Back to Journals » Journal of Multidisciplinary Healthcare » Volume 19
A Scoping Review on Genetic Mutations and Single-Nucleotide Polymorphisms Associated with Pectus Excavatum
Authors Barman S, Momin L, Islam R, Shing MS, Ranjan R ![]() , Waterhouse B, Dunning J
, Waterhouse B, Dunning J
Received 28 December 2025
Accepted for publication 17 March 2026
Published 21 March 2026 Volume 2026:19 592241
DOI https://doi.org/10.2147/JMDH.S592241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David C. Mohr
Santo Barman,1 Lana Momin,2 Rafiza Islam,3 Moung Shwe Shing,4 Redoy Ranjan,5,6 Benjamin Waterhouse,6 Joel Dunning6
1Department of Vascular Surgery, IBN Sina Specialized Hospital, Dhaka, Bangladesh; 2Sixth form, Haberdashers’ Girls’ School, Elstree, Hertfordshire, UK; 3Department of Vascular Surgery, St. Mary’s Hospital, London, UK; 4Department of Surgery, East Kent Hospital University NHS Foundation Trust, Canterbury, Kent, UK; 5Department of Cardiothoracic Surgery, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh; 6Department of Cardiothoracic Surgery, James Cook University Hospital, Middlesbrough, Cleveland, UK
Correspondence: Redoy Ranjan, Department of Cardiothoracic Surgery, James Cook University Hospital, Middlesbrough, UK, Email [email protected]
Abstract: Pectus excavatum (PE) is the most common congenital chest wall deformity, affecting approximately 1 in 400 live births, with a male predominance. While traditionally considered a primarily structural or biomechanical disorder, emerging evidence suggests significant genetic contributions to its etiology. This literature review systematically examines the current state of knowledge regarding genetic mutations, single-nucleotide polymorphisms (SNPs), and structural variants associated with pectus excavatum. A comprehensive search was conducted across multiple databases including PubMed, Google Scholar, SciSpace, and institutional repositories, yielding 14 relevant studies after de-duplication. The review reveals substantial genetic heterogeneity in PE, with identified variants predominantly affecting connective tissue genes including collagen family members (COL1A1, COL27A1, COL5A1), cartilage matrix proteins (ACAN, COMP), and signaling pathway components (SMAD4, REST). Copy number variants (CNVs), particularly the 3q29 deletion syndrome, show elevated PE prevalence. Early-onset PE demonstrates a 44% pathogenic genetic finding rate, suggesting stronger genetic contribution in childhood presentations. However, a critical gap exists in the literature: quantitative effect sizes such as odds ratios and hazard ratios are rarely reported, reflecting the predominance of case reports and small familial studies rather than large-scale genome-wide association studies. This review highlights the need for multi-center collaborative efforts to conduct adequately powered genetic epidemiological studies, establish genotype-phenotype correlations, and develop polygenic risk scores for clinical application.
Keywords: pectus excavatum, genetic mutations, single-nucleotide polymorphisms, copy number variants, connective tissue disorders, chest wall deformity
Introduction
Pectus excavatum (PE), also known as funnel chest, represents the most prevalent congenital chest wall deformity, occurring in approximately 1 in 400 live births with a marked male-to-female ratio of 3–5:1.1,2 The condition is characterized by posterior depression of the sternum and adjacent costal cartilages, resulting in a concave appearance of the anterior chest wall.3 While historically regarded as primarily a cosmetic concern, contemporary understanding recognizes PE as a condition with potential cardiopulmonary implications, psychological impacts, and significant familial clustering patterns that suggest underlying genetic contributions.4,5 The clinical presentation of PE demonstrates considerable heterogeneity in severity, age of onset, progression patterns, and associated features. Some patients present with isolated PE, while others exhibit deformity as part of recognized genetic syndromes including Marfan syndrome and Ehlers-Danlos syndrome.6,7 This phenotypic diversity, combined with observed familial aggregation up to 43%, has prompted intensive investigation into genetic architecture underlying PE susceptibility.8,9
Early studies of PE genetics focused primarily on syndromic presentations with well-characterized genetic etiologies. Marfan syndrome, caused by mutations in the FBN1 gene encoding fibrillin-1, frequently presents with PE as part of its skeletal manifestation.10 Similarly, Loeys-Dietz syndrome, resulting from mutations in transforming growth factor-beta (TGF-β) pathway genes including TGFBR1, TGFBR2, SMAD3, TGFB2, and TGFB3, commonly features chest wall deformities.11 These syndromic associations provided initial insights into potential pathogenic mechanisms, particularly implicating connective tissue integrity and extracellular matrix (ECM) homeostasis in PE development.12
More recent investigations have expanded beyond syndromic cases to examine the genetic basis of isolated, non-syndromic PE. Familial linkage studies have identified chromosomal regions of interest, including a locus on chromosome 18q identified through analysis of a large pedigree segregating both adolescent idiopathic scoliosis and PE.13 Candidate gene approaches have examined polymorphisms in genes encoding cartilage matrix components, with particular attention to aggrecan (ACAN), a major proteoglycan in cartilage tissue.14 Whole exome sequencing of multiplex families has revealed rare, family-specific variants in genes involved in connective tissue structure and regulation, though without identification of a single common causal gene across families.15
The advent of high-throughput sequencing technologies and large-scale genomic databases has enabled more comprehensive investigation of PE genetics. Copy number variant (CNV) analyses have identified structural genomic alterations associated with PE, including the 3q29 deletion syndrome which demonstrates significantly elevated PE prevalence.16 Studies of early-onset PE have revealed high rates of pathogenic genetic findings, suggesting that childhood presentations may represent a more genetically determined subset of the PE spectrum.17 Despite these advances, several critical gaps persist in our understanding of PE genetics. Most notably, literature lacks large-scale genome-wide association studies (GWAS) that could identify common variants with modest effect sizes contributing to PE risk in the general population. The predominance of case reports, small familial studies, and syndromic cohorts has limited the ability to quantify population-level genetic risk through measures such as odds ratios and hazard ratios. Additionally, the genetic architecture of PE appears highly heterogeneous, with multiple rare variants across different genes potentially converging on common pathophysiological pathways.18
Understanding the genetic basis of PE has important clinical implications. Genetic testing may aid in identifying syndromic cases requiring multidisciplinary evaluation and management. Knowledge of genetic risk factors could inform family counseling and recurrence risk assessment. Furthermore, elucidating the molecular mechanisms underlying PE pathogenesis may eventually enable development of targeted therapeutic approaches or identification of patients at higher risk for severe deformity requiring surgical intervention.17–19
This comprehensive literature review aims to systematically examine the current evidence regarding genetic mutations, single-nucleotide polymorphisms (SNPs), and structural variants associated with pectus excavatum. By synthesizing findings from diverse study designs including familial linkage analyses, candidate gene studies, whole exome sequencing investigations, and CNV analyses, this review provides a comprehensive overview of the genetic landscape of PE. We critically evaluate the strength of evidence for specific genetic associations, identify patterns across studies, and highlight methodological limitations and future research directions needed to advance the field.
