Back to Journals » Therapeutics and Clinical Risk Management » Volume 18
A Real-World Study of Recombinant Human Growth Hormone in the Treatment of Idiopathic Short Stature and Growth Hormone Deficiency
Authors Gou P, Cheng X, Leng J, Su N
Received 28 October 2021
Accepted for publication 7 December 2021
Published 16 March 2022 Volume 2022:18 Pages 113—124
DOI https://doi.org/10.2147/TCRM.S363564
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor De Yun Wang
Peng Gou, Xinran Cheng, Jie Leng, Na Su
Department of Children Genetics and Endocrinology and Metabolism, Chengdu Women and Children Center Hospital, Chengdu, Sichuan, 610074, People’s Republic of China
Correspondence: Xinran Cheng Department of Children Genetics and Endocrinology and Metabolism, Chengdu Women and Children Center Hospital, No. 1617, Riyue Avenue, Qingyang District, Chengdu, Sichuan, 610074, People’s Republic of China Tel +86 28 6186 6142 Fax +86 28 6186 6197 Email [email protected]
Objective: This study aimed to evaluate the clinical efficacy of recombinant human growth hormone (rhGH) in the treatment of children with idiopathic short stature (ISS) and growth hormone deficiency (GHD) and to explore the related factors affecting treatment efficacy.
Methods: The current research reflects a real-world study. A total of 79 patients with ISS and 95 patients with GHD (both groups pre-puberty) who had been treated with rhGH for more than one year from January 2010 to September 2019 were included in this study. The patients were divided into two groups, ie, an ISS and a GHD group, respectively. The growth indexes, such as chronological age (CA), bone age (BA), height standard deviation score (HtSDS), insulin-like growth factor-1 (IGF-1) SDS, and body mass index were recorded and compared between the two groups before and after treatment. The treatment efficacy was evaluated according to changes in HtSDS before and after treatment, and the influencing factors of clinical efficacy were analyzed using a multivariate regression model.
Results: At the start of treatment, the differences in CA, BA, height, weight, sexual development stage, HtSDS, mid-parental height SDS, and IGF-1 SDS between the two groups were not statistically significant (P > 0.05). However, the initial dose of rhGH in the GHD group was significantly lower than in the ISS group (P < 0.001). Following rhGH treatment, the differences in CA, BA, BA/CA ratio, and IGF-1 SDS measured at 6, 12, 18, and 24 months between the ISS and GHD groups were not statistically significant, while the difference in HtSDS measured at 6 months was statistically significant. With the extension of rhGH treatment time, the annual growth rate (GV) gradually decreased, and the difference between HtSDS and the baseline gradually increased; however, the differences between the ISS and GHD groups were not statistically significant. The most important factor affecting the treatment efficacy for patients with ISS was age at the start of treatment; the most important factors affecting the treatment efficacy for patients with GHD were age and IGF-1 SDS.
Conclusion: Recombinant human growth hormone treatment can significantly improve the height of patients with ISS and GHD. There was no significant difference in growth rate between patients with ISS and those with GHD at relatively high doses. The common factor affecting the treatment efficacy of the two groups was the age at the start of treatment. During treatment, monitored data indicated that rhGH treatment of GHD and ISS thyroid function showed a clinical phenomenon in the form of increased free triiodothyronine, rather than hypothyroidism, which was rarely reported in existing studies.
Keywords: growth hormone deficiency, idiopathic short stature, recombinant human growth hormone, efficacy evaluation, multivariate analysis
Introduction
Growth hormone (GH) is a single-chain polypeptide protein with 21 amino acids. It is synthesized, stored, and secreted by the anterior pituitary gland and acts through insulin-like growth factor-1 (IGF-1) to regulate tissue growth and metabolism.1,2 It also acts independently of IGF-1 in the regulation of bone, muscle, and liver tissue functions.3,4
Any factor on the growth axis that results in decreased or abnormal GH secretion can lead to short stature in childhood. Since 1985, pituitary-derived GH has been replaced by recombinant human (rh) GH. Over the past 30 years, rhGH has been recognized and accepted for its safety and efficacy and is used in the treatment of a variety of growth disorders.5
“Short stature” is defined by a height more than two standard deviation (SD) scores (SDS) below the median height for the relevant age and sex of the subject; the most common reasons for short stature are GH deficiency (GHD) and idiopathic short stature (ISS). Growth hormone deficiency is caused by a pituitary issue and is a condition in which the body does not produce enough GH. Conversely, ISS refers to a short stature of unknown origin. The term “ISS” is defined as the absence of a dysfunction in the GH/IGF axis, or due to other identifiable disorders of the endocrine, genetic, or organ systems in short-stature children.
In 2003,6 the United States Food and Drug Administration (FDA) approved rhGH for the treatment of ISS. Although GH has been used to treat short stature in GH deficiency (GHD) and other conditions for more than 40 years, the criteria for satisfactorily defining targets for GH responsiveness have never been developed. Although Ying et al7 have reported the effectiveness of rhGH treatment for ISS. Parents generally believe that using rhGH to treat GHD in children yields faster results compared with using it to treat ISS; there are even doubts as to whether rhGH treatment for children with ISS can achieve satisfactory height improvements.It suggests the importance of health education for short stature patients.
Several clinical reports compared the effects of GH treatment for children with ISS and GHD.8,9 However, only a few reports involve Chinese participants. Hou et al compared the effects of GH treatment for children with Chinese ISS and GHD have been conducted.10 In China, many ISS children’s parents are willing to observe their children growth when they are young. We need more data of Chinese population to prove the efficacy of rhGH in the treatment of ISS children, so as to dispel the doubts of Chinese parents. Accordingly, we conducted a real-world retrospective study to discover whether there was a significant difference in the related influencing factors and treatment efficacy of rhGH in the treatment of children with ISS and GHD. The factors influencing the therapeutic effect were discussed and the possible mechanisms were analyzed.
Participants and Methods
Participants
This research reflects a real-world study. The participants were 79 patients with ISS (age, 5.7 ± 1.9 years) and 95 patients with GHD (age, 5.6 ± 1.9 years) who visited the Chengdu Women’s and Children’s Central Hospital from January 2010 to September 2019. The inclusion criteria for the study were as follows: 1) children with an impaired height lower than 2.0 SD of the reference values for the average height of children of a matched age and gender. The reference value of typical children was based on the standardized growth curve data for the height and weight of Chinese children and adolescents aged 0–18 years;11 2) the testicular volume of boys was below 4 mL, the breast development of girls was at stage B1, and all children were at Tanner stage I; 3) children carried to term; 4) a birth weight above 2.5 kg; 5) the annual growth rate of children with GHD was below 4 cm; 6) body mass index (BMI) was within the range of ± 2.0 SD of children of a matched age and gender.
