")
Back to Journals » International Medical Case Reports Journal » Volume 13
A Rare Case with New Insights: Pure Sensory Guillain Barre Syndrome with Axonal Features
Authors Kofahi R , Aldabbour B , Aljezawi M
Received 3 September 2020
Accepted for publication 13 October 2020
Published 20 October 2020 Volume 2020:13 Pages 543—549
DOI https://doi.org/10.2147/IMCRJ.S280255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Raid Kofahi,1 Belal Aldabbour,1 Ma’en Aljezawi2
1Neuroscience Division, Faculty of Medicine, Jordan University of Science and Technology (JUST), Irbid, Jordan; 2Department of Community and Mental Health, Faculty of Nursing, Al Al-Bayt University, Mafraq, Jordan
Correspondence: Raid Kofahi Postal Address: P.O. Box (3030), Irbid 22110, Jordan
Tel +962796877750
Email [email protected]
Abstract: The pure sensory variant of Guillain-Barré syndrome “GBS” is controversial. Scarce case reports in the literature have described pure sensory presentations secondary to acute demyelination of peripheral sensory nerves. Pure sensory GBS secondary to axonal damage is rarer and even more controversial owing to a significant overlap with sensory neuronopathy. A 31-year-old lady with history of a recent primary varicella zoster virus (VZV) infection presented with acute onset of sensory symptoms and signs involving her four limbs and the trunk, without weakness. Examination was remarkable for severe impairment in all sensory modalities in her limbs and trunk, pseudo-athetoid limb movements, sensory ataxia, positive Romberg’s sign, and areflexia, with no motor involvement. CSF analysis showed elevated protein without pleocytosis known as albuminocytological dissociation. MRI of the spine with contrast showed multiple root enhancement. Nerve conduction studies “NCS” demonstrated absent sensory action potentials, with normal motor nerves responses. Initial electromyography was normal. After differential diagnoses were appropriately excluded, the patient was diagnosed with pure sensory axonal GBS and treated with IVIG for five days. Gradual clinical improvement was seen over the following months, with improvement in six-month GBS disability score down to two. Follow-up NCS showed findings similar to the initial study but follow-up EMG studies revealed denervation potentials in multiple levels, suggesting a subclinical axonal motor involvement and excluding sensory neuronopathy. To our best knowledge, this case represents the first case of pure sensory GBS with onset after a documented primary VZV infection. The findings in this case illustrate the difficulties in diagnosing pure sensory GBS and the significance of an early treatment. It also demonstrates the potential value of follow-up EMGs in excluding sensory neuronopathy as an important differential diagnosis for this condition.
Keywords: Guillain-Barre syndrome, acute inflammatory polyradiculoneuropathy, intravenous immunoglobulins, neurophysiology
Introduction
Guillain-Barré syndrome “GBS” initially described an acute, monophasic neurological illness marked clinically by ascending weakness, areflexia, and possible but less prominent sensory symptoms, with elevated protein but absent pleocytosis (albuminocytological dissociation) on cerebrospinal fluid “CSF” analysis and demyelination-remyelination on nerve conduction studies “NCS”.1,2 The syndrome was later recognized to be clinically, pathologically and electrophysiologically diverse, so GBS gradually became an umbrella term that includes several variants, some of which are due to peripheral axonal damage rather than demyelination.2,3 The pure sensory GBS variant refers to an acute, monophasic illness characterized by exclusive sensory symptoms and signs, with evidence of widespread sensory neuropathy, in the absence of systemic disorders that may cause an acute sensory peripheral neuropathy.4 Although it shares clinical and pathophysiological features with GBS, the pure sensory variant is still subject to debate.5
Wartenberg first discussed the possibility of a pure sensory clinical equivalent to GBS in 1958,6 and the first detailed case was described three decades later.7 The condition remains very rare to date. Uncini and Yuki identified 40 cases of sensory GBS that were published between 1980 and 2011.4 Additionally, although adequate case definitions were lacking, several case series have reported a prevalence of the pure sensory variant under one percent of all GBS cases.8–11 Identifying pure sensory GBS is complicated, with several overlapping conditions lying in the differential. Still, a timely and accurate diagnosis is important since it can prevent delays in treatment and improves the prognosis. Previous reports have described GBS onset following an acute primary Varicella Zoster Virus (VZV) infection, but their clinical presentations were predominated by motor involvement.12 Here we describe a case of pure sensory axonal GBS following a primary VZV infection in a 31-year-old lady.