Methods
The primary objective of this literature review was to comprehensively identify and synthesise all published evidence regarding genetic mutations, single-nucleotide polymorphisms, copy number variants, and other structural genomic alterations associated with pectus excavatum. Secondary objectives included cataloguing quantitative measures of genetic association, including odds ratios, hazard ratios, and other effect size estimates where reported; characterising the genetic architecture of both syndromic and non-syndromic pectus excavatum; identifying patterns across studies regarding implicated genes, pathways, and mechanisms; and evaluating the quality and strength of evidence for specific genetic associations. This study was conducted according to the declaration of Helsinki.
Search Strategy and Terms
A systematic search strategy was developed to capture all relevant literature on the genetics of pectus excavatum. The search was conducted on 30 November 2025 and included publications from database inception through the search date. The primary search string combined terms for the condition itself, including “pectus excavatum”, “funnel chest”, and “chest wall deformity”, with genetic terminology encompassing “genetic”, “genetics”, “genomic”, “mutation”, “polymorphism”, “single nucleotide polymorphism”, “copy number variant”, “chromosomal”, “familial”, “hereditary”, “genome-wide association”, “exome sequencing”, and “whole genome sequencing”. A secondary search string specifically targeting quantitative measures paired “pectus excavatum” with terms such as “odds ratio”, “hazard ratio”, “relative risk”, “association”, and “genetic risk”. Additionally, a tertiary search string focused on specific genetic elements previously implicated in chest wall development or connective tissue disorders, including ACAN, various collagen genes, COMP, SMAD4, REST, FBN1, TGFBR, and chromosomal regions, such as 3q29 deletion and 18q.
Databases and Repositories Searched
To ensure comprehensive coverage of the literature, several databases and repositories were systematically searched. PubMed served as the primary biomedical literature database, searched using both Medical Subject Headings terms and free text. Google Scholar provided broad academic coverage, capturing grey literature and institutional repositories that might otherwise be missed. ProQuest Dissertations and Theses Global enabled access to doctoral dissertations and master’s theses, whilst OpenGrey captured European grey literature. SciSpace offered comprehensive academic database coverage, including both indexed and preprint literature, with semantic search capabilities. Finally, ClinicalTrials.gov was searched to identify clinical trials that may report genetic findings.
Search Strategies
The study selection process followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for systematic reviews.20 Articles in all languages were included to minimise publication bias and ensure comprehensive global coverage. Non-English articles were translated using professional translation services when necessary. All publication types were considered eligible, encompassing peer-reviewed original research articles, case reports and case series, review articles examined for additional references, conference abstracts and proceedings, dissertations and theses, preprint manuscripts, and clinical trial reports. In addition to database searches, several manual search strategies were employed to identify relevant literature that might have been missed through electronic searching alone. Reference list screening involved examining the bibliographies of all included articles and relevant review papers. Citation tracking utilised Google Scholar and Web of Science to identify articles citing key papers through forward citation searching. The Author’s search identified prolific researchers in the field, and the subsequent examination of their complete publication lists revealed further insights. Journal hand-searching involved manual review of tables of contents for key journals, including Clinical Genetics, American Journal of Medical Genetics, European Journal of Human Genetics, and Journal of Pediatric Surgery. Conference proceedings from relevant scientific meetings, including the American Society of Human Genetics, European Society of Human Genetics, and Chest Wall International Group, were also reviewed for pertinent abstracts.
Inclusion and Exclusion Criteria
Studies were included if they reported original data on genetic mutations, polymorphisms, variants, or chromosomal alterations in individuals with pectus excavatum; included human subjects; provided sufficient detail to extract genetic findings such as gene name, variant type, or chromosomal location; clearly defined pectus excavatum diagnosis using clinical examination, imaging, or surgical confirmation; and were published or made available in any format. Conversely, studies were excluded if they discussed pectus excavatum only as an incidental finding without genetic analysis, were purely mechanistic studies without human genetic data, represented duplicate publications of the same cohort without additional data, lacked sufficient methodological detail to assess quality, or were retracted publications.
Data Extraction
Initial database searches identified 151 relevant articles. After removing duplicates and applying the inclusion criteria, 79 unique articles remained for full-text review and data extraction. Of these, 67 studies were excluded because they lacked genetic analysis data, were purely mechanistic without human genetic data, or represented duplicate publications of the same cohort without additional data; finally, 14 studies13–17,21–28 were included in this scoping review. For each included study, data elements were systematically extracted encompassing study characteristics such as authors, publication year, journal, study design, and sample size; population characteristics including age, sex, ethnicity, and whether cases represented syndromic or isolated pectus excavatum; genetic findings detailing gene name, variant identifier, and variant type; statistical measures including odds ratios, hazard ratios, relative risks, p-values, and confidence intervals; functional data describing predicted or demonstrated effects on protein function; and clinical correlations examining genotype-phenotype associations and severity measures.
Data Quality Assessment
Study quality was assessed using adapted criteria from the Newcastle-Ottawa Scale29 for observational studies and the Analytic validity, Clinical validity, Clinical utility, and Ethical implications framework for genetic test evaluation. Quality assessment considered selection bias through evaluation of appropriate case definition, representative sampling, and adequate controls; measurement quality by examining validated genetic testing methods, confirmation of variants, and quality control procedures; analytical validity through assessment of appropriate statistical methods, correction for multiple testing, and replication attempts; and reporting quality by evaluating completeness of variant reporting, availability of raw data, and adherence to reporting guidelines such as Strengthening the Reporting of Genetic Association Studies and Strengthening the Reporting of Observational Studies in Epidemiology. Studies were categorised as high, moderate, or low quality based on these criteria, with quality ratings used to inform the strength of conclusions drawn from individual studies. Given the heterogeneity in study designs, genetic testing approaches, and outcome measures, a narrative synthesis rather than a meta-analysis was employed. Data were synthesised by organising findings by gene or locus, categorising by variant type, grouping by syndromic versus non-syndromic presentations, and identifying patterns across studies regarding implicated biological pathways. Quantitative synthesis via meta-analysis was not feasible due to the predominance of case reports and the absence of comparable effect-size estimates across studies.
