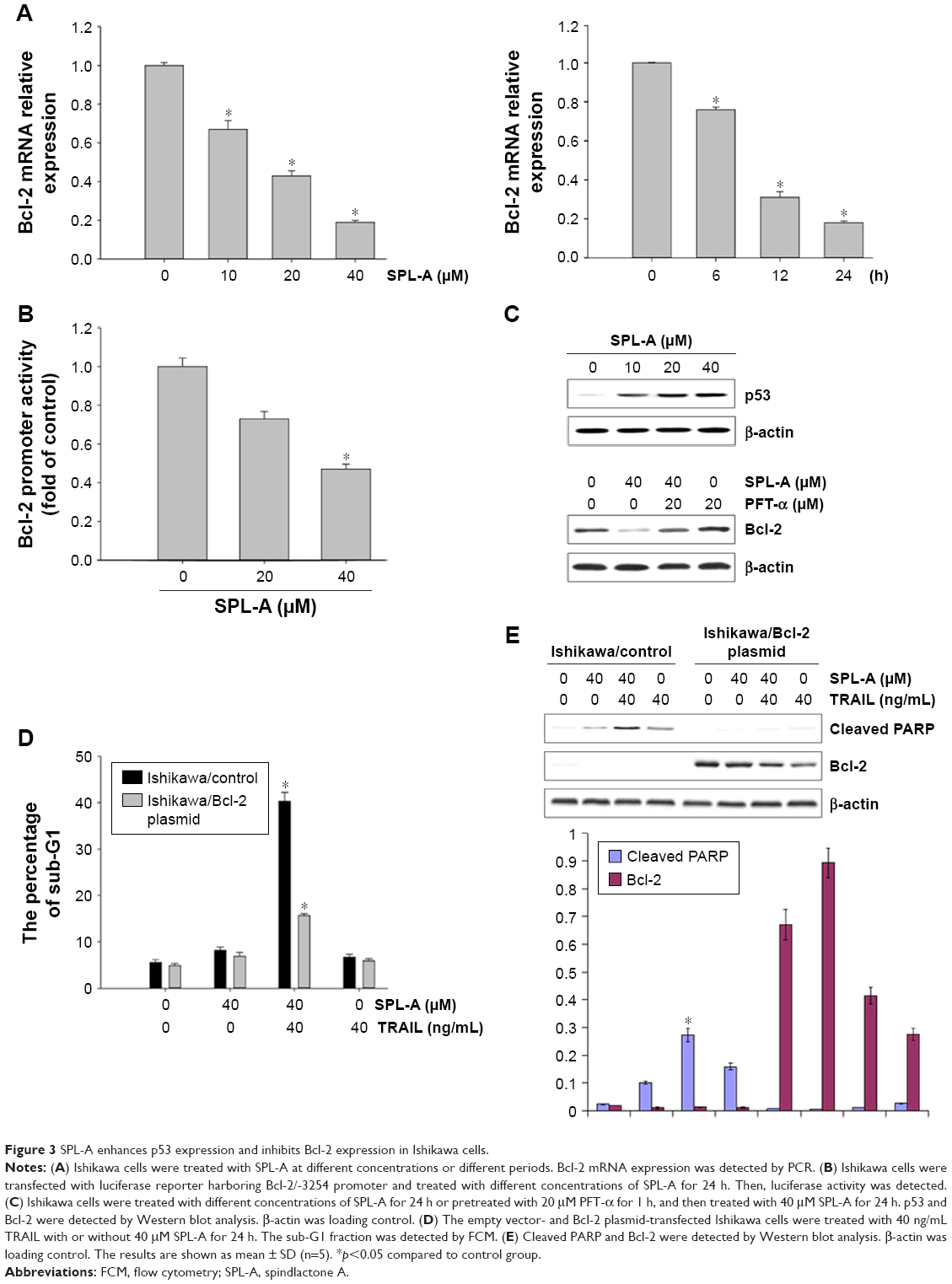 | Figure 3 SPL-A enhances p53 expression and inhibits Bcl-2 expression in Ishikawa cells. |
Previous studies showed that p53 downregulates Bcl-2 expression.14–16 Thus, we investigated whether SPL-A modulates Bcl-2 expression by p53. SPL-A increased p53 expression in a dose-dependent manner, and p53 inhibitor PFT-α could reverse the effect of SPL-A on Bcl-2 expression in Ishikawa cells (Figure 3C). Overexpression of Bcl-2 in Ishikawa cells decreased the percentage of sub-G1 cell population induced by SPL-A (Figure 3D), and inhibited PARP cleavage induced by SPL-A (Figure 3E). These results demonstrated that enhancement of TRAIL-induced apoptosis by SPL-A is associated with upregulation of p53 and downregulation of Bcl-2.
