")
Back to Journals » Patient Preference and Adherence » Volume 8
A review of denosumab for the treatment of osteoporosis
Authors Miyazaki T, Tokimura F, Tanaka S
Received 1 February 2014
Accepted for publication 6 March 2014
Published 8 April 2014 Volume 2014:8 Pages 463—471
DOI https://doi.org/10.2147/PPA.S46192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Tsuyoshi Miyazaki,1,2 Fumiaki Tokimura,1 Sakae Tanaka3
1Department of Orthopedic Surgery, 2Department of Geriatric Medicine, Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology, Itabashi-ku, Tokyo, Japan; 3Department of Orthopedic Surgery, Faculty of Medicine, University of Tokyo, Bunkyo-ku, Tokyo, Japan
Abstract: Osteoporosis is an age-related systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility. Bone remodeling involves two types of cells: osteoblasts and osteoclasts. Receptor activator of nuclear factor-κB ligand (RANKL) is a key regulator of the formation and function of bone-resorbing osteoclasts, and its cell surface receptor, receptor activator of nuclear factor-κB (RANK), is expressed by both osteoclast precursors and mature osteoclasts. Denosumab is a fully human monoclonal anti-RANKL antibody that inhibits the binding of RANKL to RANK, thereby decreasing osteoclastogenesis and bone-resorbing activity of mature osteoclasts. Although there are many medications available for the treatment of osteoporosis, inhibition of RANKL by denosumab has been shown to significantly affect bone metabolism. Denosumab appears to be a promising, highly effective, and safe parenteral therapy with good adherence for osteoporosis. Moreover, denosumab may be cost-effective therapy compared with existing alternatives. Therefore, in this review, we focus on studies of denosumab and the risks and benefits identified for this type of treatment for osteoporosis.
Keywords: bone resorption, OPG, osteoclast, RANKL
Introduction
Remodeling of bone, which begins in the early fetal stages, is a process that is maintained in the adult skeleton. It mediates the repair of microdamage while also regulating the mechanical strength and structure of bone. The bone remodeling cycle involves a series of highly regulated steps that depend on interactions between two cell lineages: the mesenchymal bone-forming osteoblastic lineage and the hematopoietic bone-resorbing osteoclastic lineage.1 The latter are differentiated from monocyte–macrophage lineage precursor cells in response to cytokines and chemokines produced by cells lining the bone surface, and these cells initiate bone remodeling.2,3 Subsequent interactions between osteoclast precursors and osteoblastic cells leads to the differentiation, migration, and fusion of large multinucleated osteoclasts.4 These mature osteoclasts then attach to a mineralized bone surface and initiate resorption by secreting hydrogen ions and lysosomal enzymes. In particular, cathepsin K is secreted, and this enzyme is able to degrade the bone matrix, including collagen, at low pH. Osteoclastic bone resorption produces irregular scalloped cavities on the trabecular bone surface, called Howship’s lacunae, or cylindrical haversian canals in cortical bone. Following this resorptive phase, the bone surface is repopulated by osteoblasts, which deposit bone matrix and eventually undergo mineralization to form a new bone surface. Generally, the same amount of bone that is removed is replaced. However, when an imbalance between these two processes leads to an increase in bone resorption, the result is focal articular bone loss and generalized osteoporosis.
Various diseases, drugs, and metabolic abnormalities adversely affect bone health and contribute to the development of osteoporosis. Activation of osteoclastic bone resorption is a common factor in the pathogenesis of bone loss and fractures,5 while estrogen deficiency during menopause or androgen deficiency in males can also lead to an unbalanced increase in bone resorption versus bone formation. As a result, bone loss can occur rather rapidly, accompanied by the destruction of bone microarchitecture.6 In older adults who commonly experience vitamin D deficiency,7 calcium absorption is impaired and secondary hyperparathyroidism can develop. Consequently, bone loss occurs and the risk of fracture increases.8 Painful vertebral fractures are the most common complication of osteoporosis and account for ~50% of reported fractures. In addition, height loss, kyphosis, back pain, and impaired physical and psychological function can occur following such fractures. The presence of a spine fracture is also the strongest risk factor for experiencing another fracture of either hip or spine,9 with the former representing the most challenging type of fracture for patient recovery. Considering that the cost of care for patients with fractures is expensive, the incidence of fractures increases progressively with advancing age, and the global population is growing older; it has been estimated that the number of fractures worldwide will double or triple by the year 2050.10
For patients at risk for osteoporosis, or those having already experienced a fracture, prevention of new or additional fractures is key. Antiresorptive (anticatabolic) drugs that are currently available include estrogen, raloxifene, and bisphosphonates. These have been shown to effectively prevent bone loss in postmenopausal women without osteoporosis.11–13 For postmenopausal women and men with osteoporosis, treatment with either an antiresorptive drug or teriparatide, an anabolic agent, has been shown to preserve or improve bone mass and substantially reduce the risk of fracture.14 Unfortunately, however, these treatments can only be safely administered for a limited period of time. For example, anabolic agents, such as teriparatide, can only be administered for a maximum of 2 years. Moreover, for bisphosphonates, prolonged administration increases the potential for rare, yet serious, adverse events such as osteonecrosis of the jaw (ONJ), atypical fractures, and esophageal cancer.15 The treatment efficacy of these drugs in clinical practice has also been limited by real or perceived intolerance, as well as poor adherence to therapy.16,17 Phase I trials of anti-sclerostin antibody, which up-regulates the interaction between Wnt ligand and LRP5/6 coreceptor on osteoblasts, showed increase in bone formation in healthy men and postmenopausal women, and Phase II trials are underway.18,19 Inhibition of sclerostin is an interesting prospect for the next generation of osteoporosis drugs. In this review, we will focus on a fully human monoclonal anti-receptor activator of nuclear factor-κB ligand (RANKL) antibody, denosumab (Figure 1), and its potential as a long-term treatment for osteoporosis with appropriate administration.