Literature Review Findings
The literature reviews identified genetic associations with pectus excavatum spanning multiple categories: single-gene mutations, single-nucleotide polymorphisms, copy number variants, and chromosomal linkage regions. The genetic architecture of PE demonstrates substantial heterogeneity, with no single common variant accounting for a large proportion of cases. Instead, the evidence points to multiple rare variants across different genes, potentially converging on common pathophysiological pathways involving connective tissue integrity, cartilage development, and extracellular matrix homeostasis. Table 1 presents a comprehensive summary of genetic variants associated with pectus excavatum identified through this scoping review. The table includes gene names, variant identifiers where available, variant types, reported statistical associations, and study populations.
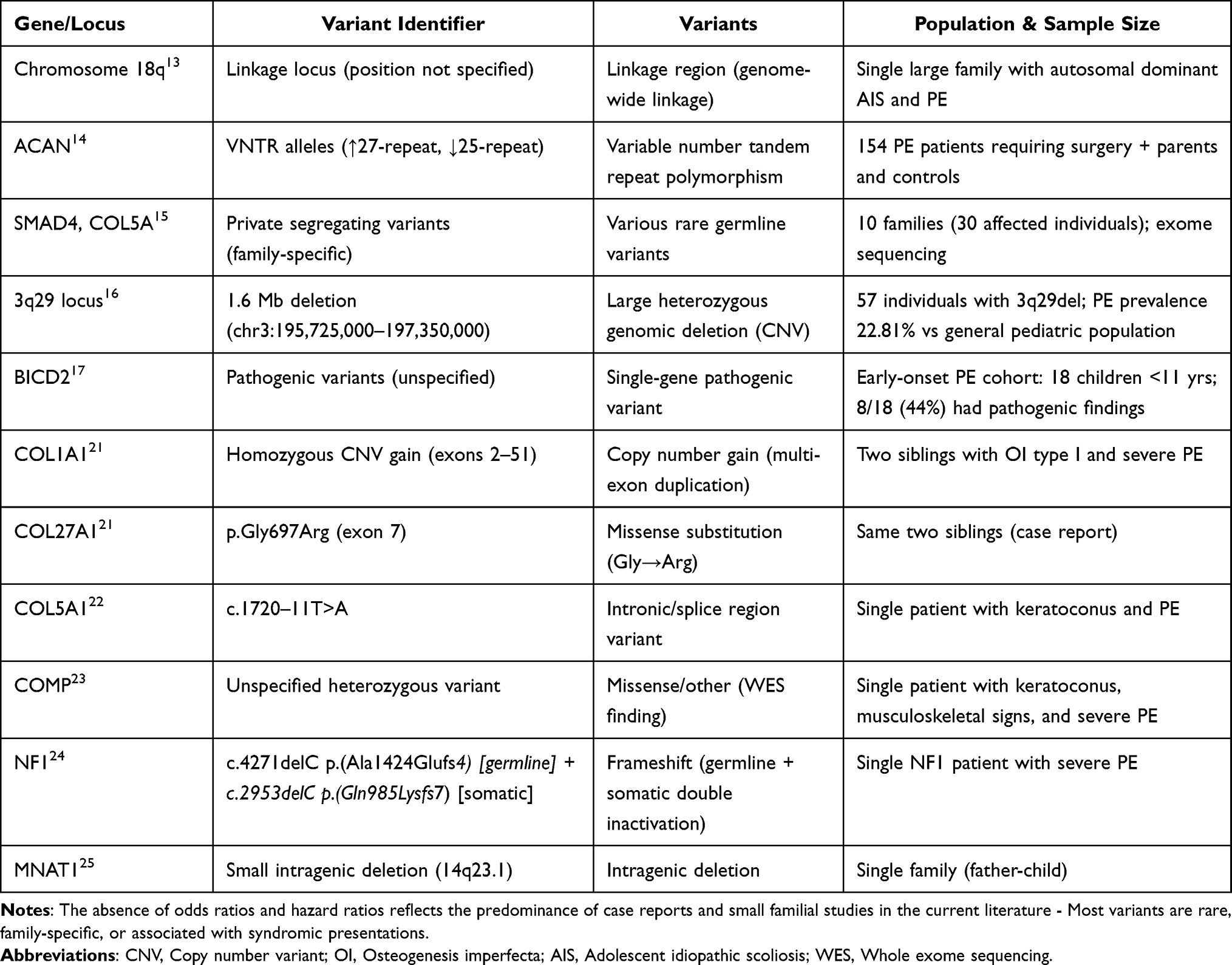 |
Table 1 Genetic Variants Associated with Pectus Excavatum |
Single-Gene Mutations and Rare Variants
Several genes harbouring rare pathogenic variants have been identified in association with pectus excavatum, predominantly through familial exome sequencing and case reports of syndromic presentations. Table 2 summarises copy number variants and structural chromosomal alterations. Multiple members of the collagen gene family have been implicated in the pathogenesis of pectus excavatum (PE). For example, a case report described two siblings with osteogenesis imperfecta type I and severe PE who carried a homozygous copy-number gain affecting exons 2–51 of COL1A1, in combination with a heterozygous missense variant in COL27A1, p.Gly697Arg.21 The authors suggested that the combined effects of these collagen gene alterations on cartilage and bone development contributed to the observed PE phenotype. Additionally, an intronic variant in COL5A1 (c.1720–11T>A) was reported in a patient with early-onset keratoconus and significant PE, suggesting a possible role for type V collagen in the connective tissue pathology underlying PE.22 The COMP gene, which encodes cartilage oligomeric matrix protein, has also been associated with PE in the context of broader musculoskeletal phenotypes. A heterozygous COMP variant was identified through whole-exome sequencing in a patient with progressive keratoconus, generalised musculoskeletal signs, and severe PE, consistent with a COMPopathy spectrum disorder involving chest wall deformity.23 Genes involved in the transforming growth factor-beta (TGF-β) signalling pathway have emerged as candidates in familial PE. Whole-exome sequencing of 10 families (30 affected individuals) identified rare, family-segregating variants in SMAD4 and REST, both of which are involved in TGF-β signalling.15 However, these variants were unique to individual families, with no single variant shared across multiple pedigrees, highlighting the genetic heterogeneity of familial PE. A unique case demonstrated somatic double inactivation of the NF1 gene in a patient with neurofibromatosis type 1 and severe PE. The patient harboured a germline frameshift mutation c.4271delC p.