")
Back to Journals » International Medical Case Reports Journal » Volume 14
A Self-Limited Childhood Epilepsy as Co-Incidental in Cerebral Palsy
Authors An O , Nagae LM , Winesett SP
Received 20 April 2021
Accepted for publication 13 July 2021
Published 5 August 2021 Volume 2021:14 Pages 509—517
DOI https://doi.org/10.2147/IMCRJ.S315550
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Olga An,1 Lidia Mayumi Nagae,2 Steven Parrish Winesett3
1Department of Children’s Diseases, Kazakhstan’s Medical University “KSPH” - School of Public Health, Almaty, Kazakhstan; 2Department of Medical Imaging, Neuroradiology, Banner University Medical Center, Tucson, AZ, USA; 3Department of Pediatrics, Division of Pediatric Neurology, University of Florida, Gainesville, FL, USA
Correspondence: Olga An
Department of Children’s Diseases, Kazakhstan’s Medical University “KSPH”, 19 a, Utepov st., Almaty, Kazakhstan
Tel +7 707 101-5023
Email [email protected]
Background: Cerebral palsy is the most frequent motor disability in childhood and is associated with a higher incidence of seizure disorders. In many instances, it is recognized that motor difficulties, as well as seizures, are from the same underlying brain lesion. However, self-limited childhood epilepsies, being a common group of epilepsy syndromes, would be expected to occur in patients with cerebral palsy merely on chance association and be unrelated to the structural brain imaging abnormality causing the motor impairment. Differential diagnosis in this case is important determining the long-term prognosis and need for anticonvulsant treatment.
Case Presentation: Here, we report two patients with cerebral palsy combined with epilepsy, whose age at onset, seizure semiology and electroclinical features were similar to children with self-limited childhood-specific seizure disorders (childhood epilepsy with centrotemporal spikes and Panayiotopoulos syndrome).
Conclusion: These cases highlight the importance of comprehensive differential diagnosis of seizures in cerebral palsy. Co-existence of age-dependent focal epilepsies with an underlying brain pathology as white matter injury, not affecting the cerebral cortex, might take place in the case of children with impaired motor skills. With health systems increasingly utilizing clinical pathways, it is important to consider the possibility of a self-limited childhood epilepsy and avoid aggressive and unnecessary medication treatment in children with cerebral palsy.
Keywords: childhood epilepsy with centrotemporal spikes, CECTS, Panayiotopoulos syndrome, cerebral palsy, white matter injury
Background
Cerebral palsy (CP) is the most common motor development disability, occurring in 2–3 children of every 1,000 newborns1 and about 40% of them have been reported to have epilepsy.2,3 In general, it is recognized that the motor difficulties as well as complicating seizures can be sequelae of the same underlying brain pathology. Likewise, the self-limited focal epilepsies are the most frequent epilepsy syndromes in childhood, including childhood epilepsy with centrotemporal spikes (CECTS) and Panayiotopoulos syndrome (PS).
The incidence of CECTS, also known as Rolandic epilepsy, is 10 to 20 per 100,000 children up to age 15 years, it makes up 8–25% of all epilepsies in children4 and approximately 13–23% of new onset childhood epilepsy syndromes.5 Furthermore, it is noteworthy that “rolandic” spikes occur in 2–3% of healthy children under 14 years of age without known history of seizures and are associated with epilepsy in only 10% of cases,6 making them a common incidental finding.