Case Report
A 31-year-old previously healthy female presented with 4 days of bilateral numbness that started distally in her feet and hands simultaneously then involved her whole four extremities and the front and back of her trunk. She also lost her balance over the same period, first becoming unable to walk then unable to sit without falling to either side. She also reported difficulty reaching for objects. She denied having double vision, slurring of speech, facial numbness or droop, tremors, limb weakness, abdominal distention, sphincteric problems, or dryness of the mouth or eyes. There was no weight loss, and no joint stiffness, swelling or pain. Family history was unremarkable. She had developed a generalized skin rash that started 10 days prior to the onset of her neurological symptoms, which was diagnosed as a primary Chickenpox infection by a dermatologist who prescribed oral acyclovir.
Neurological examination demonstrated normal higher mental functions and intact cranial nerves, with no nystagmus, ptosis, or limitation of eye movements. Muscle strength was 5/5 throughout. Sensory examination was remarkable for severe impairment in joint position and vibration sensation in both upper limbs to the elbows and lower limbs to the knees. Pain and temperature sensation were symmetrically impaired over both upper and lower limbs and the trunk, but the impairment was more pronounced over the anterior trunk than the back. There was no sensory level. Reflexes were absent all over. Babinski’s sign was negative bilaterally. Coordination revealed impaired finger to nose and heel to shin, with significant exacerbation by eye closure. She could not maintain a sitting position due to severe truncal ataxia and demonstrated pseudo-athetoid movements in the hands. Dysautonomic signs such as blood pressure and pulse fluctuations were absent, and there was no ileus.
Initial basic labs were normal. Lumbar puncture revealed CSF protein 61.1 mg/dl (reference range 15–45 mg/dl), with normal CSF white cell count (3 cells). CSF cultures and viral PCRs were negative, including HSV I and II, Mycobacterium tuberculosis (M. tb), Epstein-Barr virus (EBV), Cytomegalovirus (CMV), and enterovirus PCRs. CSF VZV PCR was unavailable. Brain MRI with and without contrast was normal. Whole spine MRI with contrast demonstrated enhancement in multiple thoracic and lumbar nerve roots (Figure 1). NCS performed on the fifth day of admission (Table 1) showed absent sensory responses in her four extremities, with normal motor responses and F-waves. Initial electromyography “EMG” using concentric needle was normal (Table 2). Serum VZV IgM was elevated (18.9). HIV screen was negative. Serum B12, folate, thyroid, zinc, vitamin E, and Creatine Kinase “CK” were within normal limits. Serum protein electrophoresis and immunofixation electrophoresis were negative. A paraneoplastic panel, including Hu, Yo, Amphiphysin, Ri, CV2, and Ma2/Ta antibodies, was negative. A comprehensive autoimmune panel was also negative. Ganglioside antibody screen also was negative. CT scan of the chest, abdomen and pelvis did not reveal any malignancy.