(Ala1424Glufs*4) and a somatic second-hit frameshift mutation c.2953delC p.(Gln985Lysfs*7) specifically in cartilage tissue, with complete loss of NF1 protein demonstrated by Western blot.24 This finding suggests that, in some cases, PE may result from somatic mutations in chest wall tissue rather than exclusively from germline variants. Furthermore, an intragenic deletion of MNAT1 at 14q23.1 was identified in a family with pectus deformities affecting a father and a child, suggesting this gene as a potential locus for familial PE.25
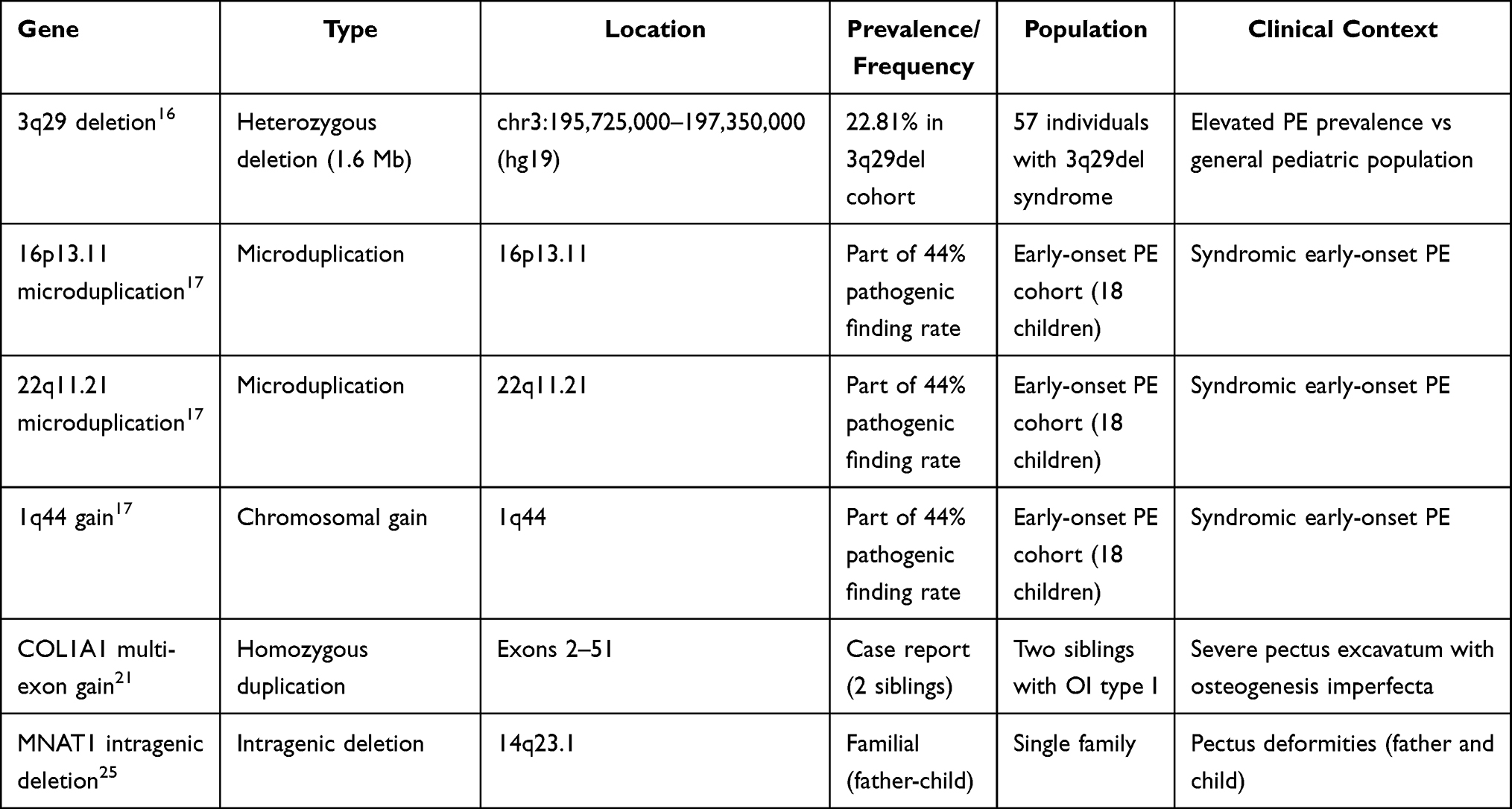 |
Table 2 Copy Number Variants (CNVs) and Structural Variants Associated with Pectus Excavatum |
Single-Nucleotide Polymorphisms and Common Variants
In contrast to the rare variant findings, few studies have examined common SNPs in PE. The most robust association reported involves variable number tandem repeat (VNTR) polymorphisms in the ACAN gene, which encodes aggrecan, a major proteoglycan component of cartilage matrix. A study of 154 PE patients requiring surgery, along with their parents and controls, identified statistically significant associations between ACAN VNTR alleles and PE, with increased 27-repeat alleles and decreased 25-repeat alleles observed in affected individuals.14 Notably, the study found no correlation between ACAN VNTR genotype and PE severity but did observe subgroup differences in patients with Marfan phenotype and female cases. While the association was reported as statistically significant, specific odds ratios were not provided in the publication.
Copy Number Variants
Copy number variants constitute a significant category of genetic variation associated with PE, especially in early-onset and syndromic cases. The most notable copy number variant (CNV) association is the 3q29 deletion syndrome, characterised by a recurrent 1.6 Mb heterozygous deletion (hg19, chr3:195,725,000–197,350,000). Analysis of 57 individuals with this deletion from a patient registry demonstrated a PE prevalence of 22.81% (13/57), which is significantly higher than that observed in the general paediatric population.16 This evidence establishes the 3q29 deletion as a high-penetrance genomic disorder for PE. In addition, a study of early-onset PE in children under 11 years of age identified pathogenic genetic findings in 44% (8/18) of cases, including chromosomal microduplications at 16p13.11, 22q11.21, and a genetic gain at 1q44.17 These results underscore the importance of CNV analysis in early-onset presentations. Furthermore, gene-specific CNVs extend beyond large chromosomal alterations; smaller gene-specific CNVs have also been reported, such as the homozygous COL1A1 exon 2–51 duplication21 and the MNAT1 intragenic deletion.25
Syndromic Associations