CECTS is characterized by seizures starting from the centrotemporal area, sometimes referred to as Rolandic area, located around the central sulcus of the brain. CECTS is classically a developmentally normal child with infrequent seizures that either consists of focal aware sensorimotor seizures in the face and arm associated with salivation or resulting in a focal to bilateral tonic-clonic seizure.4
The incidence of PS was found to be 0.8 per 100,000 children less than 16 years old.7 PS occurs in 6% of children with epilepsy between the ages of 1–15.8 Children with PS mostly have normal physical and neurocognitive development. Seizures are often prolonged (30–60 minutes), can present with an autonomic seizure manifesting as becoming pale, complaining of feeling sick and vomit with or without motor signs of a seizure, so-called “autonomic status epilepticus”. Unilateral deviation of the eyes and shaking movements can be observed. Half of patients will progress to brief focal unknown aware hemiclonic seizure or may spread to entail wider neural networks, resulting in a focal to bilateral tonic-clonic seizure. They typically last at least 6 minutes and 50% more than 30 minutes qualifying as autonomic status epilepticus.9 In most cases (up to 70%), seizures in CECTS and PS are sleep-related that manifest predominantly during drowsiness, sleep or on awakening.10,11
Self-limited focal epilepsies as the most common class of epilepsy syndromes in the pediatric age group would be expected to occur in patients with CP purely by chance and be unrelated to the structural brain imaging abnormalities, causing the motor impairments. This is very important to consider when clinical pathways are being adopted in health systems around the world. Some recent publications have speculated that co-existence of age-dependent focal epilepsies with an underlying brain lesion might be recognizable and lead to different treatment strategies.12,13
The above-mentioned sources provided a general description of the large cohorts of children with CP combined with self-limited focal epilepsies for the purpose of epidemiological assessment, without however delving into excessive clinical and neurophysiological features of the disease. The present case report aims to contribute to the literature by discussing corresponding diagnostic criteria that may lead to improved medical management for this group of patients. We describe two children with CP and co-occurring epilepsy, whose semiological and characteristic electroclinical features suggested age-dependent self-limited focal epilepsies. It can serve to emphasize the importance of this phenomenon to pediatric neurologists, general practitioners and pediatricians in the course of reviewing therapeutic strategy and advocate for avoiding anti-seizure medication (ASM) or at least early ASM weaning.
Case Presentation
Case 1