SPL-A enhances TRAIL-induced apoptosis via the inhibition of NQO1
Dicoumarol is a well-known inhibitor of NQO1 and increases ROS levels to induce apoptosis. Since SPL-A is derived from dicoumarol, the inhibition of NQO1 by SPL-A may also generate intracellular ROS and enhance TRAIL-induced apoptosis. SPL-A effectively increased intracellular ROS levels in Ishikawa cells (Figure 4A). Furthermore, pretreatment with antioxidants 6-hydroxy-2,5,7,8-tetramethychroman-2-carboxylic acid (Trolox) and N-acetyl-l-cysteine markedly decreased the apoptosis and reduced PARP cleavage in SPL-A and TRAIL treated Ishikawa cells (Figure 4B and C). These results suggest that the enhancement of ROS production contributes to the enhancement of TRAIL-induced apoptosis by SPL-A.
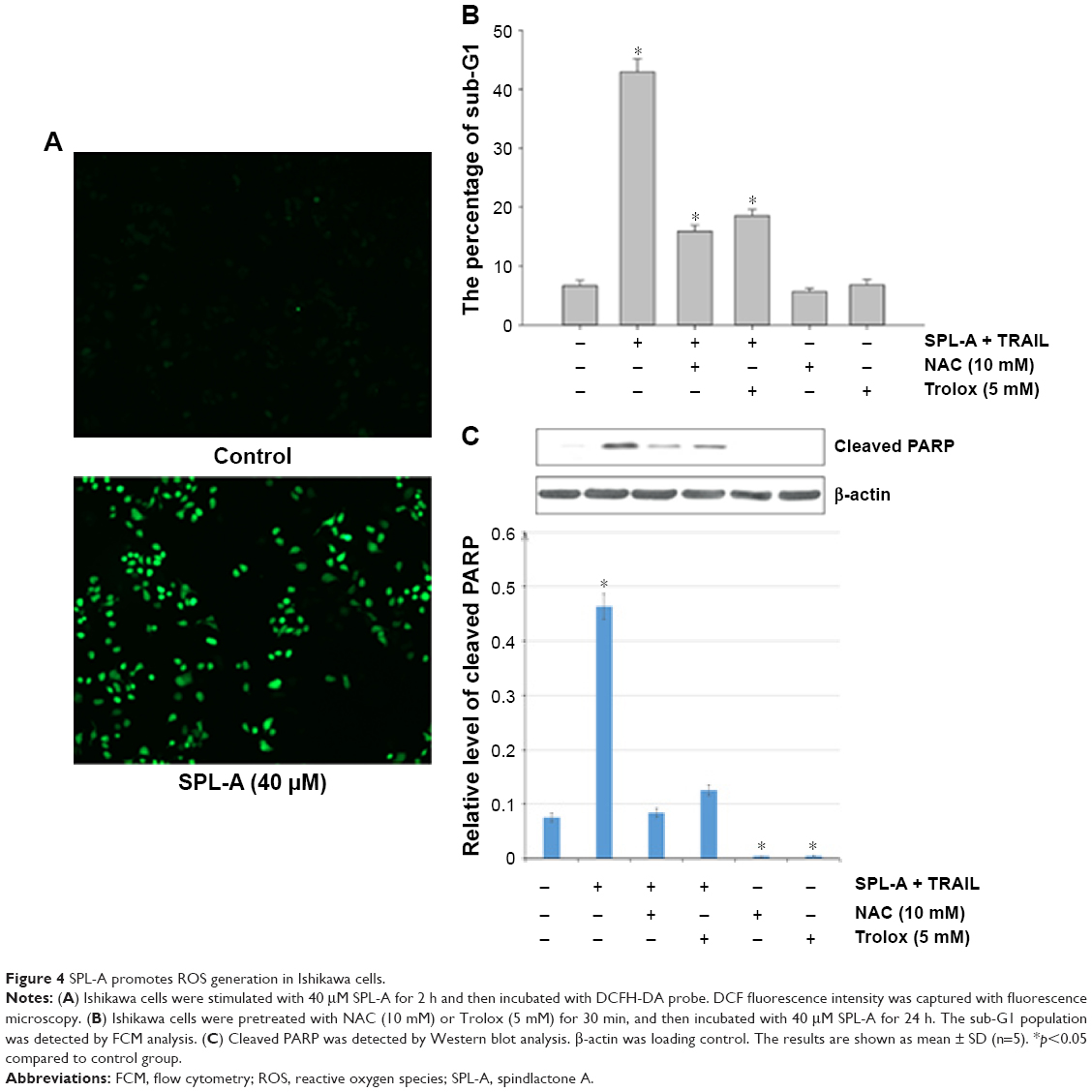 | Figure 4 SPL-A promotes ROS generation in Ishikawa cells. |
To confirm the role of NQO1 in TRAIL-induced apoptosis, we employed NQO1 inhibitor ES936. SPL-A and ES936 markedly inhibited NQO1 activity in Ishikawa cells (Figure 5A). Meanwhile, treatment with ES936 enhanced TRAIL-induced apoptosis similar to SPL-A treatment (Figure 5B). Furthermore, NQO1 siRNA and control siRNA were transfected into Ishikawa cells and then the cells were treated with TRAIL. NQO1 knockdown increased TRAIL-induced apoptosis and enhanced PARP cleavage in Ishikawa cells (Figure 5C and D). These results indicate that TRAIL-induced apoptosis mediated by SPL-A is dependent on the inhibition of NQO1.