Table 3 catalogues syndromic associations and their causative genes. PE occurs as a feature of numerous well-characterized genetic syndromes, providing insights into potential pathogenic mechanisms: Marfan syndrome, resulting from FBN1 mutations, frequently presents with pectus excavatum (PE) as part of its skeletal manifestations, highlighting the essential role of fibrillin-1 in maintaining connective tissue integrity.26 Loeys-Dietz syndrome arises from mutations in components of the TGF-β pathway, including TGFBR1, TGFBR2, TGFB2, TGFB3, and SMAD3, and is characterised by connective tissue abnormalities that often result in chest wall deformities such as pectus excavatum.17,27 Noonan syndrome, associated with mutations in genes of the RAS-MAPK pathway (PTPN11, SOS1, RAF1, KRAS, NRAS), also includes PE among its skeletal features.17,28 Pathogenic variants in BICD2 have been identified in early-onset cases of PE.17
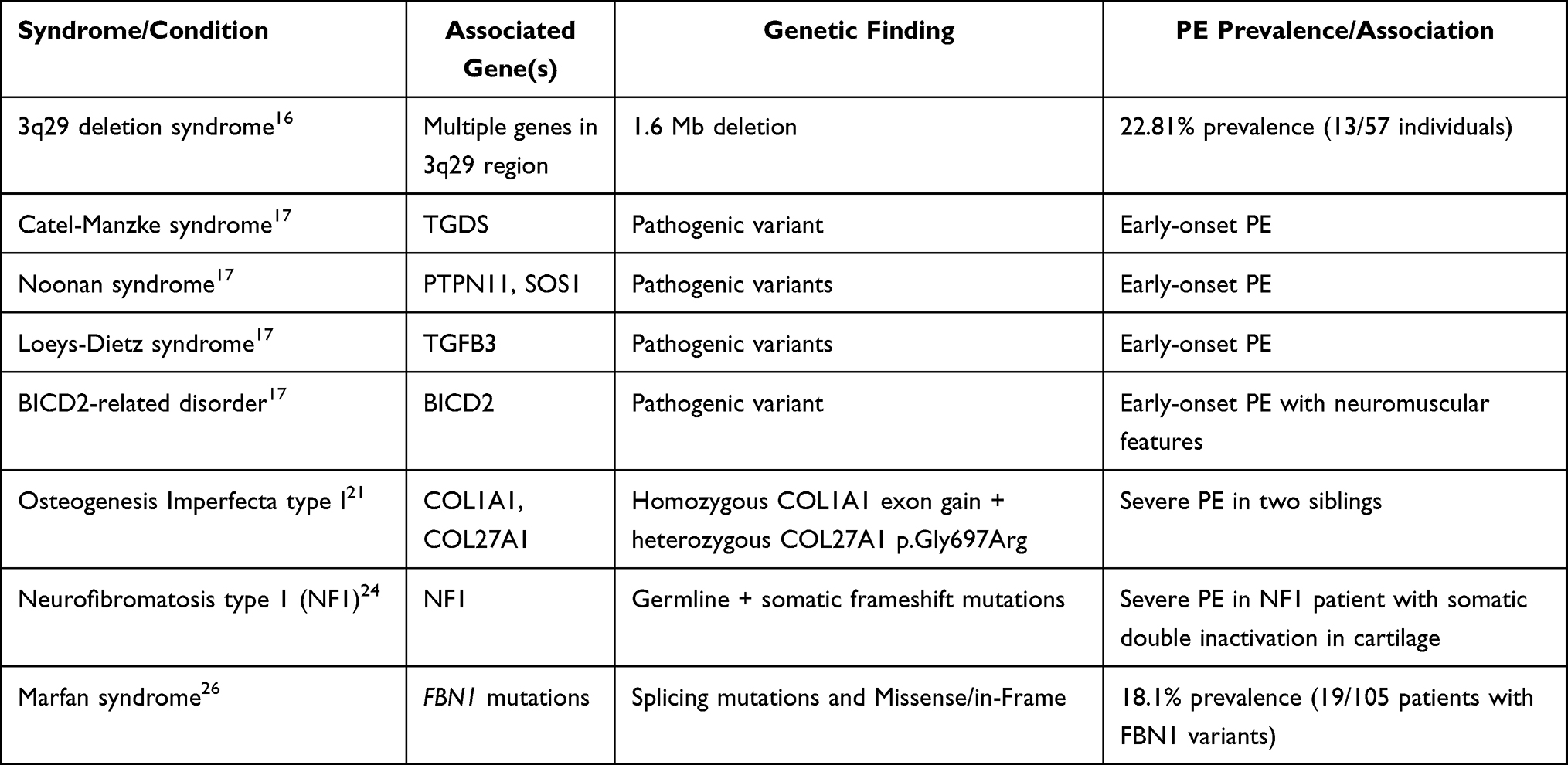 |
Table 3 Syndromic Associations and Pathogenic Variants of Pectus Excavatum |
Genetic Heterogeneity and Family-Specific Variants
A comprehensive exome sequencing study of ten families with familial PE (three affected members per family, 30 total affected individuals) revealed striking genetic heterogeneity.15 While rare variants segregating with PE were identified in each family, affecting genes including REST, SMAD4, and members of the COL5A family, these variants were private to individual families. No single causal gene or variant was shared across multiple families, suggesting that familial PE results from multiple different rare variants rather than a common founder mutation or single major gene.
Absence of Quantitative Effect Size Data
Genome-wide linkage analysis of a large family segregating both adolescent idiopathic scoliosis and PE localized a disease locus to chromosome 18q, though the specific causal gene within this region was not definitively identified.13 This finding suggests the presence of a Mendelian locus for PE in at least some families, warranting further fine-mapping and sequencing efforts. A critical finding of this literature review is the near-complete absence of reported odds ratios, hazard ratios, or other quantitative measures of effect size for genetic variants associated with PE. Of the 14 studies reviewed, none reported population-level odds ratios for identified genetic variants. This limitation reflects the predominance of case reports, small familial studies, and syndromes in the current literature, rather than large-scale case-control or cohort studies designed to quantify genetic risk. The lack of quantitative effect size estimates prevents meta-analysis and limits the ability to assess the population-level impact of identified genetic variants.
Discussion
This comprehensive literature review reveals a complex and heterogeneous genetic architecture underlying pectus excavatum, with contributions from rare single-gene mutations, copy number variants, and likely polygenic factors involving common variants of small effect. The findings have important implications for understanding PE pathogenesis, clinical genetic testing strategies, and future research directions.