Case 1 was a 7.5-year-old male, born at 39 weeks gestation with the birth weight 3.8 kg. He had signs of perinatal asphyxia and required respiratory support while in the nursery. Although he had motor developmental delay, his cognitive status remained within normal ranges. Later, he was diagnosed with hemiplegic right-sided CP, Gross Motor Function Classification System (GMFCS) level I. He was able to accomplish gross motor skills like running, jumping and walking outdoors and function with restricted balance, speed and coordination capacity in his right limbs.
At the age of 3.5 years, he presented with a prolonged focal autonomic seizure lasting up to 30–40 minutes soon after falling asleep, started with retching, ictal vomiting, staring at onset, and followed by focal to bilateral tonic-clonic impaired aware seizure. He was seizure-free for nearly three years, then at the age of 6.5 his seizure recurred twice within a month while falling asleep in the form of an autonomic seizure with pallor, pupillary dilation, staring, ictal vomiting, flaccidity and few shaking movements of his extremities lasting for 5–10 minutes. Nevertheless, the patient was normal after subsequent few hours of sleep. By clinical semiology, none of the seizures never suggested a right-sided onset. His neurocognitive status remained unchanged with excellent academic performance and diligent behavior at school.
A single sleep-EEG done at the age of 1.5-years performed as part of routine care for his CP before the first epileptic seizure did not show epileptiform abnormalities. His EEG at 3.5 and 6.5 years after the first and second events, respectively, revealed normal background activity during wakefulness and repetitive high voltage tangential dipoles with frontal/central positivity and the main negative peak bi-occipitally maximal in the left occipital area, showing marked drowsiness and sleep activation (Figures 1 and 2).14
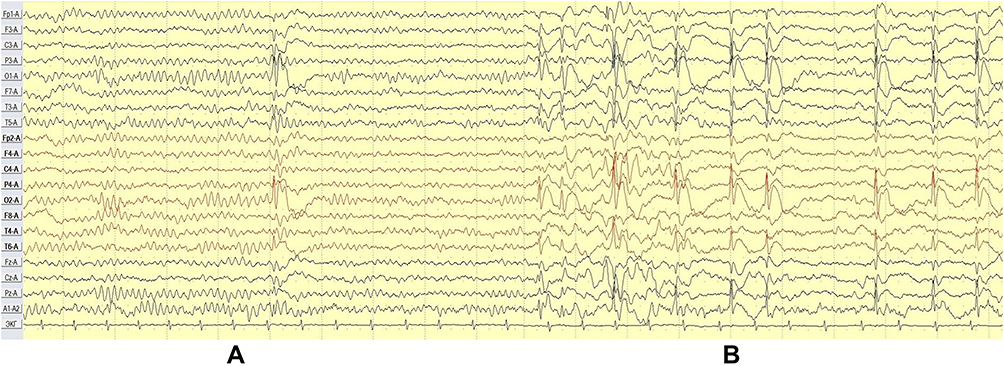 |
Figure 1 Case 1 - awake (A) vs NREM (B) (monopolar montage). |
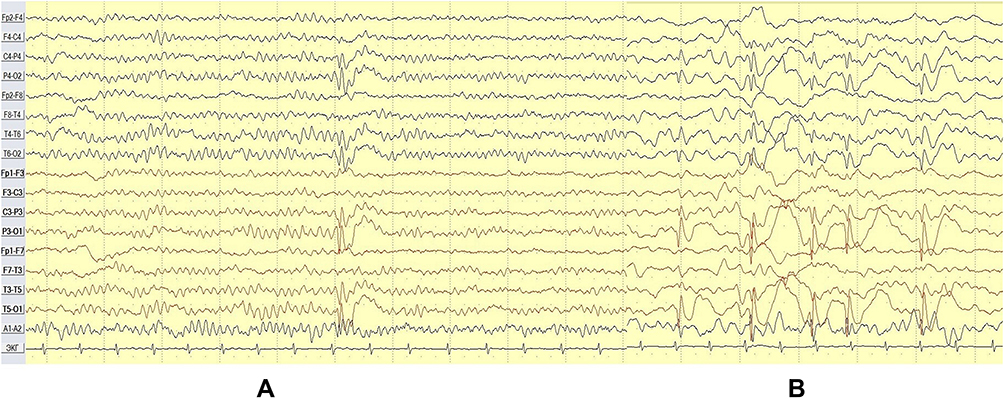 |
Figure 2 Case 1 - awake (A) vs NREM (B) (bipolar montage). |
Another aspect in favor of Panayiotopoulos syndrome was “fixation-off sensitivity” EEG-pattern most commonly encountered in patients with self-limited childhood epilepsies with occipital paroxysms (Figure 3).15
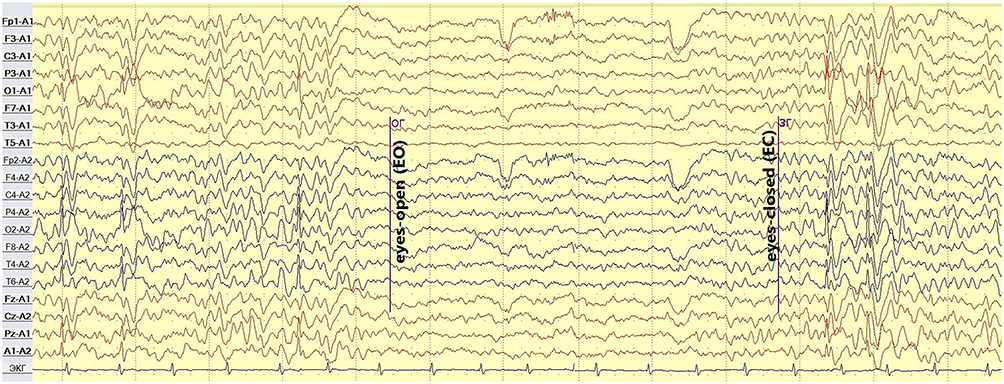 |
Figure 3 Case 1 - Fixation-off sensitivity. |
His brain magnetic resonance imaging (MRI) scans, done shortly after the second seizure, displayed asymmetric white matter lesions with mild gliosis and volume loss in periventricular regions, resulting in ex-vacuo dilatation of the left lateral ventricle (Figure 4).
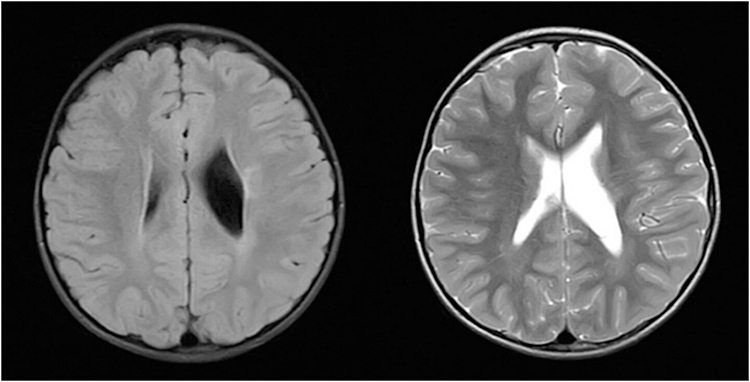 |
Figure 4 Case 1 - FLAIR ax, T2 ax. |
After the first seizure, he was diagnosed with symptomatic focal epilepsy and was prescribed valproic acid (VPA), which was later switched to levetiracetam (LEV). According to the local epilepsy clinical guidelines, he will remain on this medication for at least 2 seizure-free years and disappearance of the epileptiform focus, which typically ceases to exist around puberty for self-limited childhood epilepsies.