The exclusion criteria of the study were as follows: 1) children who were born small for gestational age (SGA); 2) children with hypothyroidism; 3) children with confirmed chromosomal abnormalities or other known genetic syndromes that affected growth including but not limited to Turner, Laron, and Prader–Willi syndromes; 4) children with psychogenic dwarfism; 5) children who failed to be reexamined on schedule; 6) children with a closed epiphyseal line.
All participants had normal routine blood and urine test results, liver and kidney function, serum electrolyte levels, fasting blood glucose, thyroid function, and insulin levels, and denied having a family history of diabetes or tumors. The diagnosis of ISS and GHD followed the guidelines of the Endocrine Genetics and Metabolism Group of the Chinese Medical Association.12 For a diagnosis of GHD, the actual age of children was older than bone age (BA), the average height increase from age 3 years to adolescence was below 5cm, intellectual development was barrier-free, the body was well-proportioned, and the peak GH measured by the GH provocation test (insulin and arginine) was below 10.0 ng/mL. In terms of ISS, body length and weight were normal at birth, and the average height increase from 3 years old to adolescence was below 5 cm; BA was normal or delayed, mental development was barrier-free, participants had a well-proportioned body type, and GH provocation test (insulin and arginine) measured patients with peak GH levels higher than 10.0 ng/mL as ISS cases.
Research Methods
All patients were treated with rhGH (Jintropin; GeneScience Pharmaceuticals, Changchun, China) through subcutaneous injection nightly before going to bed. During the treatment, the patients were interviewed every three months to observe their height, weight, BMI, growth rate, height standard deviation score, and sexual characteristics. Routine blood tests, liver and kidney function, glucose metabolism, thyroid function, IGF-1, IGF binding protein-3 (IGFBP-3), and adverse events were monitored.
Measurement Method
Trained specialist nurses used a uniform method for measuring the height of the participants, and measurements were taken before 10:00. The height measuring ruler of the measuring instrument was manufactured by an International Organization for Standardization-qualified enterprise and had been measured and checked before conducting the study. The weight scale was checked using a separate weight. Participants’ height and weight were measured three times for each individual, and the average value was noted. Serum GH, thyroid function, IGF-1, and IGFBP-3 were all measured by chemiluminescence. IGF-1 SDS was defined as (actual IGF-1 concentration-median IGF-1 concentration of normal children of the same age and sex)/ (IGF-1 concentration standard deviation for normal children of the same age and sex).
Testing Equipment
A Siemens Atellica IMI600 automatic chemiluminescence immunoassay analyzer was used, and the assay kit was provided by Siemens.
The initial therapeutic dose of rhGH was 0.26–0.44 mg/kg per week for GHD and 0.37–0.46 mg/kg per week for the ISS cases. This was adjusted every three months according to weight, the changes of height standard deviation score (ΔHtSDS), sexual development stage, BA, and IGF-1.
Statistical Methods
Measurement data were statistically described as mean ± SD and compared between the two groups using t-tests, based on the central limit theorem. Count data were expressed as the number of examples and proportions and compared between the two groups using chi-square or Fisher’s exact probability tests. After adjusting factors for important baseline imbalances by multivariate linear regression, the differences in HtSDS changes relative to the baseline at different follow-up time points after rhGH treatment were statistically tested. The least significant difference method was selected as the multiple test correction technique for conducting the comparison between groups at different follow-up time points. A multivariate stepwise linear regression model was used to explore the influencing factors for baseline changes in HtSDS at different follow-up time points in the GHD and ISS groups. All statistical tests were conducted using two-sided tests, and P ≤ 0.05 was considered statistically significant. This study adopted the SAS 9.4 software for conducting statistical analysis.
Primary Results
The Basic Conditions of the Two Groups Before Treatment
A total of 174 (79 ISS and 95 GHD) patients who had been treated for more than one year were included in this study; among them, 25 children with ISS and 48 children with GHD had been treated for more than two years. The flowchart of patient inclusion and grouping was shown in Figure 1. At the start of treatment, the differences in chronological age (CA), BA, height, weight, HtSDS, mid-parental height SDS, and IGF-1 SDS between the two groups were not statistically significant (P > 0.05). However, the initial dose of rhGH in the GHD group was significantly lower compared with the ISS group (P < 0.001). The basic characteristics of the two groups prior to the start of treatment are shown in Table 1.