Genetic Heterogeneity and Pathogenic Mechanisms
The most striking finding is the substantial genetic heterogeneity observed in PE. Unlike monogenic disorders with clear genotype-phenotype correlations, PE appears to result from diverse genetic alterations converging on common pathophysiological pathways. Figure 1 illustrates a proposed mechanism linking potential genetic mutations to the development of pectus excavatum.13,15–17,21–24 The implicated genes cluster into several functional categories: connective tissue structural proteins (collagens, fibrillin), cartilage matrix components (aggrecan, COMP), and signaling pathway regulators (TGF-β pathway genes including SMAD4, TGFBR1/2, REST).30,31 This pattern suggests that PE pathogenesis involves disruption of normal cartilage development and connective tissue homeostasis in the chest wall. During normal development, balanced growth of costal cartilages and proper extracellular matrix composition are essential for normal chest wall morphology.32 Genetic variants affecting these processes may lead to abnormal cartilage overgrowth, altered mechanical properties, or imbalanced growth patterns resulting in the characteristic sternal depression.33 The identification of somatic NF1 inactivation specifically in chest wall cartilage tissue introduces an additional layer of complexity, suggesting that in some cases, PE may result from tissue-specific somatic mutations rather than constitutional germline variants.24 Pectus excavatum appears to arise through a convergent pathway in which diverse genetic defects cause extracellular matrix dysfunction, characterised by impaired collagen fibrillogenesis and reduced mechanical integrity, leading to chondrocyte dysfunction and matrix weakness.13,15,16,21,23 These processes result in growth abnormalities such as asymmetric rib development and altered cardiac dimensions, together with loss of anterior chest wall support and rib–sternum instability, ultimately producing the characteristic sternal depression. This mechanism parallels those seen in other developmental anomalies and suggests that mosaic variants or somatic mutations may contribute to sporadic cases of pectus excavatum.34
 |
Figure 1 Putative mechanism underlying the association between genetic mutations and pectus excavatum.13,15–17,22,23 Abbreviations: COMP, Cartilage Oligomeric Matrix Protein; SMAD4, SMAD Family Member 4; BICD2, BICD Cargo Adaptor 2; ER, endoplasmic reticulum. |
Clinical Implications for Genetic Testing
The genetic findings reviewed here have several clinical implications. First, the high rate of pathogenic findings (44%) in early-onset PE (presenting before age 11 years) suggests that genetic testing has diagnostic utility in this population.17 Children presenting with PE at young ages should be evaluated for underlying genetic syndromes, as identification of syndromic diagnoses has important implications for multisystem screening and management.35 Second, the identification of PE-associated genetic variants in genes causing well-characterized syndromes (FBN1, TGFBR1/2, PTPN11) emphasizes the importance of comprehensive phenotyping in patients with PE. Features such as joint hypermobility, arachnodactyly, cardiovascular abnormalities, or dysmorphic features should prompt consideration of syndromic diagnoses and appropriate genetic testing.36 Third, for families with multiple affected individuals demonstrating apparent Mendelian inheritance, exome or genome sequencing may identify rare family-specific variants, though the genetic heterogeneity observed across families limits the clinical utility of testing in isolated cases.37 Genetic counseling should emphasize that even when a variant is identified, the lack of genotype-phenotype correlation data limits prognostic value regarding PE severity or progression.38 However, important limitations exist for genetic testing in PE. For isolated, non-syndromic PE presenting in adolescence or adulthood, the diagnostic yield of genetic testing is likely low given current knowledge. No validated gene panels or testing algorithms exist specifically for PE. Furthermore, the absence of genotype-specific treatments means that genetic testing results do not currently inform therapeutic decisions.39 The genetic findings in PE can be contextualized by comparison with pectus carinatum (PC), the second most common chest wall deformity. While less extensively studied, PC also demonstrates familial clustering and association with connective tissue disorders.40 Some families segregate both PE and PC, suggesting potential shared genetic susceptibility loci.41 Future studies examining both deformities may provide insights into common and distinct genetic mechanisms.
Methodological Limitations of Existing Studies
Several methodological limitations characterize the current literature on PE genetics. First, the predominance of case reports and small familial studies limits statistical power to detect associations and prevents quantification of effect sizes. Second, most studies lack standardized PE severity measures, limiting genotype-phenotype correlation analyses.42 Third, population stratification and ethnic diversity in study cohorts are poorly characterized, potentially confounding association studies.43 Fourth, replication studies are largely absent; most reported variants have been identified in single families or individuals without independent validation.44 The absence of large-scale genome-wide association studies represents a critical gap. GWAS have successfully identified common variants associated with numerous complex traits and diseases, but no adequately powered GWAS for PE has been published.45 Given the estimated prevalence of PE and the availability of large biobank resources, a multi-center collaborative GWAS is feasible and would likely identify common variants contributing to PE risk.46
The Need for Quantitative Effect Size Data
The near-complete absence of reported odds ratios, hazard ratios, or other quantitative effect sizes represents a major limitation preventing evidence synthesis and clinical risk assessment. Future studies should prioritize case-control or cohort designs enabling calculation of odds ratios for identified genetic variants.47 Even for rare variants, aggregated analyses examining cumulative burden of rare variants in candidate genes could provide quantitative risk estimates.48 Standardized reporting of effect sizes would enable meta-analyses combining data across studies, increasing statistical power and precision of risk estimates.49 Such quantitative data are essential for developing clinically useful genetic risk prediction models and for counseling families regarding recurrence risks.50
Pathways and Biological Insights
The convergence of genetic findings on connective tissue and cartilage-related genes provides biological insights into PE pathogenesis. The TGF-β signaling pathway emerges as a central regulator, with multiple implicated genes (TGFBR1, TGFBR2, SMAD3, SMAD4) functioning in this pathway.51 TGF-β signaling regulates chondrocyte differentiation, cartilage matrix production, and skeletal development, making it a plausible mechanistic link to chest wall morphology.52 Collagen genes (COL1A1, COL27A1, COL5A1) encode structural components of cartilage and connective tissue extracellular matrix. Variants affecting collagen structure or expression could alter the mechanical properties of costal cartilages, potentially leading to abnormal growth patterns or deformation under normal biomechanical forces.53 The identification of ACAN VNTR associations27 further implicates cartilage matrix composition, as aggrecan is the major proteoglycan providing compressive resistance in cartilage.54
These biological insights suggest potential therapeutic targets. Modulation of TGF-β signaling or interventions targeting cartilage matrix composition could theoretically prevent or ameliorate PE development, though such approaches remain speculative and would require extensive preclinical and clinical validation.55–59
Clinical Translation and Future Directions
This literature review has several important limitations. Despite comprehensive search strategies, publication bias cannot be excluded, as studies reporting negative or null findings are less likely to be published. The methodological quality of included studies varied substantially, ranging from high-quality exome sequencing analyses to single case reports, which may contribute to heterogeneity in the strength of evidence. In addition, the narrative synthesis approach, although appropriate given the heterogeneity of available data, lacks the quantitative robustness of a formal meta-analysis. Finally, the inclusion of preprints and grey literature, while enhancing coverage, may incorporate findings that have not yet undergone rigorous peer review. Future research in pectus excavatum PE genetics should extend beyond variant discovery to incorporate environmental influences, gene–environment interactions, and epigenetic mechanisms that likely modulate disease expression.56–64 Large, multi-centre GWAS and whole-genome sequencing studies with diverse populations are needed to identify both common and rare regulatory variants with adequate statistical power. Harmonised phenotyping, combined with longitudinal cohort designs, will be essential to define robust genotype–phenotype relationships and disease trajectories.63–65 Functional validation of candidate variants using experimental models should be prioritised to establish biological causality and clarify pathogenic mechanisms. Finally, integrative approaches combining genetic data with polygenic risk scores, imaging, and biomechanical analyses may enhance risk stratification and inform personalised clinical management.61–66
While current genetic findings have limited immediate clinical utility for most PE patients, the growing knowledge base is establishing foundations for future clinical translation. Future potential applications include genetic risk stratification to identify individuals requiring closer monitoring, pharmacogenetic approaches to personalize non-surgical therapies, and potentially gene-targeted interventions if specific high-penetrance variants are validated.67 The development of clinical genetic testing guidelines for PE will require additional evidence, particularly regarding clinical validity (strength of genotype-phenotype associations) and clinical utility (whether testing results inform management decisions improving outcomes).68 Professional societies may eventually develop consensus recommendations for genetic evaluation of PE, particularly for early-onset and familial cases.69 A recent systematic review highlights an intriguing paradox.70 Despite the discovery of several candidate genes for pectus excavatum in family studies, case reports, and animal models, the genetic landscape remains shrouded in uncertainty due to the absence of large-scale genome-wide association studies. In the absence of common variant associations or quantitative effect estimates, the development of genetic risk models, Mendelian randomisation analyses, and clinically validated polygenic risk scores is not currently feasible.