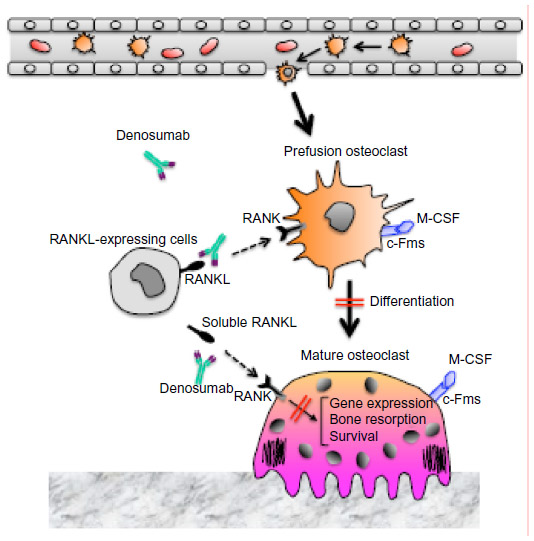 | Figure 1 The mechanism of action of denosumab on bone metabolism. |
Identification of the osteoclast differentiation factor, RANKL
Bone-resorbing osteoclasts originate from hematopoietic cells, which are hypothesized to be members of the colony forming unit-megakaryocyte-derived monocyte–macrophage family. Takahashi et al and Udagawa et al developed a mouse coculture system of hematopoietic cells and primary osteoblasts to investigate osteoclast formation in vitro.20–22 In this coculture system, several systemic and local factors were found to induce the formation of tartrate resistant acid phosphatase-positive multinucleated cells,23 and these cells exhibited a number of osteoclast characteristics. In addition, cell-to-cell contact between osteoblastic cells and osteoclast progenitors was shown to be essential for the induction of osteoclastogenesis. Based on these findings, Suda et al proposed that osteoblastic cells induce the membrane-associated osteoclast differentiation factor in response to various osteotropic factors.23 In 1997, it was first reported that RANKL and its receptor, receptor activator of nuclear factor-κB (RANK), regulate interactions between dendritic cells and T-cells.24 Furthermore, when osteoclast differentiation factor was cloned from a complimentary DNA library of mouse stromal ST2 cells treated with bone-resorbing factors,25 it was found to be identical to RANKL, to tumor necrosis factor (TNF)-related activation-induced cytokine, and to the osteoprotegerin (OPG) ligand. These results were independently validated by other research groups.26–28 In these studies, RANKL was also shown to induce osteoclast differentiation from mouse hematopoietic cells and human peripheral blood mononuclear cells in the presence of macrophage colony-stimulating factor.25,28 To date, RANK is the only signaling receptor that has been identified for RANKL for the induction of osteoclastogenesis and the activation of mature osteoclasts.4 OPG, which lacks transmembrane and cytoplasmic domains and is released in a soluble form by a variety of cells including osteoblasts, also serves as a decoy receptor for RANKL. As such, OPG competes with RANK to inhibit the differentiation and activity of osteoclasts.4
A crucial role for RANKL in bone metabolism
RANKL is a membrane-anchored molecule that is released from the cell surface following proteolytic cleavage by matrix metalloproteinases such as matrix metalloproteinases 14.29,30 Both the soluble and membrane-bound forms of RANKL function as agonistic ligands for RANK, with the membrane-bound form functioning more efficiently.29,31,32 Using a knockout mouse model of OPG, a natural inhibitor of RANKL, a link between RANKL and the development of osteoporosis was demonstrated.33 Furthermore, overexpression of OPG in mice results in lower numbers of osteoclasts and greater bone mass.34 Correspondingly, for patients experiencing estrogen deficiency, hyperparathyroidism, or other disorders that stimulate bone resorption, perturbations in the ratio of OPG to RANKL have been detected.35–37 More recently, Nakashima et al reported that purified osteocytes express higher levels of RANKL and undergo enhanced osteoclastogenesis in vitro, while osteocyte-specific RANKL knockout mice exhibit a severe osteopetrotic phenotype.38 Taken together, these results indicate that osteocytes represent a major source of RANKL for bone remodeling in vivo. Mutations in RANKL, RANK, and OPG genes have also been identified in patients with bone disorders such as autosomal recessive osteopetrosis, familial expansile osteolysis, and juvenile Paget’s disease, respectively.39 Moreover, when RANKL expression is upregulated in response to factors such as vitamin D3, prostaglandin E2, parathyroid hormone, interleukin (IL)-1, IL-6, IL-11, IL-17, and TNF-α, pathological osteoclastogenesis has been observed.34,40,41 Therefore, regulation of the RANKL/RANK/OPG axis represents a potential therapeutic target for the treatment of osteoporosis, rheumatoid arthritis, and cancer bone metastasis.