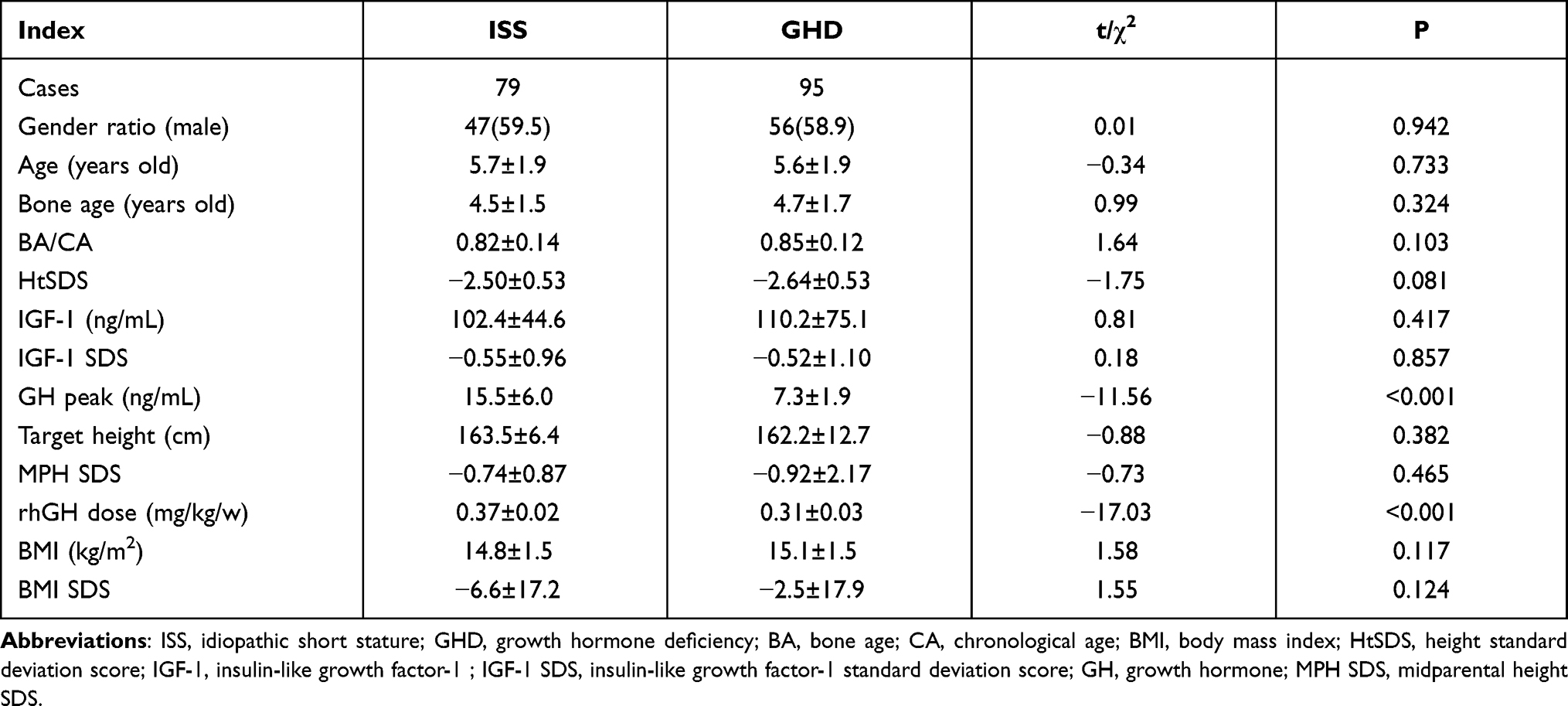 |
Table 1 Basic Characteristics of Children |
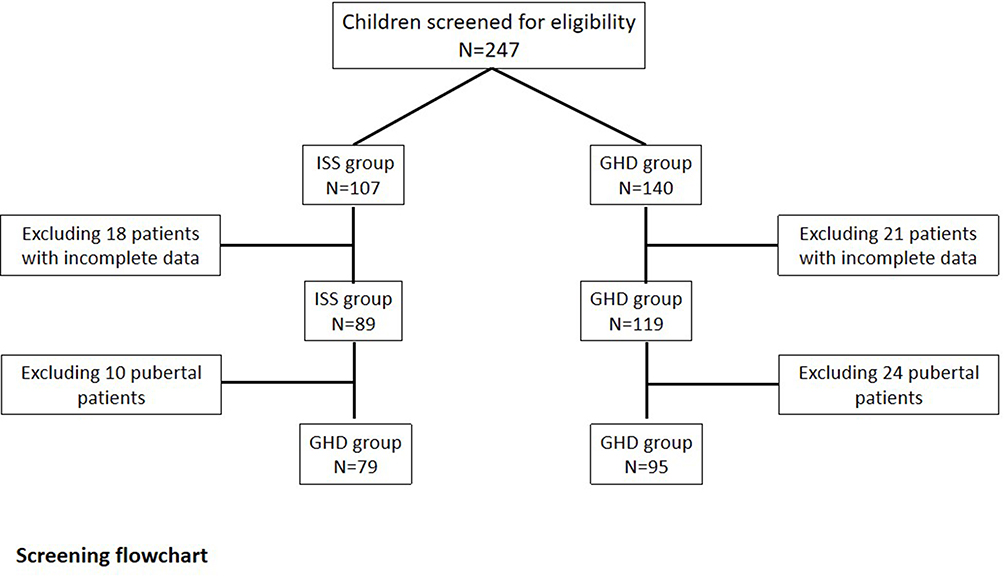 |
Figure 1 Flowchart of patient inclusion and grouping. |
Growth Index Changes Following Recombinant Human Growth Hormone Treatment
Following rhGH treatment, the differences in CA, BA, BA/CA ratio, and IGF-1 SDS measured at 6, 12, 18, and 24 months between the ISS and GHD groups were not statistically significant, while the difference in HtSDS measured at 6 months was statistically significant (Table 2). From the start and up to two years of treatment, the dose of rhGH was significantly higher in the ISS than in the GHD group. With the extension of the rhGH treatment time, the annual growth rate (GV) gradually decreased. In the second year of treatment, the GV gradually decreased to 7–8 cm/year but the difference between the ISS and GHD groups was not statistically significant (Figure 2). After treatment, the difference between HtSDS and the baseline level gradually increased, but the difference between the ISS and GHD groups was not statistically significant (Figure 3). The IGF-1 SDS in both groups increased gradually after treatment, reached a stable level one year after treatment, and was lower than +2.5 SDS (Figure 4).
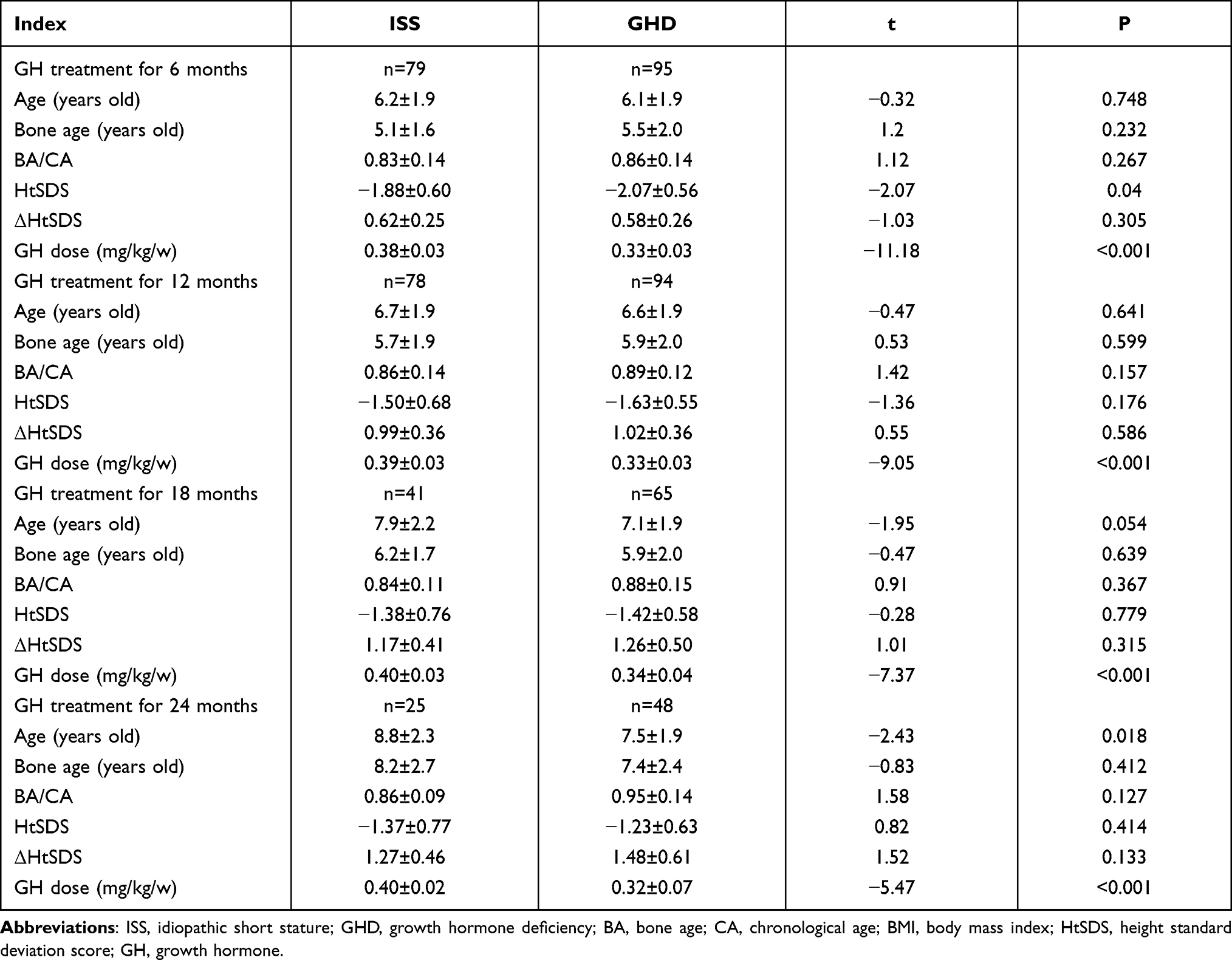 |
Table 2 Changes in Growth Indexes of ISS and GHD Children After rhGH Treatment |
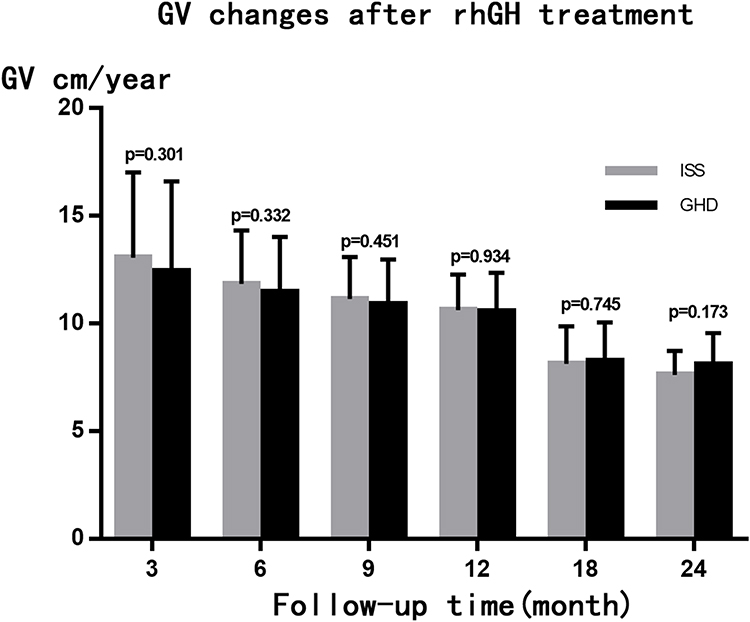 |
Figure 2 GV changes after rhGH treatment, the error bar represents standard deviation. |
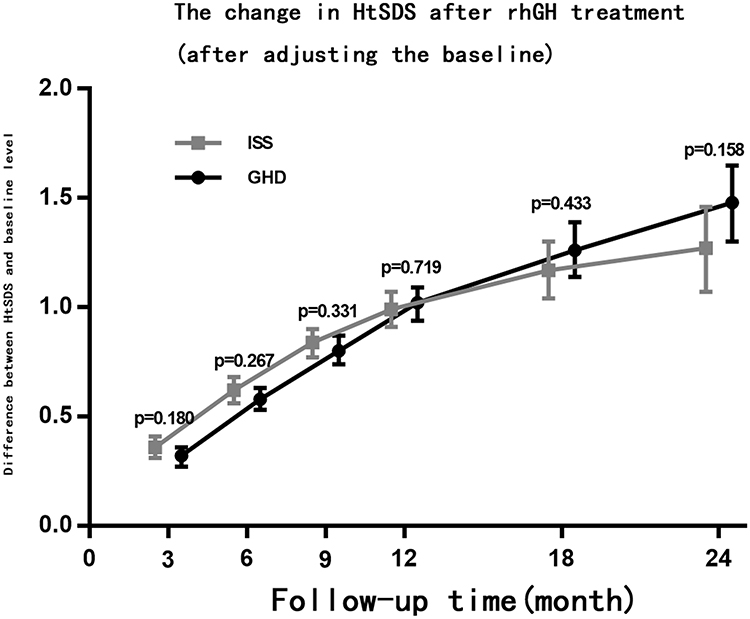 |
Figure 3 The change in HtSDS after rhGH treatment (after adjusting the baseline), the error bar represents 95% confidence interval of change in HtSDS relative to baseline. |
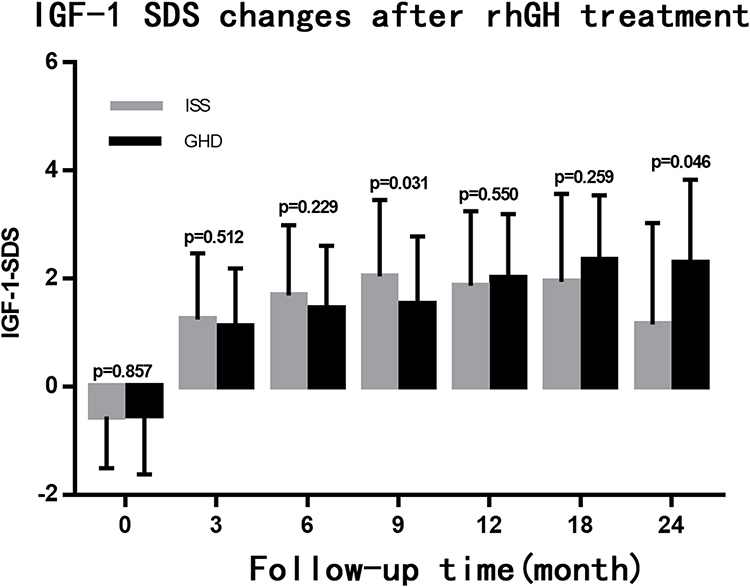 |
Figure 4 IGF-1 SDS changes after rhGH treatment, the error bar represents standard deviation. |
Analysis of the Related Factors Affecting the Baseline Changes of Height SD Score After Recombinant Human Growth Hormone Treatment
A multivariate stepwise linear regression model was used to explore the influencing factors of baseline HtSDS changes at different follow-up time points in the ISS and GHD groups. The final statistically significant variables and results are shown in Table 3.