Conclusion
This review highlights the substantial genetic heterogeneity of pectus excavatum, involving rare variants in connective tissue and cartilage genes, copy number variants, and likely polygenic influences. Despite progress in identifying candidate genes and plausible biological pathways, the absence of large-scale genome-wide association studies and quantitative effect size data continues to limit population-level risk assessment and clinical prediction. Emerging convergence on TGF-β signalling, collagen genes, and cartilage matrix components provides mechanistic insight and supports considering genetic evaluation in early-onset cases. Future research should prioritise large multi-centre GWAS and whole-genome sequencing, functional validation of candidate genes, development of polygenic risk scores, and robust genotype–phenotype mapping through standardised phenotyping to enable meaningful clinical translation in pectus excavatum.
Data Sharing Statement
All data extracted during this literature review are presented in the tables and text of this manuscript. The search strategies and inclusion/exclusion criteria are fully described in the Methods section to enable replication.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare no competing interests in this work.
References
1. Fokin AA, Steuerwald NM, Ahrens WA, Allen KE. Anatomical, histologic, and genetic characteristics of congenital chest wall deformities. Semin Thorac Cardiovasc Surg. 2009;21(1):44–14. doi:10.1053/j.semtcvs.2009.03.001
2. Cobben JM, Oostra RJ, van Dijk FS. Pectus excavatum and carinatum. Eur J Med Genet. 2014;57(8):414–417. doi:10.1016/j.ejmg.2014.04.017
3. Jaroszewski D, Notrica D, McMahon L, Steidley DE, Deschamps C. Current management of pectus excavatum: a review and update of therapy and treatment recommendations. J Am Board Fam Med. 2010;23(2):230–239. doi:10.3122/jabfm.2010.02.090234
4. Kelly RE, Lawson ML, Paidas CN, Hruban RH. Pectus excavatum in a 112-year autopsy series: anatomic findings and the effect on survival. J Pediatr Surg. 2005;40(8):1275–1278. doi:10.1016/j.jpedsurg.2005.05.010
5. Steinmann C, Krille S, Mueller A, Weber P, Reingruber B, Martin A. Pectus excavatum and pectus carinatum patients suffer from lower quality of life and impaired body image: a control group comparison of psychological characteristics prior to surgical correction. Eur J Cardiothorac Surg. 2011;40(5):1138–1145. doi:10.1016/j.ejcts.2011.02.019
6. Goretsky MJ, Kelly RE, Croitoru D, Nuss D. Chest wall anomalies: pectus excavatum and pectus carinatum. Adolesc Med Clin. 2004;15(3):455–471. doi:10.1016/j.admecli.2004.06.002
7. Kelly RE, Kelly RE, Nuss D, et al. Regional chest wall motion dysfunction in patients with pectus excavatum demonstrated via optoelectronic plethysmography. J Pediatr Surg. 2011;46(6):1172–1176. doi:10.1016/j.jpedsurg.2011.03.047
8. Creswick HA, Stacey MW, Kelly RE, et al. Family study of the inheritance of pectus excavatum. J Pediatr Surg. 2006;41(10):1699–1703. doi:10.1016/j.jpedsurg.2006.05.071
9. Kelly RE, Shamberger RC, Mellins RB, et al. Prospective multicenter study of surgical correction of pectus excavatum: design, perioperative complications, pain, and baseline pulmonary function facilitated by internet-based data collection. J Am Coll Surg. 2007;205(2):205–216. doi:10.1016/j.jamcollsurg.2007.03.027
10. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485. doi:10.1136/jmg.2009.072785
11. MacCarrick G, Black JH, Bowdin S, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16(8):576–587. doi:10.1038/gim.2014.11
12. Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81(3):454–466. doi:10.1086/520125
13. Gurnett CA, Alaee F, Bowcock A, et al. Genetic linkage localizes an adolescent idiopathic scoliosis and pectus excavatum gene to chromosome 18q. Spine. 2009;34(2):E94–E100. doi:10.1097/BRS.0b013e31818b88a5
14. Stacey MW, Neumann SA, Dooley A, et al. Variable number of tandem repeat polymorphisms (VNTRs) in the ACAN gene associated with pectus excavatum. Clin Genet. 2010;78(5):502–504. doi:10.1111/j.1399-0004.2010.01492.x
15. Farina JM, Olson RJ, Dhamija R, et al. The complexity of familial inheritance in pectus excavatum: a ten-family exome sequencing analysis. Genes. 2024;15(11):1429. doi:10.3390/genes15111429
16. Pollak RM, Tilmon J, Murphy MM, et al. Musculoskeletal phenotypes in 3q29 deletion syndrome. Am J Med Genet A. 2023;191(11):2751–2761. doi:10.1002/ajmg.a.63384
17. Billar R, Heyman S, Kant S, et al. Early-onset pectus excavatum is more likely to be part of a genetic variation. Eur J Pediatr Surg. 2024;34(4):325–332. doi:10.1055/a-2081-1288
18. Horth L, Stacey MW, Proud VK, et al. Advancing our understanding of the inheritance and transmission of pectus excavatum. J Pediatr Genet. 2012;1(3):161–173. doi:10.3233/PGE-2012-026
19. Billar RJ, Manoubi W, Kant SG, Wijnen RMH, Demirdas S, Schnater JM. Association between pectus excavatum and congenital genetic disorders: a systematic review and practical guide for the treating physician. J Pediatr Surg. 2021;56(12):2239–2252. doi:10.1016/j.jpedsurg.2021.04.016
20. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
21. Cruz-Centeno N, Saenz-Maisonet JF, López-Dones PM, Santiago-Cornier A, Ortiz-Justiniano VN. Mutations in COL1A1 and COL27A1 associated with a pectus excavatum phenotype in 2 siblings with osteogenesis imperfecta. Am J Case Rep. 2022;23:e935526. doi:10.12659/AJCR.935526
22. Bryant G, Moore P, Sathyamoorthy M. The association of a single nucleotide variant in COL5A1 to early onset keratoconus and pectus excavatum-convergence of extracellular matrix pathologies. Medicina. 2024;60(6):974. doi:10.3390/medicina60060974
23. Kounatidou NE, Kondylis G, Klavdianou O, Venkateswaran N, Fryssira E, Palioura S. Progressive keratoconus in a patient with severe pectus excavatum and a cartilage oligomeric matrix protein gene mutation: a case report. Eye Contact Lens. 2024;50(1):48–51. doi:10.1097/ICL.0000000000001053