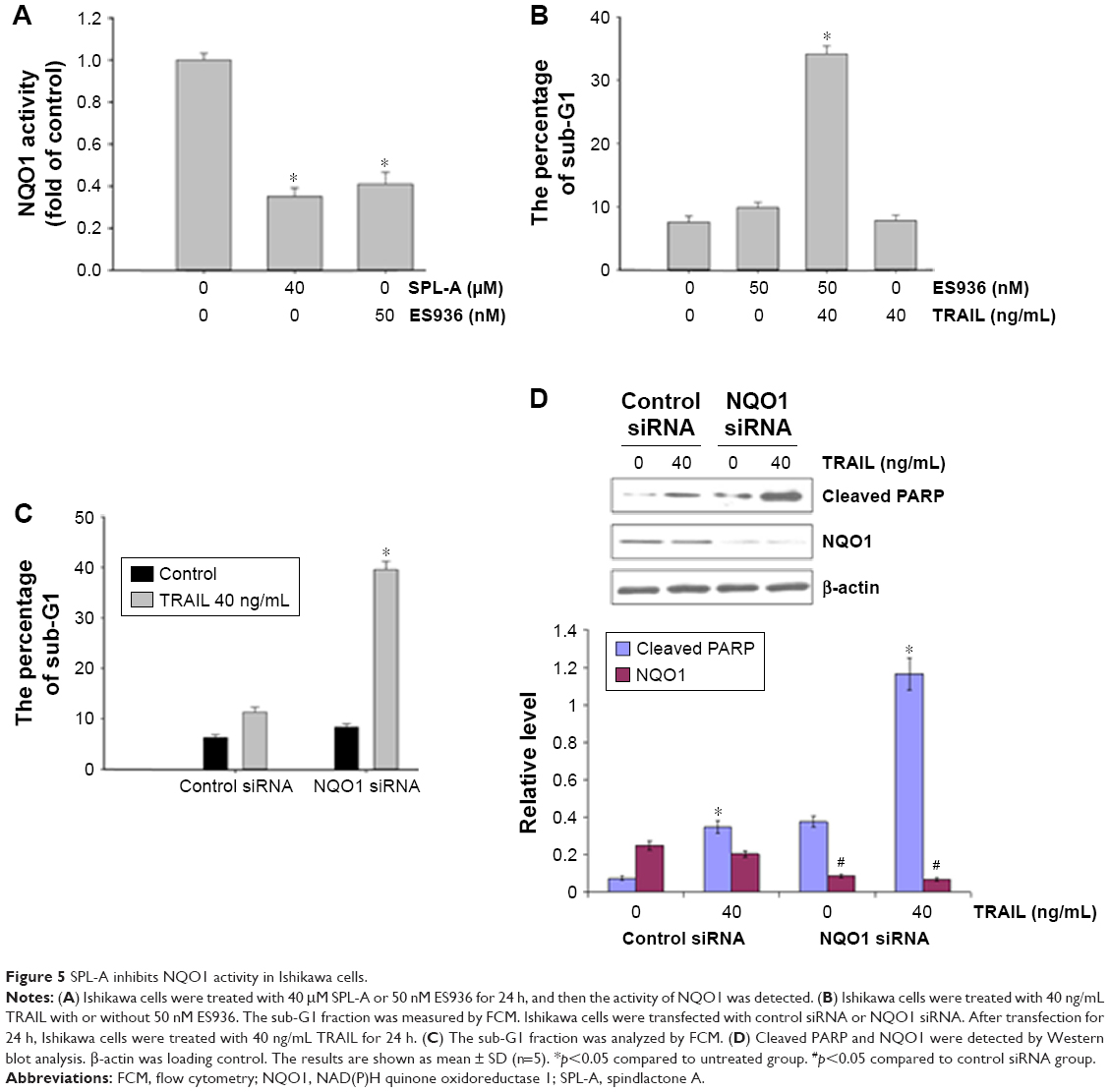 | Figure 5 SPL-A inhibits NQO1 activity in Ishikawa cells. |
Discussion
In this study, we demonstrated that SPL-A promoted TRAIL-induced apoptosis via the downregulation of Bcl-2, Bcl-xl, Mcl-1, and c-FLIP expression. Overexpression of Bcl-2 inhibited TRAIL-induced apoptosis. Inhibition of Bcl-2 expression by SPL-A is probably associated with upregulation of p53 expression in Ishikawa cells. Furthermore, SPL-A enhanced the generation of ROS, and ROS scavengers could reverse the effect of SPL-A on TRAIL-mediated apoptosis. These findings suggest that SPL-A promotes TRAIL-induced apoptosis through inducing oxidative stress in cancer cells.
The generation of intracellular ROS plays an important role in the apoptosis of various cancer cells. In the clinic, several chemotherapy drugs sensitize cancer cells to TRAIL-induced apoptosis by inducing intracellular ROS generation. It has been reported that dicoumarol can bind to albumin and induce oxidative stress through the inhibition of mitochondria electron transport. In this study, we found that dicoumarol derivative SPL-A enhanced oxidative stress and increased ROS levels in Ishikawa cells. In addition, we found that dicoumarol derivative SPL-A decreased Bcl-2 mRNA and protein expression while increasing p53 expression, and p53 inhibitor