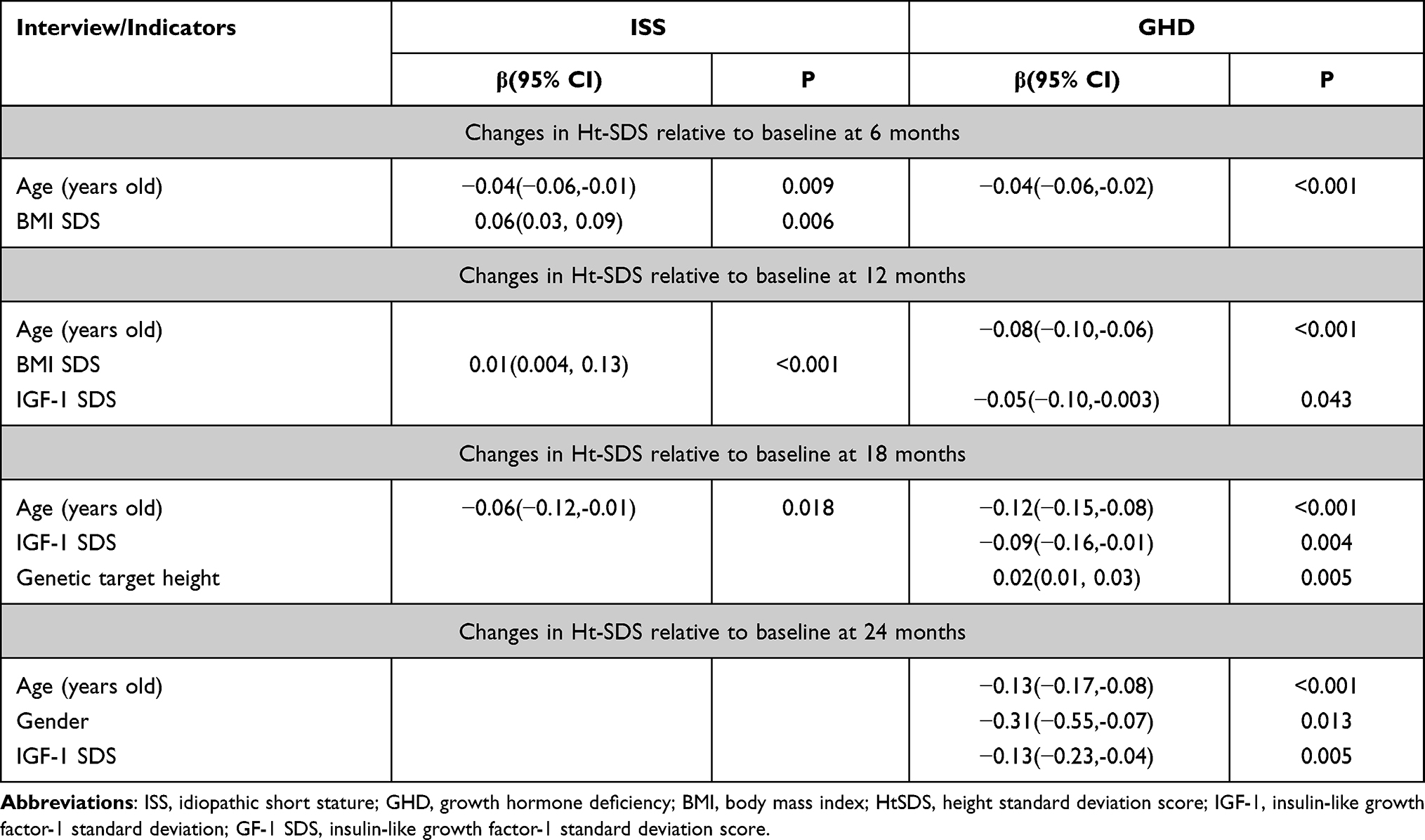 |
Table 3 Factors Influencing the Changes of HtSDS After Treatment |
For patients with GHD, the age at the beginning of treatment (the first 6 months) was the main factor influencing HtSDS changes following rhGH treatment. With the extension of treatment time, the factors that determined HtSDS changes also changed. The IGF-1 SDS at the beginning of treatment was also a key influencing factor. For patients with ISS, changes in HtSDS at 6 months were mainly related to age and BMI SDS at the beginning of treatment. Changes in HtSDS at 12 months were only related to the BMI SDS at the start of treatment. Changes in HtSDS at 18 months were only related to the participant’s age at the start of treatment.
The Effect of Recombinant Human Growth Hormone Treatment on Thyroid Function
During the treatment, thyroid-stimulating hormone increased in four children with GHD and four children with ISS. Free triiodothyronine (FT3) serum increased in 14 children with GHD and 24 children with ISS. There was no significant difference in the incidence of increased FT3 between the two groups at each follow-up time point (Table 4).The changes in thyroid function indexes of ISS and GHD children during rhGH treatment were shown in Table 5.
