")
Back to Journals » International Medical Case Reports Journal » Volume 16
A Rare Case Report of Neonatal Neurofibromatosis Type 1 Presented with Giant Faciocervical Mass and Complicated with HIE
Authors Liu B, Wang W , Bi J , Huo R
Received 10 November 2023
Accepted for publication 8 December 2023
Published 15 December 2023 Volume 2023:16 Pages 833—839
DOI https://doi.org/10.2147/IMCRJ.S446981
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Boce Liu,1 Wenjing Wang,1 Jianhai Bi,2 Ran Huo1,2
1Department of Plastic Surgery, Shandong Provincial Hospital, Shandong University, Jinan, People’s Republic of China; 2Department of Plastic Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, People’s Republic of China
Correspondence: Ran Huo, Department of Plastic Surgery, Shandong Provincial Hospital, Shandong University, Jinan, Shandong, People’s Republic of China, Tel +86-15168889001, Fax +86-0531-68778153, Email [email protected]
Abstract: A newborn with giant faciocervical mass and presented with asphyxia during birth was admitted to the hospital. After stabilizing her vital sign, we provided the patient with image examinations and whole-exome sequencing, which revealed a heterozygous variation of neurofibromatosis type 1 (NF1). The final diagnosis of the patient was NF1 complicated with neonatal hypoxic-ischemic encephalopathy (NHIE). During hospitalization, the patient received comprehensive and systematic care. There was no reports of similar cases in the literature. So, this report aimed to elucidate the special clinical manifestations, diagnosis, treatment and prognosis of NF1 complicated with NHIE by analyzing the clinical data of the patient and her family and reviewing relevant literature.
Keywords: neurofibromatosis type 1, neonatal hypoxic-ischemic encephalopathy, NF1 gene variation, whole-exome sequencing
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disease caused by mutations of the NF1 gene that can affect the entire nervous system and manifest as neurocutaneous syndromes, including café-au-lait macules (CALMs), skin and subcutaneous neurofibromas, malignant tumors of the central and peripheral nervous systems, and rare manifestations such as learning disabilities and skeletal system abnormalities.1,2 Neonatal hypoxic-ischemic encephalopathy (NHIE) refers to brain damage in fetuses and infants caused by various perinatal factors such as hypoxia and reduced or suspended cerebral blood flow. NHlE is a common acute and critical illness during the neonatal period and an important cause of early neonatal death and intellectual developmental disorders in children.3 Patient’s birth difficulties led to NHIE. There was a huge mass in the faciocervical region at birth. The mass enlarged and caused compression symptoms including difficult breathing. The final diagnosis was NF1 complicated with NHIE based on the patient’s medical history, image examinations and whole-exome sequencing. The patient received comprehensive and systematic care. This case suggests that NF1 has multiple clinical manifestations and requires multidisciplinary management as well as social and psychological interventions. Therefore, it is necessary to strengthen the understanding of physicians and attach great importance to prenatal screening.
Case Report
A female infant was delivered via a perineal lateral incision at local hospital at 39 weeks plus 1 day of gestation because of difficulties with vaginal delivery. She presented with asphyxia after birth. The computed tomography (CT) examination results showed a decrease in the cerebral white matter density in both the frontal and parietal lobes (CT value of 12 HU). The ventricles of the brain were slightly widened, the sulcus fissure was widened, and the midline structure was centered. Soft tissues of the patient’s head, jaw, and neck were swollen. These findings were consistent with the manifestations of NHIE observed using CT. The infant was born with a 10-cm × 5-cm mass in the faciocervical region and had slightly pale skin that was tough to the touch. NHIE, facial developmental malformations, and occipital hematoma were diagnosed at the local hospital. The patient was transferred to the neonatal intensive care unit after birth. After treatment, all vital signs remained stable. She was transferred to our department at age 28 days because of the faciocervical mass (Figure 1A). Compression symptoms manifesting as coughing while eating and obvious snoring while sleeping occurred with enlargement of the mass. Brown spots of varying sizes were scattered on the surface of the mass (Figure 1B). More than six CALMs were visible throughout the patient’s body. Color Doppler ultrasound revealed a diffuse solid hypoechoic nodule around the left parotid gland and left neck with unclear boundaries, irregular morphology, and uneven internal echoes (Figure 2). Rich arterial and venous blood flow signals were detected within the mass and accounted for 30% of the entire mass area. The subcutaneous fat layer in the neck was diffusely thickened, and the soft tissue structure was disrupted by the neck mass pressing forward against the left common carotid artery. The volume of the left parotid gland increased slightly, and its structure was plump.