Immunological function of RANKL
Prior to their identification in bone cells, RANKL and RANK were found to effect T-cell activation and dendritic cell survival.24,27,42 Moreover, during early development, RANKL signaling regulates the microenvironment of the thymus, thereby facilitating the deletion of self-reactive T-cells to provide self-tolerance and prevent autoimmunity.43 These results imply that inhibition of RANKL by denosumab may alter immune function, or increase susceptibility to infections. In studies of mice deficient in RANKL or RANK, an absence of lymph nodes and significantly smaller Payer’s patches were observed.28,43 These findings demonstrate the critical role that RANK activation has in the early stages of lymphoid tissue inducer cell development in peripheral lymphoid organs. A pathological model of inflammatory bowel disease has also demonstrated a role for RANKL in the stimulation of dendritic cells,44,45 suggesting that RANKL may mediate the activation of dendritic cells under certain autoimmune conditions. On the other hand, inhibition of RANKL by OPG has not been found to alter cellular or humoral immunity, nor does it render mice susceptible to bacterial challenge.46 Thus, although dendritic cells and T lymphocytes express RANK and RANKL, it would appear that they play a minor or redundant role in the mammalian immune response. However, these results do not guarantee the safety of denosumab treatments.
In vitro studies of denosumab
Direct binding assays have demonstrated that denosumab is able to bind human RANKL, yet does not bind murine RANKL, human TNF-related apoptosis-inducing ligand, and other human TNF family members.47 Denosumab also does not suppress bone resorption in normal mice or rats, although it prevented a resorptive response in mice challenged with human RANKL (huRANKL). huRANKL knock-in mice have been generated, and these mice exclusively express chimeric (human/murine) RANKL47 and are responsive to denosumab. In studies of young huRANKL mice treated with denosumab, trabecular osteoclast surfaces were reduced by 95% and bone density and volume increased.47 In contrast, adult huRANKL mice treated with denosumab exhibited reduced bone resorption, increased cortical and cancellous bone mass, and improved trabecular microarchitecture.47 The same group also reported that subcutaneous administration of denosumab (25 or 50 mg/kg/month) for up to 16 months prevented the loss of cancellous bone and preserved indices of bone strength for adult ovariectomized cynomolgus monkeys.48
Clinical development of denosumab as a prophylactic and/or therapeutic agent for osteoporosis
To evaluate whether inhibition of RANKL has clinical utility, 52 healthy postmenopausal women were given single doses of an osteoprotegerin-immunoglobulin Fc segment complex (Fc:OPG) (0.1, 0.3, 1.0, or 3.0 mg/kg) in a Phase I randomized placebo-controlled study.49 Urinary levels of the cross-linked N-telopeptide of type I collagen (NTX), a specific marker of bone resorption, and bone-specific alkaline phosphatase (BSAP), an index of bone formation, were subsequently monitored for 84 days. Within 12 hours of receiving Fc:OPG, a dose-dependent decrease in NTX/creatinine ratios was observed. Furthermore, this ratio decreased by 70% to 80% within 5 days for the highest doses of Fc:OPG. After several weeks, levels of NTX/creatinine returned to baseline. A significant decrease in levels of BSAP were also observed for the 1.0 mg/kg and 3.0 mg/kg doses. For the latter group, inhibition of BSAP occurred more slowly, with levels 30% below baseline observed after 60 days. There were also no serious adverse events reported in this study. In one patient, a transient neutralizing antibody to OPG was detected, although this did not have any obvious clinical effect. These data provide evidence that inhibition of RANKL by its natural inhibitor, OPG, can result in clinically measurable effects. However, the development of OPG as a therapy for osteoporosis was not further pursued due to its potential immunogenicity, and because immunologic resistance to OPG could have negative effects on the skeleton.50