PFT-α could reverse the effect on Bcl-2 expression in SPL-A-treated Ishikawa cells. Additionally, SPL-A treatment led to downregulation of Mcl-1 and c-FLIP. Although SPL-A obviously inhibits the expression of Bcl-2 family members at a transcriptional level, further investigations are needed to elucidate the detailed mechanism responsible for the inhibition of Bcl-2 family members expression by SPL-A.
Numerous studies have shown that dicoumarol could disrupt pyrimidine biosynthesis and inhibit the activities of several enzymes such as glutathione transferase and UDP glucuronosyltransferase.17,18 NQO1 has been shown to regulate many biological processes.19–21 However, a study reported that dicoumarol sensitized renal cell carcinoma Caki cells to TRAIL-induced apoptosis through the downregulation of Bcl-2, Mcl-1, and c-FLIP in an NQO1-independent manner.22 In this study, we found that ES936 (NQO1 inhibitor) and NQO1 siRNA could enhance TRAIL-induced apoptosis in endometrial carcinoma Ishikawa cells, demonstrating that the effect of SPL-A on TRAIL-induced apoptosis is dependent on the inhibition of NQO1 activity. The exact role of NQO1 in the sensitization of cancer cells to TRAIL-induced apoptosis by SPL-A needs to be further dissected. We speculate that the function of NQO1 may be different depending on the cancer type.