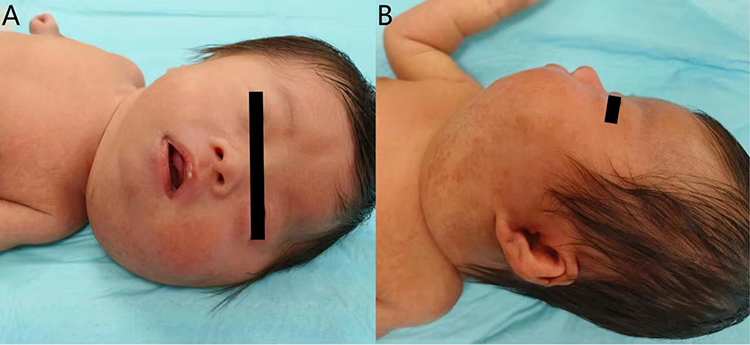 |
Figure 1 Lesional pictures of the patient. (A)The faciocervical mass was about 10-cm × 5-cm in size with a slightly pale surface and a hard texture to the touch. (B)Brown spots of varying sizes were scattered on the surface of the mass. |
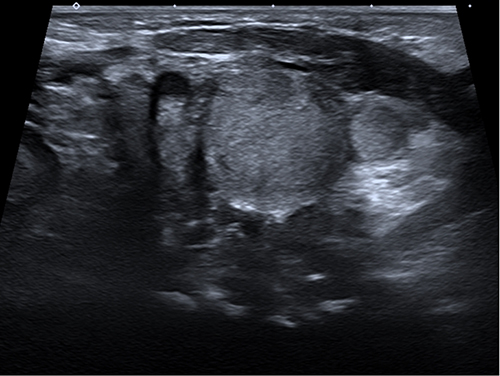 |
Figure 2 Ultrasound image of the patient. Ultrasound revealed a diffuse solid hypoechoic nodule around the left parotid gland and left neck with unclear boundaries, irregular morphology, and uneven internal echoes. |
During pregnancy, the mother of the child underwent genetic testing to determine whether there were abnormalities in trisomy 21, trisomy 18, and trisomy 13 in the fetus. A high-throughput sequencing analysis of free fetal DNA in the maternal peripheral blood revealed a low probability of disease. As a result of an examination during the second month of pregnancy, the mother was diagnosed with hypothyroidism; therefore, she received oral administration of sodium levothyroxine during pregnancy. The child’s paternal lineage has familial intellectual disability (Figure 3). Whole-exome sequencing of the patient and her parents was performed, and a heterozygous variation of the NF1 gene of the patient (Table 1), which was a de novo variant that was not detected in her patients, was found. The chromosomal position of the NF1 gene variation was Chr17:29527570_ 29527571, and the nucleic acid changed to c.1019_ 1020del. Further Sanger sequencing confirmed that the patient’s NFl gene underwent mutations (Figure 4). According to the American College of Medical Genetics and Genomics guidelines (2015), this mutation of NF1 was pathogenic and associated with NF1, neurofibromatosis-Noonan syndrome, familial spinal neurofibromatosis, Watson syndrome, and juvenile myelomonocytic leukemia. Based on the patient’s medical history, clinical manifestations, imaging examination results, and genetic testing results, a preliminary diagnosis of NF1 could be made. However, the final diagnosis should be based on the pathological examination results. During hospitalization, the patient underwent a comprehensive and systematic examination and essential care. The vital signs of the child were stable, and there were no neurological sequelae such as convulsions or seizures. Because the patient had NHIE, we recommended careful observation and early rehabilitation. Because of the abundant blood supply to the facial and cervical lesions, low body weight, and poor nutritional status of the patient, surgery was considered risky. Therefore, the patient’s parents refused surgery, and careful observation and follow-up were performed.