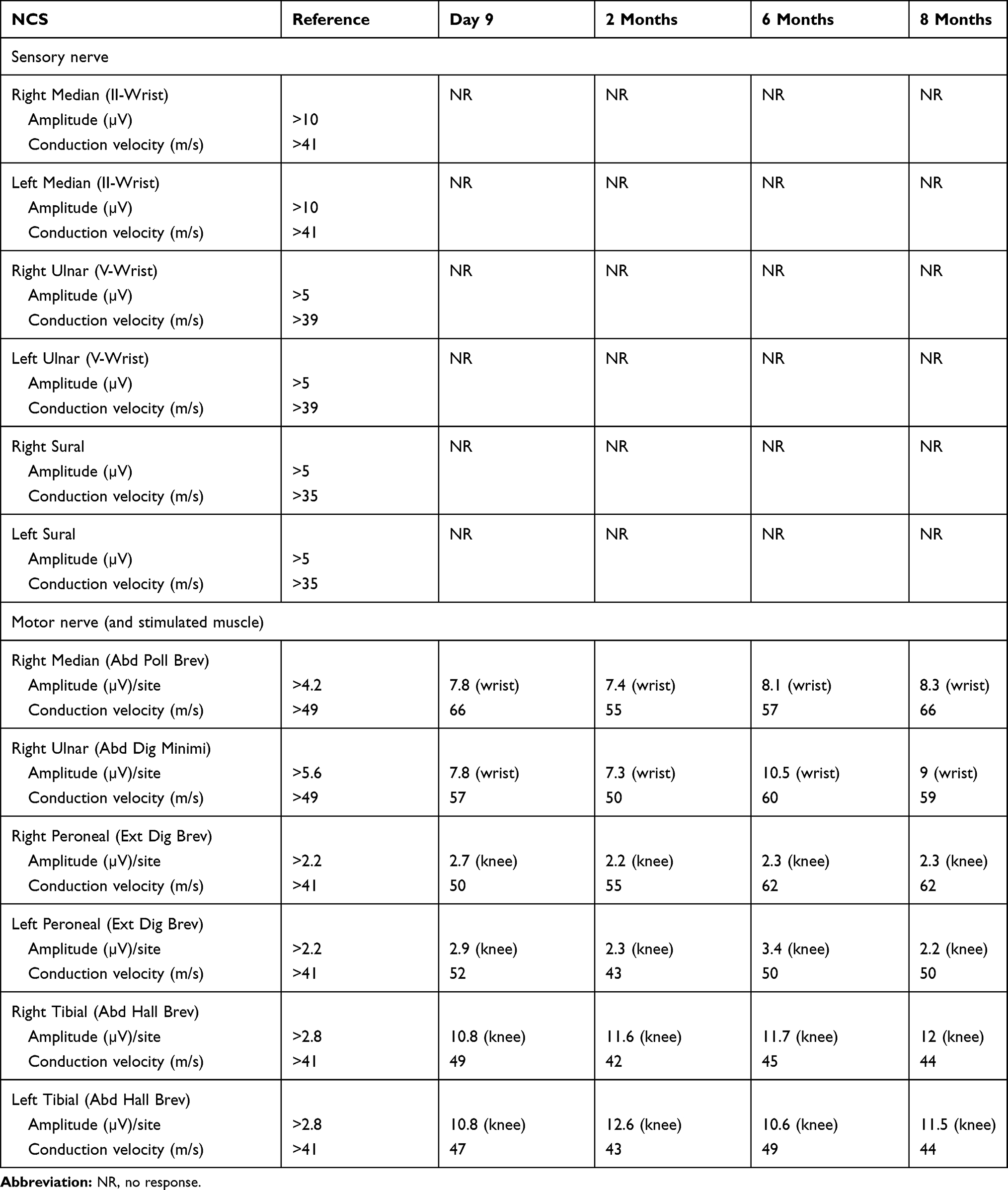 |
Table 1 Nerve Conduction Study Results |
 |
Table 2 Electromyography Results |
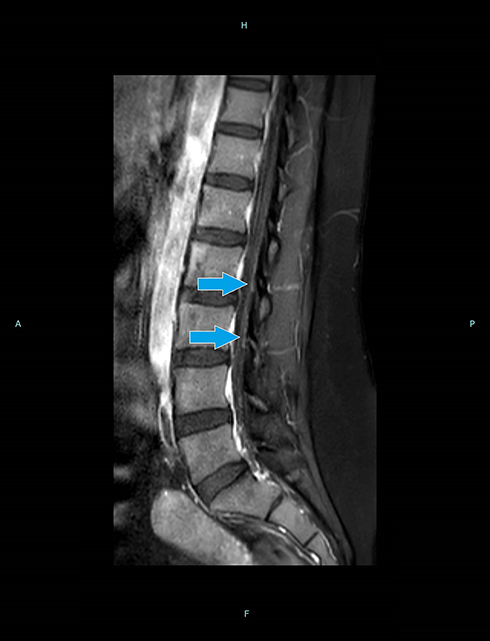 |
Figure 1 Lumbar MRI T1W image with contrast (sagittal view) demonstrating enhancement of multiple nerve roots (blue arrows). |
Symptoms peaked two weeks from onset. A diagnosis of sensory GBS was made based on clinical presentation, supportive investigations, and the exclusion of alternative diagnoses. The patient received intravenous immunoglobulin “IVIG” 0.4 gm/kg daily for five days. At discharge, she had a GBS disability score of 4 (bedridden or chair bound). On follow up six weeks later she reported improvement in numbness and unsteadiness. She could stand unsupported with her eyes open but fell if they were closed and could not walk independently. On examination, her dysmetria showed improvement but was still worsened with eye closure. Improvement was noted in all sensory modalities and the pseudo-athetoid movements. Repeat NCS was similar (Table 1). EMG was remarkable for minimal denervation activity in the right anterior tibialis, gastrocnemius, and lower lumbar paraspinal muscles (Table 2). Clinical examination six months after admission demonstrated significant improvement in pinprick and temperature sensation, and proprioception impairment was limited to fingers and toes. She walked independently. Dysmetria was mild but still exacerbated with eye closure. Trace deep tendon reflexes reemerged. She had a GBS disability score of two (able to walk 10 meters or more without assistance but unable to run). Further improvement was evident at her eight-month follow up. EMG studies performed at months six and eight demonstrated persistence of minimal denervation in several upper and lower extremity muscles (Table 2).
Discussion