Since denosumab specifically binds RANKL,47 it is less likely to affect the immune system or other regulatory systems. Moreover, denosumab does not have the potential for autoimmunization against a vital regulatory protein and is characterized by a longer half-life, which permits less frequent dosing.51 Each of these attributes makes denosumab a more attractive therapeutic agent than forms of OPG. To evaluate the safety, pharmacokinetics (PK), and possible bone resorption effects of denosumab, a Phase I study was conducted. Subcutaneous administration of various concentrations of denosumab (0.01 mg/kg to 3.0 mg/kg) were administered to 49 healthy postmenopausal women.51 The PK of denosumab were found to be nonlinear with dose. A prolonged absorption phase also occurred, with maximum serum concentrations reached between 5 days and 21 days after the women received the initial dose. Conversely, the disappearance of denosumab from the serum occurred in two phases: a slow phase and a fast phase. The initial slow phase was associated with half-lives of approximately 20 days for the lower doses of denosumab, and approximately 32 days for the higher doses. When circulating levels of denosumab were ~1,000 ng/mL, clearance occurred more rapidly. Urinary NTX levels were also found to decrease within 12 hours of dosing. Overall, the magnitude of the initial response was similar among the doses, although the duration of the effect was dose-dependent. These results are consistent with the pharmacokinetic data. By the end of the 9-month follow-up period, NTX levels had returned to baseline for all of the doses. Alternatively, serum levels of BSAP remained stable for the first two weeks following dosing, then decreased in a dose-dependent manner. Taken together, these results suggest that the effect of denosumab on bone formation is indirect.
Optimizing the dose of denosumab for osteoporosis
To evaluate the safety, tolerability, PK, and pharmacodynamics (PD) of denosumab, a randomized double-blind dose-escalation study was conducted. For a group of healthy postmenopausal Japanese women, denosumab was administered subcutaneously at doses of 0.03, 0.1, 0.3, 1.0, or 3.0 mg/kg, and was compared with a placebo.52 Suppression of bone turnover markers (BTM) was rapidly detected (within 2 days of dosing) and the duration of suppression was dose-dependent. Moreover, there was no marked differences in the PK and PD profiles between Japanese52 and non-Japanese subjects,51 and denosumab was well tolerated. In another study, the efficacy and safety of three doses of denosumab (14, 60, and 100 mg) were compared with a placebo over 12 months for a group of postmenopausal Japanese women with osteoporosis. The results associated with the 60 mg dose of denosumab were consistent with the results of a similar Phase II study of osteoporosis in a Caucasian population that was conducted in the United States.53–56
Reduced fracture risk with denosumab
A total of 7,868 women between the ages of 60 and 90 years who had a bone mineral density (BMD) T-score <−2.5 and >−4.0 at the lumbar spine or total hip received either 60 mg denosumab or a placebo subcutaneously every 6 months for 36 months.57 In this study, it was observed that denosumab reduced the risk of new radiographic vertebral fractures by 68% (P<0.001), with the risk of hip fractures and nonvertebral fractures decreasing by 40% and 20%, respectively. Moreover, this effect did not significantly differ for any of the nine subgroups analyzed according to patient age, body mass index, femoral neck BMD T-score, prevalent vertebral fracture, prior nonvertebral fracture, estimated creatinine clearance, geographic region, ethnicity, and prior use of osteoporosis medications.58
Long-term denosumab treatment
To evaluate denosumab efficacy and safety for up to 10 years of treatment, participants who completed the FREEDOM (Fracture REduction Evaluation of Denosumab in Osteoporosis every 6 Months) trial57 were eligible to receive an additional 2 years of denosumab treatment (the long-term group). For comparison, patients from the FREEDOM placebo group could receive 2 years of denosumab treatment (the cross-over group). A total of 4,550 women elected to participate in the extended trial, with 2,343 women in the long-term group and 2,207 women in the cross-over group.59 In the former group, BMD for the lumbar spine and the total hip further increased, resulting in 5-year gains of 13.7% and 7.0%, respectively. BMD for the lumbar spine and the total hip also increased for the latter group, with values of 7.7% and 4.0%, respectively, over the 2-year denosumab treatment period. Regarding adverse events, the number did not increase for the long-term group. However, for the cross-over group, two adverse events consistent with ONJ were reported. In one woman, healing occurred within the 6-month dosing interval, and she continued to receive denosumab (two further doses) without any additional oral events. For the other woman, healing occurred after the 6-month dosing interval, and denosumab was subsequently discontinued.