Case 2
Case 2 was a 6.5-year-old female born prematurely at 33 weeks gestation with the birth weight 2.0 kg. She had perinatal asphyxia and required respiratory support while in the nursery. Her motor skills milestones were delayed so that she was diagnosed with hemiplegic right-sided CP, GMFCS level I. Currently, she can walk at home, school, outdoors, and in the community by herself without hand-held mobility devices but with hemiplegic gait patterns and slower walking speed. She sits, runs and jumps with limited balance, poor motor coordination and stability. Her intelligence and oral language abilities developed within the normal ranges.
At the age of 3.0 and 3.2 years, she presented with seizures during sleep starting with making gargling laryngeal sounds and focal clonic seizure involving the right arm, then progressing to a focal to bilateral tonic-clonic seizure. Two years later her parents described the third nocturnal seizure with twitching of the child’s facial muscles on both sides and subsequent focal right-sided hemiclonic seizure with rhythmic jerking of the limbs. She was diagnosed with symptomatic focal epilepsy after the second seizure and was prescribed VPA.
The EEG was not done before the onset of epilepsy. The first interictal EEG-video-monitoring at the age of 3 after the first seizure showed a normal background activity with 2 independent foci (left centro-temporo-parieto-occipital was presented while awake and with increasing representation of spikes when falling asleep and right centro-parieto-occipital appearing only at sleep). In repeated EEG examinations 2 years later the prior left centro-temporo-parieto-occipital focus shifted forward and was localized at centro-temporal area while right centro-parieto-occipital one was not visualized (Figure 5).
These EEG-patterns consisted of high amplitude stereotyped characteristic horizontal dipoles with maximum negativity in centrotemporal regions, and positivity in frontal regions followed by slow waves with noticeable drowsiness and sleep activation (Figure 6).16
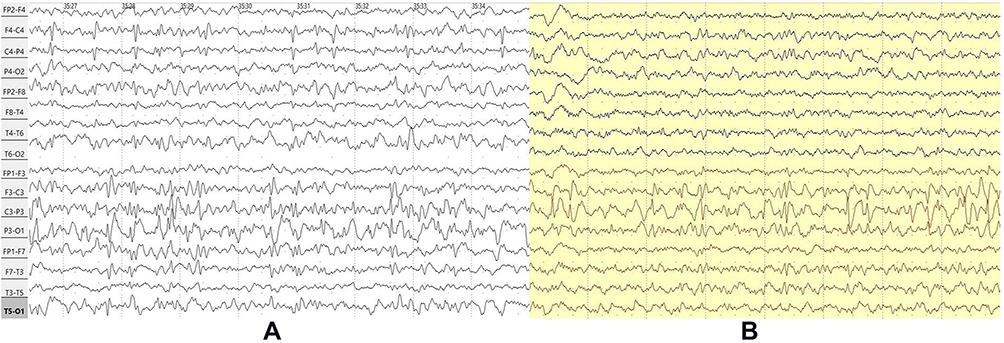 |
Figure 5 Case 2 - NREM 3 y.o. (A) vs 5 y.o. (B) (bipolar montage). |
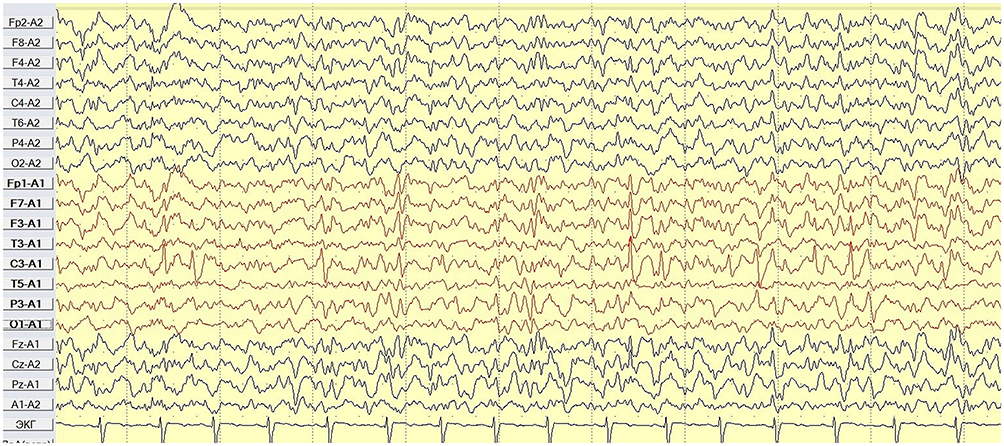 |
Figure 6 Case 2 - awake 5 y.o. (ipsilateral mastoid reference montage). |
Her brain MRI done after the third seizure at the age of 5 represented symmetric periventricular white matter lesions with gliosis and volume loss, consistent with periventricular leukomalacia (PVL) (Figure 7).