The scarcity and heterogeneity of cases has made it difficult to classify pure sensory GBS or establish universal diagnostic criteria for the condition.4,5 This scarcity is due to most cases with predominant sensory symptoms also manifesting some weakness clinically hence they are ultimately excluded. Also, there is significant clinical and neurophysiological overlap between pure sensory GBS and acute sensory neuronopathy, and only few cases have undergone nerve biopsy or postmortem examination to establish their diagnoses.4,13 The first diagnostic criteria for a sensory loss and areflexia variant of GBS were proposed by Asbury in 1981.1 They included a rapid onset, widespread and symmetrical symptoms, near complete or total recovery, albuminocytological dissociation in the CSF, and NCS findings consistent with demyelination. An updated set of criteria proposed by Oh et al, in 2001 also necessitated evidence of demyelination in at least two peripheral nerves on NCS.14 However, demonstration of this criterion cannot be always fulfilled as the pathophysiology of some cases apparently involves axonal damage rather than demyelination. Also, the F-wave study, an important marker frequently used to detect demyelination, depends on identifying abnormalities in motor conduction so it may be normal in some cases with a motor-sparing pattern on NCS.4
In their attempt to resolve some of the uncertainty surrounding pure sensory GBS, Uncini and Yuki classified cases into three separate groups based on the possible site of primary damage: Acute Sensory Demyelinating Polyneuropathy, Acute Sensory Small-Fiber Neuropathy–Ganglionopathy, and Acute Sensory Large-Fiber Axonopathy–Ganglionopathy.4 Out of 13 cases in the latter cohort, an antecedent infection was documented in 11 patients, of which none were a primary VZV infection. Albuminocytological dissociation was demonstrated in eight of the 13 cases. Patients in this cohort presented with numbness, paresthesia, gait unsteadiness, and loss of deep tendon reflexes, and a subset of patients within this group manifested sensory ataxia, impaired vibration sensation, positive Romberg’s sign, and reduced or absent Sensory Nerve Action Potential “SNAP” with normal motor conduction on NCS. This subset was termed “acute sensory ataxic neuropathy (ASAN)”. The clinical and electrophysiological features of our case are highly consistent with ASAN.