In a Phase II study, denosumab treatment was administered for up to 8 years to postmenopausal women with low bone mass.60 For the subjects who received continuous administration of denosumab over that period, BMD for the lumbar spine (n=88) and for the total hip (n=87) increased by 16.5% and 6.8%, respectively, compared with the baseline of the parent study, and increased by 5.7% and 1.8%, respectively, compared with the baseline of the extension study. At the end of year 8, serum levels of C-terminal telopeptide of type I collagen (CTX) and BSAP remained below the parent study baseline, and median reductions of 65% and 44%, respectively, were observed. Overall, the results of this Phase II study and its extension demonstrate that denosumab therapy mediated a progressive and substantial increase in BMD over 8 years for postmenopausal women with low bone mass. In addition, treatment was well tolerated and the adverse event profile was similar to what has been reported previously.
Effects of discontinuing denosumab on BMD and levels of BTM
For 256 postmenopausal women, 60 mg denosumab or a placebo was administered every 6 months for 2 years, followed by 2 years of discontinued treatment.61 After this 4 year period, the group that initially received denosumab was found to maintain a higher BMD than the placebo group (P≤0.05). Furthermore, levels of BTM were found to increase above baseline within 3 months (for the serum C-terminal telopeptide of type 1 collagen) or 6 months (for the N-terminal propeptide of type 1 procollagen) of the initial 2 year treatment period. By the end of the 4 year period, the levels of the BTM had returned to baseline. Adverse event rates during the nontreatment phase were found to be similar between the two groups. For the 60 mg denosumab dose that was administered for 24 months, levels of BMD and BTM were found to be reversible upon discontinuation, thereby reflecting the biological mechanism of action for denosumab. However, residual BMD measurements did remain greater than those of the placebo group.61
Effects of denosumab on bone histomorphometry
Iliac crest bone biopsies were collected 24 and/or 36 months from the first diagnosis of osteoporosis for 45 postmenopausal women who received a placebo and 47 postmenopausal women who received denosumab in the FREEDOM study.57 Biopsies were also collected from postmenopausal women who had been treated for 12 months with alendronate in the STAND (Study of Transitioning from AleNdronate to Denosumab) study.62 Of this latter group, 21 continued to receive alendronate while 15 received denosumab upon entry into the FREEDOM trial. Indices of bone turnover tended to be lower for the women who received denosumab compared to alendronate alone. Moreover, the women who received denosumab maintained normal bone microarchitecture and there were no adverse effects associated with the mineralization or formation of lamellar bone. In future studies, a longer follow-up period will be necessary to determine the duration during which such low turnover is safe.
A cohort study was also conducted to evaluate the effects of discontinuing denosumab at the tissue level.63 The mean period of discontinued osteoporosis treatment was 25.1 months (range, 21–29 months). Bone histomorphometry studies showed normal histology and bone remodeling similar to that observed for untreated postmenopausal women with osteoporosis. Furthermore, all of the biopsy specimens from women who had discontinued treatment showed evidence of tetracycline labels. Assays of biochemical markers also found levels to be comparable with pretreatment levels. Taken together, these data confirm that the effects of denosumab on bone turnover at the tissue level are reversible.
Renal function does not significantly affect the PK or PD of denosumab
Chronic kidney disease (CKD) has been identified as a potential independent risk factor for bone loss.64–66 Correspondingly, CKD is also more common among older adults. To evaluate whether treatment with denosumab affects renal function, subjects were enrolled in one of five renal function groups based on glomerular filtration rates (GFRs) as follows: normal renal function (GFR >80 mL/minute/1.73 m2) (n=12); mild CKD (GFR 50–80 mL/minute/1.73 m2) (n=13); moderate CKD (GFR 30–49 mL/minute/1.73 m2) (n=13); severe CKD (GFR <30 mL/minute/1.73 m2) (n = 9); or kidney failure requiring hemodialysis (n=8).67 Data collected for these groups indicated that renal function did not have a significant effect on the PK or PD of denosumab, and dose adjustments were not needed for these patients. However, the potential for developing hypocalcemia was found to be higher for subjects with severe CKD and kidney failure compared with subjects with mild or moderate CKD or subjects with normal renal function. Two subjects who experienced kidney failure (one symptomatic and one asymptomatic) were hospitalized for intravenous calcium gluconate treatment. Thus, it is recommended that patients with impaired renal function who receive denosumab, particularly those with severe kidney disease (GFR <30 mL/minute/1.73 m2), should receive calcium and vitamin D supplements and should be monitored for secondary hyperparathyroidism.
Safety
Although denosumab has been shown to be safe in the collective data from Phase II and III clinical trials,68 the clinical concern was the potential risk for infections or neoplasms due to the ubiquitous presence of RANKL throughout many tissues. In a meta-analysis of randomized placebo-controlled trials involving denosumab, including the large FREEDOM registration trial,57 a borderline increased risk of serious infection was observed (risk ratio =1.25, 95% confidence interval: 1.00–1.54) for women with postmenopausal osteoporosis when intention-to-treat analysis was used.69 However, a nonsignificant risk ratio of 2.1 was observed when a per-protocol analysis was employed.70 Thus, the incidence of infection and neoplasms in ongoing larger Phase III trials will be of interest.