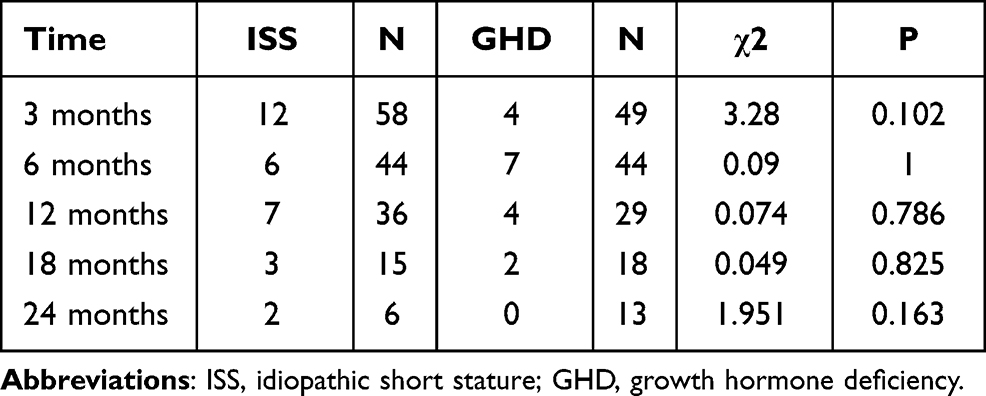 |
Table 4 Comparison of Increase in FT3 During rhGH Treatment |
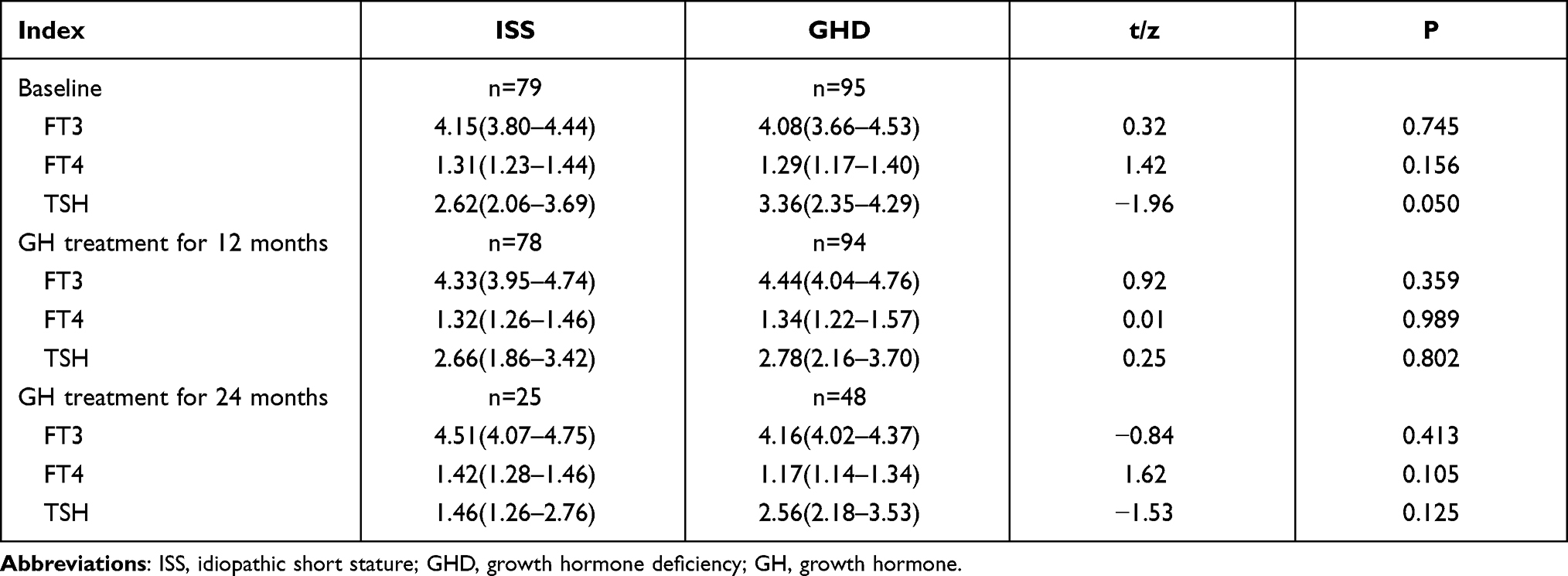 |
Table 5 Changes in Thyroid Function Indexes of ISS and GHD Children During rhGH Treatment |
Discussion
In 1985, the first variety of rhGH was approved by the US FDA. Compared with biological GH, the availability and safety of recombinant GH have brought revolutionary changes to the field of GH, and its application scope and indications continued to expand in the 30 years following its introduction. In addition to many known diseases, such as being small for gestational age, Turner syndrome, Noonan syndrome, and genetic diseases such as SHOX and NPR2 mutations, due to the clinical heterogeneity and complex etiology of ISS, the application of rhGH in children with this condition has always been an area of concern. The recommended dose for ISS treatment in the Chinese “Guidelines for Diagnosis and Treatment of Short Stature Children” (2008) is 0.35–0.46 mg/kg per week.10 The main goal of rhGH treatment for ISS and GHD is to allow children to reach the standard growth parameters according to their age, as well as the target height (TH) SDS closely matching the adult TH.13
The range of responses to GH is broad, and differences can be attributed to diagnosis, age, GH dose, parental height, compliance, intercurrent illness, other (endocrine) therapies, and poorly defined molecular and biochemical factors. An existing study revealed that following rhGH treatment for ISS, the treatment response indicated a significant dose-dependent pattern, where a higher unit dose was more effective than a lower unit dose.14 Alzahrani et al15 retrospectively analyzed 110 children (76 with GHD and 34 with SGA). Their research considered the starting age, treatment time, and treatment dose as the main influencing factors on growth rate. Several studies also showed that rhGH could improve the height gain and growth rate of children with ISS. Alzahrani et al16 conducted a retrospective study in this regard, and the results revealed that short-term use of the same unit dose of GH did not yield a significant difference in height growth rate between children with ISS and those with GHD. Dommelen et al17 showed that in the first two years of growth- hormone therapy, poor compliance negatively impacted growth response.
The investigators in the present study observed that in the first year of treatment, the primary growth variables of the two groups showed significant improvement. The GV and HtSDS were increased, and there were no significant differences in BA, BA/CA ratio, HtSDS, and IGF-1 SDS between the GHD and ISS groups. During treatment, IGF-1 remained within +2.5 SD; this result was consistent with the results of the bulk of clinical trials.5 However, the present study revealed that children with ISS needed a higher unit dose to reach a growth rate similar to that of children with GHD, thus confirming that GH had a dose/effect correlation in terms of improving the growth rate of children with a non-GH deficiency.7 This was consistent with the fact that the dose of GH for patients with ISS approved by the FDA (2003) was generally 0.15 IU/(kg · d), which is higher compared with the dose for GHD treatment at 0.1 IU/(kg · d) and is different from the rhGH unit dose used for 19 children with ISS and 36 children with GHD as reported by Alzahrani et al.15
From the start of and up to two years of treatment, the rhGH dose was significantly higher in the ISS group compared with the GHD group. With the extension of the rhGH treatment time, the GV of the two groups at the second year of treatment was lower than at the first but the difference between the ISS and GHD groups was not statistically significant. This clinical phenomenon was consistent with the conclusion presented by Zhang et al.18 The majority of existing studies19 found that with prolongation of the rhGH treatment time, the therapeutic effect was gradually weakened but the growth rate was higher compared with before treatment.