NCS findings in cases with sensory GBS are diverse.4,7,13–18 Classical demyelinating features such as increased SNAP latency, slow conduction velocity, temporal dispersion, and/or delayed or absent F-waves, with or without motor involvement, have been demonstrated in many cases. Less frequently, other cases have demonstrated markedly low-amplitude or absent SNAPs with normal motor studies and F-wave latencies, suggesting axonal injury.4
On the other hand, EMG studies were reported infrequently in sensory GBS cases. In our literature review, we identified nine cases where EMG was performed once at presentation and reported normal,14,16 but a single case reported by Miralles et al in 1992 was exceptional in that initial EMG was normal but a follow up study was performed a month after discharge and showed changes compatible with minimal denervation.19 Denervation potentials appear 1–4 weeks after nerve injury and therefore they are not expected to be seen during EMG performed at presentation.20 In our case, follow up EMG performed two months after presentation revealed changes suggestive of denervation in different segments, which persisted six and eight months later despite evident clinical improvement. Oh et al, argued that absent SNAP and normal motor NCS indicate sensory neuronopathy rather than peripheral neuropathy.14 The denervation potentials on follow up EMG in our case and the case by Miralles signify that a peripheral neuropathy was the underlying pathophysiology for the clinical presentation, and strongly disfavor sensory neuronopathy as motor fibers should be spared in the latter.
Pathologically, it has been proposed that some forms of pure sensory GBS overlap with and may represent an incomplete form of Miller Fisher Syndrome “MFS”.3,4,21 This suggestion is prompted by clinical similarities between the two conditions and the detection of ganglioside antibodies (GM1, GQ1b, GD1b) in some cases with pure sensory GBS.4,8,15 However, these antibodies were demonstrated only in a few sensory GBS cases. Also, pure sensory GBS cases are very rare and heterogenous which limits the scope for clinical comparisons with MFS. In addition, several previous reports have proposed evidence of direct cerebellar involvement in Miller Fisher syndrome, adding to the ambiguity surrounding this debate which remains unsettled.22,23
The most important differential diagnosis of pure sensory GBS is sensory neuronopathy (ganglionopathy). Several findings in our case make this diagnosis much less likely. Sensory involvement in acute sensory neuronopathy is usually universal and involves the face and scalp.4,24,25 Pain and dysesthesia can be prominent with sensory neuronopathy, and progression subacute or chronic.24,25 The GBS disability score assesses the functional status of patients with GBS, with scores ranging from 0 (normal) to 6 (death).26 Our patient had a favorable outcome (defined as GBS disability score at six months of two or less), while recovery in sensory neuronopathy is usually poor and response to IVIG is much less frequent.24,25 Additionally, the various autoimmune, paraneoplastic, drugs and toxic conditions that may predispose for sensory neuronopathy were excluded by the lack of suggestive history and the absence of their clinical, laboratory and radiological markers. Post-varicella neuronopathy and post-varicella radiculitis are late complications. They usually present unilaterally and affect a single or very few levels, manifesting as dermatomal pain and vesicular eruption (post-herpetic neuralgia) years after the primary infection and therefore unlikely to be related to our patient’s presentation.27,28 Also, radiculitis may be associated with weakness unlike our case.29 Finally, as stated above, the denervation potentials seen on follow up EMG indicate subclinical involvement of motor axons, therefore they exclude sensory neuronopathy.