While accumulating evidence indicates that denosumab is a safe treatment, there remains the potential for side effects from this treatment. For bisphosphonate therapy, ONJ has recently emerged as an adverse side effect,71,72 although the nature and cause of ONJ remains controversial. Given the capacity for denosumab to strongly inhibit osteoclastic bone resorption similar to bisphosphonates, it will be important for future studies of denosumab to monitor the incidence and clinicopathologic characteristics of ONJ. Another potential side effect to consider is the so-called frozen bone process, whereby complete inhibition of remodeling leads to an accumulation of microfractures and an increased risk for atypical femoral fractures. This complication was considered in an analysis of postmenopausal women receiving bisphosphonate therapy based on the findings of animal studies.73 In trials that have continuously administered denosumab for up to 5 years, there have been no reports of atypical femoral fractures.57,59 However, two cases of atypical femoral fracture have been confirmed in patients receiving denosumab 60 mg for 2.5 years or more participating in the ongoing open-label extension study of the pivotal Phase III fracture trial in postmenopausal osteoporosis (FREEDOM).74 It is possible that differences in shorter half-life of denosumab compared with bisphosphonates (5 years or longer) can account for less incidence of atypical femoral fractures. Tsai et al also recently reported that a combination treatment of teriparatide and denosumab increased BMD to a greater extent than either agent alone.75 Teriparatide is an effective anabolic (bone growing) agent that might help prevent frozen bone caused by denosumab-induced oversuppression of bone turnover. Furthermore, although denosumab has been found to be completely cleared from the body following its discontinuation, the frozen bone process may still be an issue for long-term denosumab treatments.
Since RANK and RANKL are also expressed by endothelial cells and lymphocytes,76 additional studies are needed to evaluate the potential effects of denosumab therapy on the cardiovascular and immune systems of the body. Continued documentation and quantification of the efficacy of denosumab for large numbers of patients will also be important. Recently, RANKL signaling was implicated in the pathogenesis of hepatic insulin resistance and type 2 diabetes mellitus.77 This may provide a link between inflammation and disrupted glucose homeostasis, and may also contribute to pharmacological strategies being developed for the treatment of RANKL-related diseases.
Finally, Freemantle et al reported that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with subcutaneous injections of denosumab every 6 months than with once-weekly alendronate tablets in a 2-year randomized crossover study.78 In addition, the women expressed greater satisfaction with injectable denosumab and preferred it over oral alendronate. Thus, preferences in the administration of denosumab may influence patient persistence and adherence to therapy, and this represents an important consideration for the treatment of chronic conditions that require long-term therapy.
Conclusion
The inhibition of RANKL by denosumab has been shown to significantly affect bone metabolism. Correspondingly, this highly specific antibody for RANKL appears to be a promising treatment for osteoporosis and other bone diseases characterized by increased bone turnover. Freemantle et al showed that denosumab was more effective at reducing the occurrence of vertebral fractures than raloxifene, risedronate, and alendronate.79 The cost-effectiveness of denosumab in postmenopausal osteoporotic women has been evaluated by estimating expected cost and quality-adjusted life-years. Analyses have shown that denosumab represented good value-for-money in postmenopausal women with low bone mass compared with no treatment 80 or treatment with oral bisphosphonates,81–83 and, therefore, has the potential to be a first-line treatment for postmenopausal osteoporotic women. In addition, the cost-effectiveness of denosumab is favorable, particularly for patients at high risk of fracture and low expected adherence to oral treatments.84 The long-term efficacy and toxicity of denosumab remains to be confirmed with studies that include longer follow-up periods. This is particularly relevant since postmenopausal women are increasingly experiencing a longer life expectancy, and, thus, the potential for anti-osteoporosis therapy to span multiple decades is a growing consideration.
Disclosure
The authors report no conflicts of interest in this work.