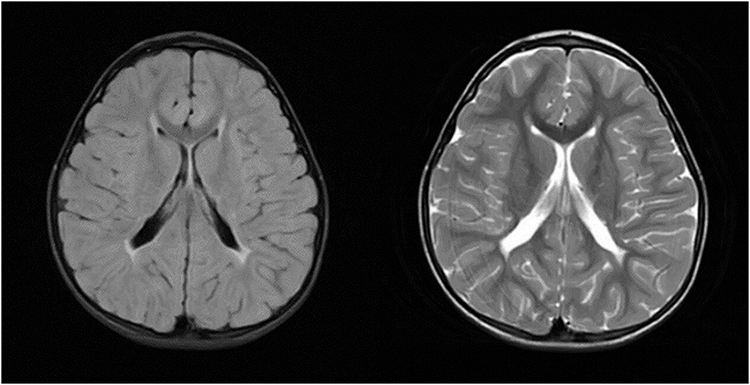 |
Figure 7 Case 2 - FLAIR ax, T2 ax. |
In this paper, we will explore if these cases could be PS and CECTS respectively and avoid long-term ASM therapy.
Discussion
CECTS was first conceptualized as a syndrome by Cesare Lombroso in the 1960s, who reported that children with epilepsy and mid-temporal spikes had a more benign prognosis than those with anterior temporal foci.17 Subsequently, other age-related partial childhood epilepsies were defined. The accepted self-limited focal epileptic syndromes include CECTS and occipital epilepsies of childhood, divided into the early onset type (Panayiotopoulos syndrome) and the late onset type (Gastaut syndrome).18 Initially, a benign disease course was suggested for this relatively frequent group of epilepsies, emerging in neurologically normal children and remitting prior to adulthood.9,17
CECTS is much more common than the other self-limited focal epilepsies. In most cases, seizures resolve by adolescence, and are often not treated.4,6 About 75% of children present with their first seizure between 7 and 10 years and the majority have less than 10 episodes during their course.11 ASM are considered in case of frequent daytime seizures or if associated with language and neurocognitive decline and increased risk of atypical evolution of CECTS. Treatment is usually of short duration and can be discontinued after 1 to 2 years of seizure freedom.6,10
The typical and irreplaceable EEG-pattern for the diagnosis of CECTS is the presence of high voltage “rolandic spikes” which typically have a centrotemporal negative maximum with frontal positivity and a triphasic configuration.16 The foci are unstable and may shift with time towards and away from the centrotemporal region, change one side to another or occur simultaneously in various locations in the same child.4 Young patients (<5 years) tend to have the discharges in occipital location, whereas older school age children more commonly have centrotemporal foci. This goes with concept that occipital foci in children less than 5 years of age often migrate forward to Rolandic area.6 This fact can explain the presence of both occipital and centrotemporal spikes in patients with CECTS or PS.
One of the most distinctive characteristics of rolandic spikes is their marked activation with non-rapid eye movement sleep (NREM). Not infrequently sharp waves appear only during sleep.10 The spikes ordinarily disappear in patients over 15–18 years of age at the latest.9 According to current studies, waiting for the EEG to normalize is not helpful since it is often abnormal well after the seizures stop, therefore, persistence of spikes at the time of ASM withdrawal is not associated with seizure recurrence. Moreover, other researchers found evidence that anticonvulsant treatment does not shorten the period of spike persistence.19
PS has similar high-voltage spikes with triphasic configuration and sleep activation over the occipital region in approximately 2/3 of the patients. PS is unusual in that the epileptiform discharges can also appear in multiple different areas of the brain in the same individual receiving multiple EEGs.6 The autonomic seizures seen in this syndrome are felt to be activated by an immature autonomic circuit from overlying ictal discharges which are not necessarily the occipital lobe and include centrotemporal, parietal and frontal locations.8 Similar to CECTS, seizures generally resolve without ASM treatment in 1–2 years and only a minority have more than 4 episodes.9
Epilepsy in patients with CP is regularly encountered in pediatric epilepsy clinics. The risk of such combination depends on the pattern of injury and etiology. Gray matter injury is thought to be much more likely, than pure white matter injury (WMI), to cause epilepsy.20 Data has shown that 35% of CP patients manifest with spastic diplegia, particularly preterm infants, who often have preferential WMI due to the predilection of damage to the periventricular white matter.1 They have less risk for epilepsy as compared to CP with spastic quadriplegia or hemiplegia (particularly from embolic strokes) which often have injury involving both white and grey matter. However, isolated WMI does not exclude grey matter abnormalities, which can be too subtle to see on routinely available MRI sequences.