Another differential is peripheral neuropathy due to causes such as toxic, metabolic, paraneoplastic, autoimmune, hereditary diseases, and drugs, which were excluded in the same manner as sensory neuronopathy. Also, although ataxia after a primary VZV infection can be due to cerebellitis, the ataxia in our patient was proportional to the severe sensory deficit, and it was significantly accentuated by eye closure, which is the hallmark characteristic of sensory ataxia. (Additionally) Cerebellar imaging was unremarkable.
On the other hand, the acute and monophasic course, progression to nadir within two weeks, the demonstration of CSF albuminocytological dissociation, root enhancement on MRI, and response to IVIG are all characteristic of GBS. Additionally, the difference in sensory impairment noted between the anterior and posterior trunk indicates a length-dependent process. Alternative diagnoses were reasonably excluded, including sensory ganglionopathy, post-varicella radiculitis and cerebellitis, peripheral neuropathies due to other causes, and disorders of the spinal cord. The different clinical criteria proposed by Asbury and by Oh were ultimately fulfilled. The authors believe that the neurophysiological insights of this case reflect how the different proposed criteria are still having difficulty encompassing the different aspects and presentations of the pure sensory GBS variant.
Conclusions
Early recognition of pure sensory GBS is of paramount importance as it carries a better prognosis than sensory neuronopathy, and since early treatment with IVIG can result in good outcome in sensory GBS. To our knowledge, this is the first case with onset after a documented primary VZV infection. We also feature the clinical and neurophysiological outcomes on longitudinal follow up of a severe case. Finally, we believe that repeating EMG studies can help differentiate difficult cases as detecting denervation potentials on follow up EMG indicates peripheral neuropathy rather than sensory neuronopathy. We also note that clinical improvement does not correlate with electrophysiologic improvement on EMG/NCS.
Ethics Approval and Informed Consent
IRB approval is not required by the institution for case reports where anonymization of data is guaranteed. The patient has provided informed consent for the case details and accompanying images to be published.
Acknowledgments
The authors would like to acknowledge Dr. Wael M. Marashdeh from the Department of Diagnostic Radiology and Nuclear Medicine for his valuable contribution to this report.
Disclosure
All the authors declare that they have no conflicts of interest in this work.
References
1. Asbury AK. Diagnostic considerations in Guillain-Barre syndrome. Ann Neurol. 1981;9(Suppl):1–5. doi:10.1002/ana.410090703
2. Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31(2):491–510. doi:10.1016/j.ncl.2013.01.005
3. Sejvar JJ, Kohl KS, Gidudu J, et al. Guillain–Barré syndrome and fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011;29(3):599–612. doi:10.1016/j.vaccine.2010.06.003
4. Uncini A, Yuki N. Sensory Guillain-Barré syndrome and related disorders: an attempt at systematization. Muscle Nerve. 2012;45(4):464–470. doi:10.1002/mus.22298
5. Leonhard SE, Mandarakas MR, Gondim FAA, et al. Diagnosis and management of Guillain–Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671–683. doi:10.1038/s41582-019-0250-9
6. Neuritis, sensory neuritis, neuralgia: a Clinical Study with review of the Literature. J Am Med Assoc. 1958;167(8):1052. doi:10.1001/jama.1958.02990250124023
7. Dawson DM, Samuels MA, Morris J. Sensory form of acute polyneuritis. Neurology. 1988;38(11):1728. doi:10.1212/WNL.38.11.1728
8. Wakerley BR, Kokubun N, Funakoshi K, Nagashima T, Hirata K, Yuki N. Clinical classification of 103 Japanese patients with Guillain-Barre syndrome. J Neurol Sci. 2016;369:43–47. doi:10.1016/j.jns.2016.08.002
9. Kalita J, Misra UK, Goyal G, et al. Guillain-Barre syndrome: subtypes and predictors of outcome from India. J Peripher Nerv Syst. 2014;19(1):36–43. doi:10.1111/jns5.12050
10. Yadegari S, Nafissi S, Kazemi N. Comparison of electrophysiological findings in axonal and demyelinating Guillain-Barre syndrome. Iran J Neurol. 2014;13(3):138–143.