References
Eriksen EF. Normal and pathological remodeling of human trabecular bone: three dimensional reconstruction of the remodeling sequence in normals and in metabolic bone disease. Endocr Rev. 1986;7(4):379–408. | |
Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem. 1994;55(3):273–286. | |
Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. | |
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–357. | |
Robbins JA, Schott AM, Garnero P, Delmas PD, Hans D, Meunier PJ. Risk factors for hip fracture in women with high BMD: EPIDOS study. Osteoporos Int. 2005;16(2):149–154. | |
Dufresne TE, Chmielewski PA, Manhart MD, Johnson TD, Borah B. Risedronate preserves bone architecture in early postmenopausal women in 1 year as measured by three-dimensional microcomputed tomography. Calcif Tissue Int. 2003;73(5):423–432. | |
Gloth FM 3rd, Gundberg CM, Hollis BW, Haddad JG Jr, Tobin JD. Vitamin D deficiency in homebound elderly persons. JAMA. 1995;274(21):1683–1686. | |
Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001;22(4):477–501. | |
Johnell O, Kanis JA, Odén A, et al. Fracture risk following an osteoporotic fracture. Osteoporos Int. 2004;15(3):175–179. | |
Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int. 1997;7(5):407–413. | |
No authors listed. Effects of hormone therapy on bone mineral density: results from the postmenopausal estrogen/progestin interventions (PEPI) trial. The Writing Group for the PEPI. JAMA. 1996;276(17):1389–1396. | |
Delmas PD, Bjarnason NH, Mitlak BH, et al. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337(23):1641–1647. | |
McClung MR, Wasnich RD, Hosking DJ, et al. Prevention of postmenopausal bone loss: six-year results from the Early Postmenopausal Intervention Cohort Study. J Clin Endocrinol Metab. 2004;89(10):4879–4885. | |
Delmas PD. Treatment of postmenopausal osteoporosis. Lancet. 2002;359(9322):2018–2026. | |
Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95(4):1555–1565. | |
Recker RR, Gallagher R, MacCosbe PE. Effect of dosing frequency on bisphosphonate medication adherence in a large longitudinal cohort of women. Mayo Clin Proc. 2005;80(7):856–861. | |
Caro JJ, Ishak KJ, Huybrechts KF, Raggio G, Naujoks C. The impact of compliance with osteoporosis therapy on fracture rates in actual practice. Osteoporos Int. 2004;15(12):1003–1008. | |
Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26. | |
Costa AG, Bilezikian JP. Sclerostin: therapeutic horizons based upon its actions. Curr Osteoporos Rep. 2012;10(1):64–72. | |
Takahashi N, Yamana H, Yoshiki S, et al. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988;122(4):1373–1382. | |
Udagawa N, Takahashi N, Akatsu T, et al. The bone marrow-derived stromal cell lines MC3T3-G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology. 1989;125(4):1805–1813. | |
Udagawa N, Takahashi N, Akatsu T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87(18):7260–7264. | |
Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13(1):66–80. | |
Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390(6656):175–179. | |
Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597–3602. | |
Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. | |
Wong BR, Rho J, Arron J, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272(40):25190–25194. | |
Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–323. | |
Nakashima T, Kobayashi Y, Yamasaki S, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275(3):768–775. | |
Schlöndorff J, Lum L, Blobel CP. Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor alpha convertase. J Biol Chem. 2001;276(18):14665–14674. | |
Miyamoto T, Arai F, Ohneda O, Takagi K, Anderson DM, Suda T. An adherent condition is required for formation of multinuclear osteoclasts in the presence of macrophage colony-stimulating factor and receptor activator of nuclear factor kappa B ligand. Blood. 2000;96(13):4335–4343. | |
Hikita A, Yana I, Wakeyama H, et al. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281(48):36846–36855. | |
Bucay N, Sarosi I, Dunstan CR, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12(9):1260–1268. | |
Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. | |
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999;140(9):4367–4370. | |
Locklin RM, Khosla S, Turner RT, Riggs BL. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89(1):180–190. | |
Hofbauer LC, Gori F, Riggs BL, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140(10):4382–4389. | |
Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. | |
Danks L, Takayanagi H. Immunology and bone. J Biochem. 2013; 154(1):29–39. | |
Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. | |
Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304–309. | |
Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y. TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med. 1999;189(7):1025–1031. | |
Desanti GE, Cowan JE, Baik S, et al. Developmentally regulated availability of RANKL and CD40 ligand reveals distinct mechanisms of fetal and adult cross-talk in the thymus medulla. J Immunol. 2012;189(12):5519–5526. | |
Moschen AR, Kaser A, Enrich B, et al. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut. 2005;54(4):479–487. | |
Ashcroft AJ, Cruickshank SM, Croucher PI, et al. Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity. 2003;19(6):849–861. | |
Stolina M, Guo J, Faggioni R, Brown H, Senaldi G. Regulatory effects of osteoprotegerin on cellular and humoral immune responses. Clin Immunol. 2003;109(3):347–354. | |
Kostenuik PJ, Nguyen HQ, McCabe J, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res. 2009;24(2):182–195. | |