Etiology matters with CP from PVL and intraventricular hemorrhage (IVH), seen more often in preterms, being far less likely to cause epilepsy than other etiologies, such as watershed and basal ganglia patterns of hypoxic ischemic injury, meningitis, non-accidental trauma, and cerebrovascular accidents (more common in term and postnatal CP). A current European survey showed 60% of postnatal CP having epilepsy versus 30–35% in preterm and term CP.21 It has been observed that most CP cases tend to have an earlier onset of epilepsy, with studies showing higher incidence of seizures in quadriplegic patients presenting in the first 2 years of life and somewhat later in diplegic and hemiplegic children. According to one population-based longitudinal study, 75% of epileptic seizures in diplegic patients occurred by 4 years and 95% by 6 years of age, while in hemiplegic patients the epilepsy was relatively delayed in onset with 55% by 4 years and 90% by 6 years.22
Since the 1970s, some researchers have occasionally put forward that self-limited epileptic disorders can be diagnosed in children with CP and a more favorable prognosis can be predicted.23 Even though most clinicians assume a high risk of intractable epilepsy in CP patients, one study from 1996 looked at 65 motor-disabled children with epilepsy after 2 years of seizure freedom who discontinued ASM treatment and 58.5% (38 patients) could be successfully weaned. It was emphasized that the remission rates were greater for children than for adults, presumably because some may have been benign age-dependent epilepsies.24
Comorbidity is defined as the coincidence of more than one disorder, unrelated in etiology or causality to the principal diagnosis, in the same individual. There are many causes of CP, which give a high likelihood of seizures often in the same regions where CECTS occurs, including embolic strokes, cortical dysplasia, IVH, meningitis, and brain trauma.3,20 These lesions, particularly those which lead to epilepsy and interictal discharges in the centrotemporal and parietal regions, can lead to the dilemma of whether the epilepsy is symptomatic of the lesion causing CP or is a self-limited focal epilepsy of childhood.20,25 Initially, CECTS was first noted in children with normal brain MRI; however, a number of reports have indicated that rolandic spikes can also be seen in children with static brain lesions.6,26,27
Recent publications have looked retrospectively at whether age-related partial seizure disorders can be diagnosed in the presence of CP despite the presence of structural changes in the brain causing motor impairments. For example, Wanigasinghe et al12 reported that out of 29 patients with focal seizures and hemiplegic CP, resulting from perinatal arterial ischemic strokes, 20 of them had clinical features, EEG findings, and remission typical for idiopathic self-limited focal epilepsy. According to the authors, this inference was made on the basis of the characteristic age presentation and early resolution of their epilepsy. However, they noted the difficulties with using EEG since the middle cerebral artery location of the perinatal stroke made the discharges “atypical” with a more posterior location than typical centrotemporal spikes.
Similarly, studying 166 children with epilepsy and CP due to isolated WMI Cooper et al13 proposed that among them 17 patients were diagnosed with CECTS and 4 with PS. The authors support the idea that the temporary coexistence of age-related focal epilepsies of childhood should be considered in patients with CP, so that ASM therapy might not be necessary or be prolonged by reason of concern about the structural brain abnormalities or persistence of spikes on EEG during childhood.
Here we report two patients with CP combined with epilepsy, whose age at onset, seizure semiology and characteristic electroclinical features were similar to children with self-limited focal seizure disorders despite abnormal MRI scans. We felt comfortable with these children having PS and CECTS instead of symptomatic epilepsy due to the lesions being confined to white matter without significant cortical injury, as well as, having spike morphology and locations of foci characteristic for the self-limited focal epilepsies of childhood.
In the first case, the presence of occipital focus of high-amplitude sharp-slow-wave complexes amplifying during NREM sleep in a boy with specific autonomic seizures, normal cognitive development and brain MRI without significant cortical injury allowed us to suggest PS.