Growth hormone promotes growth by binding with the GH receptor and stimulating two signaling pathways. One of these is the Janus kinase signal transducer and activator of transcription (JAK-STAT) pathway which, in turn, stimulates IGF-1 synthesis. The second is the MARK/extracellular signal-regulated protein kinase pathway, which stimulates chondrocyte proliferation.20
Insulin-like GF-1 is synthesized in the liver. Using different doses of rhGH given to BALB/c mice, Zhao21 revealed that long-term rhGH stimulation inhibited the JAK-STAT signaling pathway in the liver, decreasing the sensitivity of the liver to GH. This may explain why many clinical studies have found a prolonged rhGH treatment time to correlate with a decrease in growth rate. In addition, age at the start of rhGH treatment negatively correlated with the changes of HtSDS after rhGH treatment in children with ISS and GHD. This may reflect the fact that height improvement is generally more significant when the start of treatment age is younger.22,23
Recombinant human GH stimulates IGF-1 production in the liver and forms ternary complexes with IGFBP and the acid instability subunit (AIS). These complexes can prolong the half-life of IGF-1 and promote the linear growth of bone. Regular monitoring of serum IGF-1 concentration during rhGH treatment plays an invaluable role for evaluating the safety and effectiveness of rhGH treatment in children. Furthermore, IGF-I levels may help assess compliance and GH sensitivity, and levels that are consistently elevated (>2.5 SDS) should prompt the consideration of a reduction in GH dose.24
In this study, the IGF-1 SDS of the two groups were higher than before treatment and were maintained within +2.5 SD of the matched age and gender. In addition, the IGF-1 SDS was maintained between 1SD ~ 2 SD, and HtSDS improved significantly. At months 12, 18, and 24 of treatment, IGF-1 SDS in the GHD group was higher than in the ISS group. This suggested that children with GHD were more sensitive to rhGH, and a higher IGF-1 SDS increase could be achieved with lower doses to obtain a growth rate similar to children with ISS. Meanwhile, ISS represented a partial GH-insensitive state that manifested during treatment with higher GH doses.25 Li et al11 compared the efficacy and safety of rhGH in the treatment of children with GHD and ISS. The increase of IGF-1 SDS was more significant in children with ISS, which differed from the conclusions of the present study. The difference may be due to the fact that ISS patients were given smaller doses and GHD patients were given larger doses in the present study compared to their study. In our center, the IGF-1 SDS of the two groups was between +1 and +2.5 SD. Therefore, the dose was not reduced during treatment. The European Society of Pediatric Endocrinology and the National Society of Pediatric Endocrinology believe there is no evidence to support the safety value of IGF-1 monitoring in children, and there is no data to indicate a safe upper limit of serum IGF-1 concentration. Guidelines established by the Lawson Wilkins Pediatric Endocrinology Society26 recommend that when the concentration of IGF-1 is higher than the normal range, the dose of rhGH should be reduced. Regarding IGF-1-based GH dose regulated correlation, Cohen25 pointed out that IGF-based GH dosing is clinically feasible for both GHD and ISS patients, although GH dose requirements and auxological outcomes are distinct between these groups. This suggests a degree of both GH and IGF insensitivity in subjects with ISS that requires specific management strategies to optimize growth during GH therapy.
This study revealed that overall, adverse reactions in 174 children of short stature who were treated with rhGH were rare. This result is similar to the primary safety outcomes of a 15-year study involving a total of 22,311 children receiving GH treatment in 30 countries published by The Journal of Clinical Endocrinology & Metabolism in 2019.27 One child had local skin redness, swelling, and itching, which did not reoccur after changing the injection site. No increased fasting blood glucose was detected at any follow-up time points. Two children had transient IGF elevation but after three weeks of discontinuation, a reexamination revealed a normal result, and the medication was restored. There was no compensatory hypothyroidism in children with GHD during treatment. This differed from results reported in studies stating that hypothyroidism occurred.28 Contrastingly, the results of the present real-world research revealed that of the 79 patients with ISS and 95 patients with GHD treated with rhGH, the monitoring of thyroid function in months 3, 6, and 12 of treatment indicated a higher FT3 in some children than before treatment, as well as levels higher than the upper limit of children of the same age (detection instrument, Siemens Atellica IMI600 automatic chemiluminescence immunoassay analyzer). There was no significant difference in the incidence of increased FT3 between the ISS and GHD groups at each follow-up time point.
Growth hormone has multiple functions in the promotion of growth and regulating metabolic activity. Several studies have revealed the effect of GH replacement therapy on thyroid function, where GH replacement altered the peripheral interconversion of thyroxine (T4) and triiodothyronine (T3).29 Additionally, GH treatment increased serum T3 or free FT330,31 and decreased T4 or free T4. Yamauchi32 revealed that GH mediated and stimulated increased FT3 secretion by up-regulating iodothyronine deiodinase type-2.
In this retrospective study, the number of included GHD and ISS cases was similar. The incidence rate of ISS in clinical short stature was approximately 70%. The reason for the similar number of cases in the two groups of this study was analyzed, and the results showed that the subjective treatment willingness of the parents of children with GHD was higher compared with the parents of children with ISS. In ISS families, parents subjectively considered that their children’s growth axis assessment showed “no GH deficiency” and, accordingly, accepted that there was “no need for treatment,”; alternatively, they may have subjectively considered that “no GH deficiency, may be the treatment effect is not good” and as a result did not engage in treatment. This resulted in the proportion of ISS treatment cases in our hospital being relatively low. Our hospital data revealed that the subjective treatment willingness of parents of children with GHD was higher compared with the parents of children with ISS. This emphasizes the importance of health education.
Conclusion
Recombinant human GH did not bring about significant differences in the growth patterns of children with GHD and those with ISS. The treatment of ISS required a higher unit dose, and no obvious side effects were observed during the treatment. The common factor determining the height benefit of children with GHD and ISS was children’s age at the start of treatment. Meanwhile, Treatment with rhGH minimized the risk of increasing IGF-I levels above the normal range. Some children showed increased FT3, suggesting the effect of GH replacement therapy on thyroid function. Growth hormone replacement also altered the peripheral interconversion of T4 and T3. Moreover, GH may improve the biological action of thyroid hormone on bone; this finding was not previously reported in cases involving children.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Chengdu Women and Children Center Hospital. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all patient guardians.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
Sichuan Province Science and Technology Program Funded Project: Mobile Internet Service Platform for Children’s Genetic Metabolic Disease Screening, Diagnosis and Treatment and Referral (2019JDPT0034).
Disclosure
The authors declare that they have no competing interests.