Kostenuik PJ, Smith SY, Jolette J, Schroeder J, Pyrah I, Ominsky MS. Decreased bone remodeling and porosity are associated with improved bone strength in ovariectomized cynomolgus monkeys treated with denosumab, a fully human RANKL antibody. Bone. 2011;49(2):151–161. | |
Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16(2):348–360. | |
McClung M. Role of RANKL inhibition in osteoporosis. Arthritis Res Ther. 2007;9 Suppl 1S3. | |
Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19(7):1059–1066. | |
Kumagai Y, Hasunuma T, Padhi D. A randomized, double-blind, placebo-controlled, single-dose study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of denosumab administered subcutaneously to postmenopausal Japanese women. Bone. 2011;49(5):1101–1107. | |
Nakamura T, Matsumoto T, Sugimoto T, Shiraki M. Dose-response study of denosumab on bone mineral density and bone turnover markers in Japanese postmenopausal women with osteoporosis. Osteoporos Int. 2012;23(3):1131–1140. | |
McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354(8):821–831. | |
Lewiecki EM, Miller PD, McClung MR, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22(12):1832–1841. | |
Miller PD, Bolognese MA, Lewiecki EM, et al. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43(2):222–229. | |
Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361(8):756–765. | |
McClung MR, Boonen S, Torring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res. 2012;27(1):211–218. | |
Papapoulos S, Chapurlat R, Libanati C, et al. Five years of denosumab exposure in women with postmenopausal osteoporosis: results from the first two years of the FREEDOM extension. J Bone Miner Res. 2012;27(3):694–701. | |
McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int. 2013;24(1):227–235. | |
Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab treatment and discontinuation on bone mineral density and bone turnover markers in postmenopausal women with low bone mass. J Clin Endocrinol Metab. 2011;96(4):972–980. | |
Reid IR, Miller PD, Brown JP, et al. Effects of denosumab on bone histomorphometry: the FREEDOM and STAND studies. J Bone Miner Res. 2010;25(10):2256–2265. | |
Brown JP, Dempster DW, Ding B, et al. Bone remodeling in postmenopausal women who discontinued denosumab treatment: off-treatment biopsy study. J Bone Miner Res. 2011;26(11):2737–2744. | |
Jassal SK, von Muhlen D, Barrett-Connor E. Measures of renal function, BMD, bone loss, and osteoporotic fracture in older adults: the Rancho Bernardo study. J Bone Miner Res. 2007;22(2):203–210. | |
Ishani A, Paudel M, Taylor BC, et al. Renal function and rate of hip bone loss in older men: the Osteoporotic Fractures in Men Study. Osteoporos Int. 2008;19(11):1549–1556. | |
Jamal SA, Swan VJ, Brown JP, et al. Kidney function and rate of bone loss at the hip and spine: the Canadian Multicentre Osteoporosis Study. Am J Kidney Dis. 2010;55(2):291–299. | |
Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res. 2012;27(7):1471–1479. | |
Miller PD. A review of the efficacy and safety of denosumab in postmenopausal women with osteoporosis. Ther Adv Musculoskelet Dis. 2011;3(6):271–282. | |
Toulis KA, Anastasilakis AD. Increased risk of serious infections in women with osteopenia or osteoporosis treated with denosumab. Osteoporos Int. 2010;21(11):1963–1964. | |
von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum. 2011;41(2):178–186. | |
Woo SB, Hellstein JW, Kalmar JR. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med. 2006;144(10):753–761. | |
Capsoni F, Longhi M, Weinstein R. Bisphosphonate-associated osteonecrosis of the jaw: the rheumatologist’s role. Arthritis Res Ther. 2006;8(5):219. | |
Allen MR, Iwata K, Phipps R, Burr DB. Alterations in canine vertebral bone turnover, microdamage accumulation, and biomechanical properties following 1-year treatment with clinical treatment doses of risedronate or alendronate. Bone. 2006;39(4):872–879. | |
MHRA. Denosumab 60 mg (Prolia▾): rare cases of atypical femoral fracture with long-term use. [webpage on the Internet]. London: MHRA; 2013. Available from: http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON239411. Accessed March 31, 2014. | |
Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet. 2013;382(9886):50–56. | |
Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276(23):20659–20672. | |
Kiechl S, Wittmann J, Giaccari A, et al. Blockade of receptor activator of nuclear factor-kappaB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. 2013;19(3):358–363. | |
Freemantle N, Satram-Hoang S, Tang ET, et al. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int. 2012;23(1):317–326. | |
Freemantle N, Cooper C, Diez-Perez A, et al. Results of indirect and mixed treatment comparison of fracture efficacy for osteoporosis treatments: a meta-analysis. Osteoporos Int. 2013;24(1):209–217. | |
Hiligsmann M, Reginster JY. Potential cost-effectiveness of denosumab for the treatment of postmenopausal osteoporotic women. Bone. 2010;47(1):34–40. | |
Hiligsmann M, Reginster JY. Cost effectiveness of denosumab compared with oral bisphosphonates in the treatment of post-menopausal osteoporotic women in Belgium. Pharmacoeconomics. 2011;29(10):895–911. | |
Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy. 2013;11(5):485–497. | |
Chau D, Becker DL, Coombes ME, Ioannidis G, Adachi JD, Goeree R. Cost-effectiveness of denosumab in the treatment of postmenopausal osteoporosis in Canada. J Med Econ. 2012;15 Suppl 1:3–14. | |
Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of Denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int. 2011;22(3):967–982. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.