In case 2, a girl at the age of 3 had typical dipoles occurring both centrotemporally, as well as more posterior which is common for self-limited focal epilepsies. She had bilateral independent spikes which moved forward with age, suggesting that she was predisposed to an age-dependent focal epilepsy. Arguments suggesting that it was not CECTS include that she had hemiplegic CP and the fact that the focus did occur on the same side as the hemisphere producing her hemiplegia. Although, taking into account the specific semiology of infrequent seizures, normal neurocognitive development and WMI on brain MRI, we felt that this with high probability was CECTS.
They presented to the outpatient pediatric neurology department for a routine medical examination several months after one and two seizures, respectively. By then, they were prescribed ASM therapy because of “symptomatic epilepsy”. According to the boy’s parents, they intentionally stopped taking ASM each time after a few months due to their own concerns about long-term drug side-effects. In the second case, parents forgot to administer medication and noted their daughter did not feel any different when they missed her medication for one or more days. The issues of impaired quality of life and medication side effects remain a fear for parents when following a prolonged treatment. Their uncertainty about clinician’s recommendations increased over time, due to the fact that convulsions did not recur for a relatively long period (3 and 2 years, respectively), even without drug therapy. According to local treatment protocols, they are prevented from ASM discontinuation owing to the presence of epileptiform spikes on EEG and abnormal MRI despite prolonged drug-free remission in history and rare frequency of seizures.
These children were followed up until 7–8 years old, about 2 years after the last seizure. Further observation with an assessment of evolution of the EEG-patterns over time and a tendency for recurrent seizures will help in the differential diagnosis.
Conclusions
As demonstrated in this report, co-existence of self-limited focal childhood epilepsies with an underlying brain lesions not affecting the cerebral cortex might take place in the case of motor-disabled children. Defining who should be involved in a pathway for self-limited focal childhood seizure disorders has important implications for treatment and we would argue that properly screened patients without significant cortical injury (ie, patient with CP and exclusively white matter lesions) may be included in this clinical pathway and not be subjected to excessive ASM. These examples might help pediatric neurologists to review the therapeutic strategy regarding such patients and advocate for either no ASM or consideration of early weaning after 2 years of seizure freedom.
Abbreviations
CECTS, childhood epilepsy with centrotemporal spikes; PS, Panayiotopoulos syndrome; CP, cerebral palsy; GMFCS, gross motor function classification system; WMI, white matter injury; PVL, periventricular leukomalacia; IVG, intraventricular hemorrhage; EEG, electroencephalography; NREM, nonrapid eye movement; MRI, magnetic resonance imaging; FLAIR, fluid-attenuated inversion recovery; ASM, anti-seizure medication; VPA, valproic acid; LEV, levetiracetam.
Data Sharing Statement
All available data can be obtained by contacting the corresponding author.
Informed Consent/Institutional Approval
A written informed consent has been provided by the patients’ parents to have the case details and any accompanying images published. Institutional approval was not required to publish the case details.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Patel DR, Neelakantan M, Pandher K, Merrick J. Cerebral palsy in children: a clinical overview. Transl Pediatr. 2020;9(Suppl 1):S125–S135. doi:10.21037/tp.2020.01.01
2. Pavone P, Gulizia C, Le Pira A, et al. Cerebral palsy and epilepsy in children: clinical perspectives on a common comorbidity. Children (Basel). 2020;8(1):16. doi:10.3390/children8010016
3. Tokatly Latzer I, Blumovich A, Sagi L, Uliel-Sibony S, Fattal-Valevski A. Prediction of drug-resistant epilepsy in children with cerebral palsy. J Child Neurol. 2020;35(3):187–194. doi:10.1177/0883073819883157
4. Dryżałowski P, Jóźwiak S, Franckiewicz M, Strzelecka J. Benign epilepsy with centrotemporal spikes - current concepts of diagnosis and treatment. Neurol Neurochir Pol. 2018;52(6):677–689. doi:10.1016/j.pjnns.2018.08.010
5. Bourel-Ponchel E, Mahmoudzadeh M, Adebimpe A, Wallois F. Functional and structural network disorganizations in typical epilepsy with centro-temporal spikes and impact on cognitive neurodevelopment. Front Neurol. 2019;10:809. doi:10.3389/fneur.2019.00809