In summary, our results demonstrate that dicoumarol derivative SPL-A sensitized endometrial carcinoma cells to TRAIL-mediated apoptosis through the downregulation of Bcl-2 family members Mcl-1 and c-FLIP. Also, ROS scavengers reversed, while NQO1 inhibitor or siRNA promoted, TRAIL-induced apoptosis in endometrial carcinoma cells. These findings suggest that SPL-A might be a potential adjuvant that can be used in combination with TRAIL for endometrial carcinoma treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhou Y, Shen J, Xia L, Wang Y. Estrogen mediated expression of nucleophosmin 1 in human endometrial carcinoma clinical stages through estrogen receptor-α signaling. Cancer Cell Int. 2014;14(1):540. | ||
Bezerra AL, Batista TP, Martins MR, Carneiro VC. Surgical treatment of clinically early-stage endometrial carcinoma without systematic lymphadenectomy. Rev Assoc Med Bras (1992). 2014;60(6):571–576. | ||
Zhang DW, Li HY, Lau WY, et al. Gli2 silencing enhances TRAIL-induced apoptosis and reduces tumor growth in human hepatoma cells in vivo. Cancer Biol Ther. 2014;15(12):1667–1676. | ||
Karpel-Massler G, Pareja F, Aimé P, et al. PARP inhibition restores extrinsic apoptotic sensitivity in glioblastoma. PLoS One. 2014;9(12):e114583. | ||
Chen X, Gu Y, Singh K, Shang C, Barzegar M, Jiang S, Huang S. Maduramicin inhibits proliferation and induces apoptosis in myoblast cells. PLoS One. 2014;9(12):e115652. | ||
Shi YL, Feng S, Chen W, Hua ZC, Bian JJ, Yin W. Mitochondrial inhibitor sensitizes non-small-cell lung carcinoma cells to TRAIL-induced apoptosis by reactive oxygen species and Bcl-X(L)/p53-mediated amplification mechanisms. Cell Death Dis. 2014;5:e1579. | ||
Yao R, Kondoh Y, Natsume Y, et al. A small compound targeting TACC3 revealed its different spatiotemporal contributions for spindle assembly in cancer cells. Oncogene. 2014;33(33):4242–4252. | ||
Kobayashi K, Kajiwara E, Ishikawa M, et al. Cytotoxic effects of benzbromarone and its 1′-hydroxy metabolite in human hepatocarcinoma FLC4 cells cultured on micro-space cell culture plates. Drug Metab Pharmacokinet. 2013;28(3):265–268. | ||
Siegel D, Kepa JK, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PLoS One. 2012;7(9):e44861. | ||
Timson DJ. Dicoumarol: a drug which hits at least two very different targets in vitamin K metabolism. Curr Drug Targets. 2017;18(5):500–510. | ||
Zhang W, Su J, Xu H, et al. Dicumarol inhibits PDK1 and targets multiple malignant behaviors of ovarian cancer cells. PLoS One. 2017;12(6):e0179672. | ||
Fourie J, Oleschuk CJ, Guziec F Jr, et al. The effect of functional groups on reduction and activation of quinone bioreductive agents by DT-diaphorase. Cancer Chemother Pharmacol. 2002;49(2):101–110. | ||
Krause D, Lyons A, Fennelly C, O’Connor R. Transient activation of Jun N-terminal kinases and protection from apoptosis by the insulin-like growth factor I receptor can be suppressed by dicumarol. J Biol Chem. 2001;276(22):19244–19252. | ||
Liu F, Yu G, Wang G, et al. An NQO1-initiated and p53-independent apoptotic pathway determines the anti-tumor effect of tanshinone IIA against non-small cell lung cancer. PLoS One. 2012;7(7):e42138. | ||
Lien YC, Kung HN, Lu KS, Jeng CJ, Chau YP. Involvement of endoplasmic reticulum stress and activation of MAP kinases in β-lapachone-induced human prostate cancer cell apoptosis. Histol Histopathol. 2008;23(11):1299–1308. | ||
Qiu X, Forman HJ, Schönthal AH, Cadenas E. Induction of p21 mediated by reactive oxygen species formed during the metabolism of aziridinylbenzoquinones by HCT116 cells. J Biol Chem. 1996;271(50):31915–31921. | ||
Cullen JJ, Hinkhouse MM, Grady M, et al. Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003;63(17):5513–5520. | ||
Karczewski JM, Peters JG, Noordhoek J. Quinone toxicity in DT-diaphorase-efficient and -deficient colon carcinoma cell lines. Biochem Pharmacol. 1999;57(1):27–37. | ||
Bey EA, Bentle MS, Reinicke KE, et al. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by β-lapachone. Proc Natl Acad Sci U S A. 2007;104(28):11832–11837. | ||
Zheng S, Byrd AS, Fischer BM, Grover AR, Ghio AJ, Voynow JA. Regulation of MUC5AC expression by NAD(P)H:quinone oxidoreductase 1. Free Radic Biol Med. 2007;42(9):1398–1408. | ||
Asher G, Lotem J, Cohen B, Sachs L, Shaul Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc Natl Acad Sci U S A. 2001;98(3):1188–1193. | ||
Park EJ, Min KJ, Choi KS, Kwon TK. Dicoumarol sensitizes renal cell carcinoma Caki cells to TRAIL-induced apoptosis through down-regulation of Bcl-2, Mcl-1 and c-FLIP in a NQO1-independent manner. Exp Cell Res. 2014;323(1):144–154. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.