24. Chelleri C, Scala M, De Marco P, et al. Somatic double inactivation of NF1 associated with NF1-related pectus excavatum deformity. Hum Mutat. 2023;2023:3160653. doi:10.1155/2023/3160653
25. Heithaus JL, Davenport S, Twyman KA, Torti EE, Batanian JR. An intragenic deletion of the gene MNAT1 in a family with pectus deformities. Am J Med Genet A. 2014;164A(5):1293–1297. doi:10.1002/ajmg.a.36445
26. Stark VC, Hensen F, Kutsche K, et al. Genotype-phenotype correlation in children: the impact of FBN1 variants on pediatric marfan care. Genes. 2020;11(7):799. doi:10.3390/genes11070799
27. Schepers D, Tortora G, Morisaki H, et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum Mutat. 2018;39(5):621–634. doi:10.1002/humu.23407
28. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–342. doi:10.1016/S0140-6736(12)61023-X
29. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25(9):603–605. doi:10.1007/s10654-010-9491-z
30. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22(4):233–241. doi:10.1080/08977190412331279890
31. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi:10.1038/nrm3434
32. Cohen MM. The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet A. 2006;140(23):2646–2706. doi:10.1002/ajmg.a.31368
33. Feng J, Jing J, Li J, et al. BMP signaling orchestrates a transcriptional network to control the fate of mesenchymal stem cells in mice. Development. 2017;144(14):2560–2569. doi:10.1242/dev.150136
34. Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14(5):307–320. doi:10.1038/nrg3424
35. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
36. Loeys BL, Dietz HC. Loeys-Dietz syndrome. In: Adam MP, Bick S, Mirzaa GM, editors. GeneReviews. Seattle: University of Washington; 2008.
37. Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi:10.1001/jama.2014.14601
38. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
39. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American college of medical genetics and genomics. Genet Med. 2017;19(2):249–255. doi:10.1038/gim.2016.190
40. Fonkalsrud EW, Beanes S. Surgical management of pectus carinatum: 30 years’ experience. World J Surg. 2001;25(7):898–903. doi:10.1007/s00268-001-0048-x
41. Koumbourlis AC. Pectus excavatum: pathophysiology and clinical characteristics. Paediatr Respir Rev. 2009;10(1):3–6. doi:10.1016/j.prrv.2008.12.002
42. Daunt SW, Cohen JH, Miller SF. Age-related normal ranges for the Haller index in children. Pediatr Radiol. 2004;34(4):326–330. doi:10.1007/s00247-003-1116-1
43. Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361(9357):598–604. doi:10.1016/S0140-6736(03)12520-2
44. Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29(3):306–309. doi:10.1038/ng749
45. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22. doi:10.1016/j.ajhg.2017.06.005
46. Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi:10.1038/s41586-018-0579-z
47. Altman DG, Bland JM. How to obtain the confidence interval from a P value. BMJ. 2011;343(aug08 1):d2090. doi:10.1136/bmj.d2090
48. Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95(1):5–23. doi:10.1016/j.ajhg.2014.06.009
49. Evangelou E, Ioannidis JP. Meta-analysis methods for genome-wide association studies and beyond. Nat Rev Genet. 2013;14(6):379–389. doi:10.1038/nrg3472
50. Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet. 2013;14(7):507–515. doi:10.1038/nrg3457
51. Chen PY, Qin L, Simons M. TGFβ signaling pathways in human health and disease. Front Mol Biosci. 2023;10:1113061. doi:10.3389/fmolb.2023.1113061
52. van der Kraan PM, Blaney Davidson EN, van den Berg WB. A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther. 2010;12(1):201. doi:10.1186/ar2896
53. Eyre DR, Weis MA, Wu JJ. Advances in collagen cross-link analysis. Methods. 2008;45(1):65–74. doi:10.1016/j.ymeth.2008.01.002
54. Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Res. 2002;12(1):19–32. doi:10.1038/sj.cr.7290106
55. Akhurst RJ, Hata A. Targeting the TGFβ signaling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi:10.1038/nrd3810
56. Rappaport SM. Genetic factors are not the major causes of chronic diseases. PLoS One. 2016;11(4):e0154387. doi:10.1371/journal.pone.0154387
57. Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–484. doi:10.1038/s41576-019-0127-1
58. Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112(17):5473–5478. doi:10.1073/pnas.1418631112
59. Robinson PN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33(5):777–780. doi:10.1002/humu.22080
60. MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–476. doi:10.1038/nature13127
61. Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19(9):581–590. doi:10.1038/s41576-018-0018-x
62. Loos RJ. 15 years of genome-wide association studies and no signs of slowing down. Nat Commun. 2020;11(1):5900. doi:10.1038/s41467-020-19653-5
63. Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–350. doi:10.1038/nature15817
64. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538(7624):161–164. doi:10.1038/538161a
65. Xie L, Cai S, Xie L, et al. Development of a computer-aided design and finite-element analysis combined method for customized Nuss bar in pectus excavatum surgery. Sci Rep. 2017;7(1):3543. doi:10.1038/s41598-017-03622-y
66. Knoppers BM, Harris JR, Budin-Ljøsne I, Dove ES. A human rights approach to an international code of conduct for genomic and clinical data sharing. Hum Genet. 2014;133(7):895–903. doi:10.1007/s00439-014-1432-6
67. Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi:10.1038/gim.2013.73
68. Grosse SD, Khoury MJ. What is the clinical utility of genetic testing? Genet Med. 2006;8(7):448–450. doi:10.1097/01.gim.0000227935.26763.c6
69. Rehm HL, Berg JS, Brooks LD, et al. ClinGen–the clinical genome resource. N Engl J Med. 2015;372(23):2235–2242. doi:10.1056/NEJMsr1406261
70. Ranjan R, Imtiaz N, Waterhouse B, Paul I, Brunswicker A, Dunning J. Genetic associations with pectus excavatum: a systematic review. Curr Issues Mol Biol. 2026;48(1):122. doi:10.3390/cimb48010122
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.