6. Panayiotopoulos C. A clinical guide to epileptic syndromes and their treatment.
7. Weir E, Gibbs J, Appleton R. Panayiotopoulos syndrome and benign partial epilepsy with centro-temporal spikes: a comparative incidence study. Seizure. 2018;57:66–69. doi:10.1016/j.seizure.2018.03.002
8. Graziosi A, Pellegrino N, Di Stefano V, Raucci U, Luchetti A, Parisi P. Misdiagnosis and pitfalls in Panayiotopoulos syndrome. Epilepsy Behav. 2019;98(Pt A):124–128. doi:10.1016/j.yebeh.2019.07.016
9. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain. 2008;131(Pt 9):2264–2286. doi:10.1093/brain/awn162
10. Kumar J, Solaiman A, Mahakkanukrauh P, Mohamed R, Das S. Sleep related epilepsy and pharmacotherapy: an insight. Front Pharmacol. 2018;9:1088. doi:10.3389/fphar.2018.01088
11. Amrutkar C, Riel-Romero RM. Rolandic epilepsy seizure. Treasure Island (FL): StatPearls Publishing LLC; 2020.
12. Wanigasinghe J, Reid SM, Mackay MT, Reddihough DS, Harvey AS, Freeman JL. Epilepsy in hemiplegic cerebral palsy due to perinatal arterial ischaemic stroke. Dev Med Child Neurol. 2010;52(11):1021–1027. doi:10.1111/j.1469-8749.2010.03699.x
13. Cooper MS, Mackay MT, Fahey M, et al. Seizures in children with cerebral palsy and white matter injury. Pediatrics. 2017;139(3):e20162975. doi:10.1542/peds.2016-2975
14. Datta AN, Wallbank L, Micallef J, Wong PKH. Pediatric occipital spikes at a single center over 26 years and the significance of tangential dipole. J Child Neurol. 2021;36(7):530–536. doi:10.1177/0883073820984042
15. Panayiotopoulos CP. Inhibitory effect of central vision on occipital lobe seizures. Neurology. 1981;31(10):1330–1333. doi:10.1212/wnl.31.10.1331
16. Gregory DL, Wong PK. Clinical relevance of a dipole field in rolandic spikes. Epilepsia. 1992;33(1):36–44. doi:10.1111/j.1528-1157.1992.tb02280.x
17. Lombroso CT. Sylvian seizures and midtemporal spike foci in children. Arch Neurol. 1967;17(1):52–59. doi:10.1001/archneur.1967.00470250056005
18. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–521. doi:10.1111/epi.13709
19. Kim H, Kim SY, Lim BC, et al. Spike persistence and normalization in benign epilepsy with centrotemporal spikes - implications for management. Brain Dev. 2018;40(8):693–698. doi:10.1016/j.braindev.2018.04.011
20. Rasmussen T. Localizational concepts in epilepsy: past, present and future. Appl Neurophysiol. 1987;50(1–6):355–358. doi:10.1159/000100739
21. Sellier E, Uldall P, Calado E, et al. Epilepsy and cerebral palsy: characteristics and trends in children born in 1976–1998. Eur J Paediatr Neurol. 2012;16(1):48–55. doi:10.1016/j.ejpn.2011.10.003
22. Carlsson M, Hagberg G, Olsson I. Clinical and aetiological aspects of epilepsy in children with cerebral palsy. Dev Med Child Neurol. 2003;45(6):371–376. doi:10.1017/s0012162203000719
23. Lerman P, Kivity S. Benign focal epilepsy of childhood. A follow-up study of 100 recovered patients. Arch Neurol. 1975;32(4):261–264. doi:10.1001/archneur.1975.00490460077010
24. Delgado MR, Riela AR, Mills J, Pitt A, Browne R. Discontinuation of antiepileptic drug treatment after two seizure-free years in children with cerebral palsy. Pediatrics. 1996;97(2):192–197.
25. Salanova V, Andermann F, Olivier A, Rasmussen T, Quesney LF. Occipital lobe epilepsy: electroclinical manifestations, electrocorticography, cortical stimulation and outcome in 42 patients treated between 1930 and 1991. Surgery of occipital lobe epilepsy. Brain. 1992;115(Pt 6):1655–1680. doi:10.1093/brain/115.6.1655
26. Santanelli P, Bureau M, Magaudda A, Gobbi G, Roger J. Benign partial epilepsy with centrotemporal (or rolandic) spikes and brain lesion. Epilepsia. 1989;30(2):182–188. doi:10.1111/j.1528-1157.1989.tb05452.x
27. Sarkis R, Wyllie E, Burgess RC, Loddenkemper T. Neuroimaging findings in children with benign focal epileptiform discharges. Epilepsy Res. 2010;90(1–2):91–98. doi:10.1016/j.eplepsyres.2010.03.012
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.