11. Gibbels E, Giebisch U. Natural course of acute and chronic monophasic inflammatory demyelinating polyneuropathies (IDP). A retrospective analysis of 266 cases. Acta Neurol Scand. 1992;85(4):282–291. doi:10.1111/j.1600-0404.1992.tb04045.x
12. Islam B, Islam Z, GeurtsvanKessel CH, et al. Guillain-Barré syndrome following varicella-zoster virus infection. Eur J Clin Microbiol Infect Dis. 2018;37(3):511–518. doi:10.1007/s10096-018-3199-5
13. Yang J, Huan M, Jiang H, et al. Pure sensory Guillain-Barré syndrome: a case report and review of the literature. Exp Ther Med. 2014;8(5):1397–1401. doi:10.3892/etm.2014.1955
14. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain–Barré syndrome. Neurology. 2001;56(1):82–86. doi:10.1212/WNL.56.1.82
15. Bashtawi M, Abuabada A, Aldabbour B. Somatic cough syndrome: a report of two cases and review of literature. Egypt J Neurol Psychiatry Neurosurg. 2020;56(1):83. doi:10.1186/s41983-020-00215-x
16. Lee -S-S, Lee S-H. Does sensory Guillain–Barré syndrome exist without any abnormalities in motor nerve conduction? Neurology 2006;66(6):947–948.
17. Semproni M, Gibson G, Kuyper L, Tam P. Unusual sensory variant of Guillain-Barré syndrome. BMJ Case Rep. 2017;2017;bcr–2016.
18. Kalita J, Misra UK, Das M. Neurophysiological criteria in the diagnosis of different clinical types of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2008;79(3):289–293. doi:10.1136/jnnp.2007.118000
19. Miralles F, Montero J, Reñe R, Matos JM. Pure sensory Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 1992;55(5):411–412. doi:10.1136/jnnp.55.5.411-a
20. Feinberg J. EMG: myths and facts. HSS J. 2006;2(1):19–21. doi:10.1007/s11420-005-0124-0
21. Ito M, Matsuno K, Sakumoto Y, et al. Ataxic Guillain-Barre syndrome and acute sensory ataxic neuropathy form a continuous spectrum. J Neurol Neurosurg Psychiatry. 2011;82(3):294–299. doi:10.1136/jnnp.2010.222836
22. Kim YK, Kim JS, Jeong S-H, et al. Cerebral glucose metabolism in Fisher syndrome. J Neurol Neurosurg Psychiatry. 2009;80(5):512–517. doi:10.1136/jnnp.2008.154765
23. Kornberg AJ, Pestronk A, Blume GM, et al. Selective staining of the cerebellar molecular layer by serum IgG in Miller-Fisher and related syndromes. Neurology. 1996;47(5):1317–1320. doi:10.1212/WNL.47.5.1317
24. Kuntzer T, Antoine J-C, Steck AJ. Clinical features and pathophysiological basis of sensory neuronopathies (ganglionopathies). Muscle Nerve. 2004;30(3):255–268. doi:10.1002/mus.20100
25. Martinez AR, Nunes MB, Nucci A, França MC. Sensory neuronopathy and autoimmune diseases. Autoimmune Dis. 2012;2012:873587.
26. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical prognostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589–594. doi:10.1016/S1474-4422(07)70130-8
27. Bennett GJ, Watson CP. Herpes zoster and postherpetic neuralgia: past, present and future. Pain Res Manag. 2009;14(4):275–282. doi:10.1155/2009/380384
28. Drazin D, Hanna G, Shweikeh F, et al. Varicella-zoster-mediated radiculitis reactivation following cervical spine surgery: case report and review of the literature. Case Rep Infect Dis. 2013;2013:647486.
29. Claflin B, Thomas M, Wilson AJ. Polyradiculopathy and herpes zoster. Proc (Bayl Univ Med Cent). 2009;22(3):223–225. doi:10.1080/08998280.2009.11928521
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.