References
1. Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord. 2006;7(4):225–235. doi:10.1007/s11154-007-9025-5
2. Chia DJ. Minireview: mechanisms of growth hormone-mediated gene regulation. Mol Endocrinol. 2014;28(7):1012–1025. doi:10.1210/me.2014-1099
3. Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. 2008;29(5):535–559. doi:10.1210/er.2007-0036
4. Chikani V, Ho KK. Action of GH on skeletal muscle function: molecular and metabolic mechanisms. J Mol Endocrinol. 2014;52(1):R107–R123. doi:10.1530/JME-13-0208
5. Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, Society of Pediatrics, Chinese Medical Association The Editorial Board, Chinese Journal of Pediatrics. Recommendations for the clinical use of recombinant human growth hormone in children. Zhonghua Er Ke Za Zhi. 2013;51:426–432.
6. Park P, Cohen P. Insulin-like growth factor I (IGF-I) measurements in growth hormone (GH) therapy of idiopathic short stature (ISS). Growth Hormone Igf Res. 2005;15(supp–S):13–20. doi:10.1016/j.ghir.2005.06.011
7. Ying YQ, Hou L, Liang Y, et al. Efficacy and safety of recombinant human growth hormone in treating Chinese children with idiopathic short stature. Growth Hormone IGF Res. 2018;42:80–85. doi:10.1016/j.ghir.2018.09.003
8. Kang JC, Choi YS, Choi IK, Kim HS, Kim DH. The effect of growth hormone on patients with growth hormone deficiency and idiopathic short stature. Korean J Pediatr. 2004;47:310–318.
9. Kim SA, Choe YR, Yang EM, Kim CJ. Comparison of growth hormone treatment in patients with idiopathic short stature and idiopathic growth hormone deficiency. Chonnam Med J. 2014;50:63–66. doi:10.4068/cmj.2014.50.2.63
10. Hou L, Liang Y, Wu W, et al. Comparison of the efficacy and safety of recombinant human growth hormone in treating idiopathic short stature and growth hormone deficiency in children. Growth Hormone IGF Res. 2020;53-54:101331. doi:10.1016/j.ghir.2020.101331
11. Li H, Ji CY, Zong XN, et al. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Chin J PED. 2009;47(7):487–492.
12. Endocrinology G, Group M. Pediatrics Branch of Chinese Medical Association. Guidelines for the diagnosis and treatment of short stature children. Chin J Ped. 2008;6:428–430.
13. Heo SH, Choi JH, Kim YM, et al. Comparative proteomic analysis in children with idiopathic short stature(ISS)before and after short-term recombinant human growth hormone (rhGH)therapy. Proteomics. 2013;13(7):1211–1219.
14. de Arriba MA, Cancela MV, Juan Alcón SJ, et al. Impact of adherence on growth response during the first 2 years of growth hormone treatment.Endocrine. 2020;1–11:513.
15. Alzahrani AK, Algethami AK, Barnawi GM, et al. Differences in response to recombinant growth hormone therapy on height gain in patients with idiopathic short stature vs. patients with growth hormone deficiency. Cureus. 2020;12(3):e7319.
16. Ranke Michael B, Anders L, KIGS International Board. Prediction models for short children born small for gestational age (SGA) covering the total growth phase. Analyses based on data from KIGS (Pfizer International Growth Database). BMC Med Inform Decis Mak. 2011;11:38.
17. Dommelen PV, Koledova E, Wit JM, et al. Effect of adherence to growth hormone treatment on 0–2 year catch-up growth in children with growth hormone deficiency. PLoS One. 2018;13(10):e0206009. doi:10.1371/journal.pone.0206009
18. Zhang X, Xiong F, Zhu M, et al. Efficacy of recombinant human growth hormone therapy for children with different causes of short stature. J Third Mil Med Univ. 2016;38(16):1889–1894.
19. Bakker B, Frane J, Anhalt H, et al. J Clin Endocrinol Metab. 2008;93(2):352–357. doi:10.1210/jc.2007-1581
20. Zhengyan L, Wang Q, Bofei L, et al. Influence of enhanced recovery after surgery programs on laparoscopy-assisted gastrectomy for gastric cancer: a systematic review and meta-analysis of randomized control trials. World J Surg Oncol. 2017;15:207.
21. Zhao QF. Long-Term Stimulation of Growth Hormone Leads to Insensitivity to Growth Hormone in the Live. Southern Medical University; 2016.
22. Ross Judith L, Lee Peter A, Robert G, et al. Increased height standard deviation scores in response to growth hormone therapy to near-adult height in older children with delayed skeletal maturation: results from the ANSWER Program. Int J Pediatr Endocrinol. 2015;2015:1.
23. Rim JH, Byul KE, Seok SY, et al. Comparative study of growth hormone treatment in children with idiopathic short stature and growth hormone deficiency. Curr Drug Metab. 2015;16:940–946.
24. Cohen P, Rogol AD, Deal CL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008;93:4210–4217. doi:10.1210/jc.2008-0509
25. Pinchas C, John G, Rogol Alan D, et al. Variable degree of growth hormone (GH) and insulin-like growth factor (IGF) sensitivity in children with idiopathic short stature compared with GH-deficient patients: evidence from an IGF-based dosing study of short children. J Clin Endocrinol Metab. 2010;95:2089–2098. doi:10.1210/jc.2009-2139
26. Lee MM; Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Guidelines for the Use of Growth Hormone in Children with Short Stature; 2016.
27. Child Christopher J, Zimmermann Alan G, Chrousos George P, et al. Safety outcomes during pediatric GH therapy: final results from the prospective genesis observational program. J Clin Endocrinol Metab. 2019;104:379–389. doi:10.1210/jc.2018-01189
28. Bell J, Parker KL, Swinford RD, et al. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab. 2010;1:167.
29. Glynn N, Halsall DJ, Boran G, et al. Growth hormone replacement may influence the biological action of thyroid hormone on liver and bone tissue. Growth Hormone IGF Res. 2021;57–58:101393. doi:10.1016/j.ghir.2021.101393
30. Glynn N, Kenny H, Quisenberry L, et al. The effect of growth hormone replacement on the thyroid axis in patients with hypopituitarism: in vivo and ex vivo studies. Clin Endocrinol (Oxf). 2016;86(5):747–754. doi:10.1111/cen.13272
31. Jørgensen JO, Møller J, Laursen Tet al. Growth hormone administration stimulates energy expenditure and extrathyroidal conversion of thyroxine to triiodothyronine in a dose-dependent manner and suppresses circadian thyrotrophin levels: studies in GH-deficient adults. Clin Endocrinol (Oxf). 2010;41(5):609–614.
32. Ichiro Y, Yoriko S, Takafumi Y, et al. Effects of growth hormone on thyroid function are mediated by type 2 iodothyronine deiodinase in humans. Endocrine. 2018;59:353–363.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.