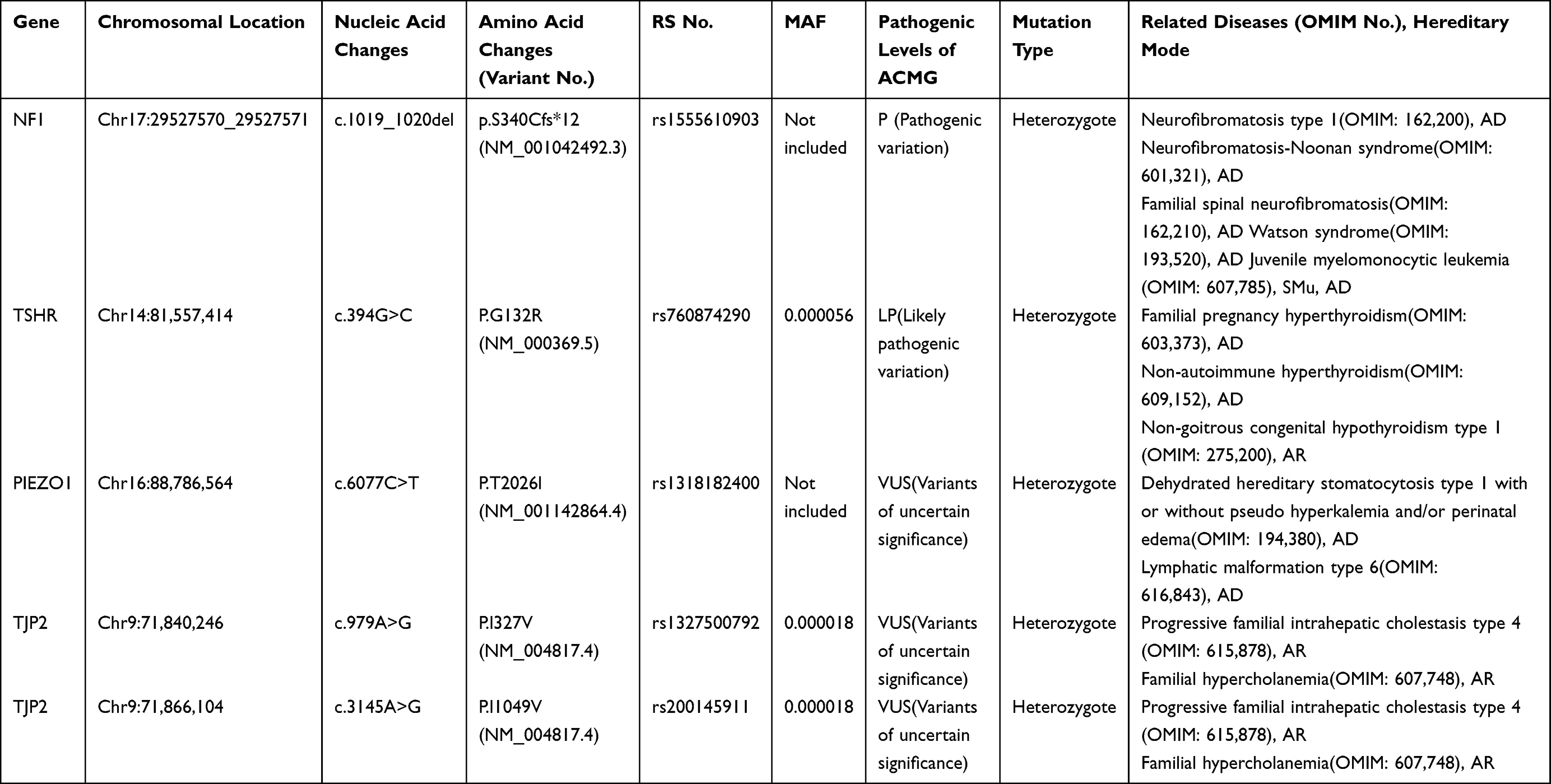 |
Table 1 We Identified Five Gene Mutations in NF1, TSHR, PIEZO1 and TJP2 Through the Whole-Exome sequencing. A Heterozygous Variation of the NF1 Gene of the Patient Was Found, Which Was a de Novo Variant That Was Not Detected in Her Patients. The Chromosomal Position of the NF1 Gene Variation Was Chr17:29527570_ 29527571, and the Nucleic Acid Changed to C.1019_ 1020del |
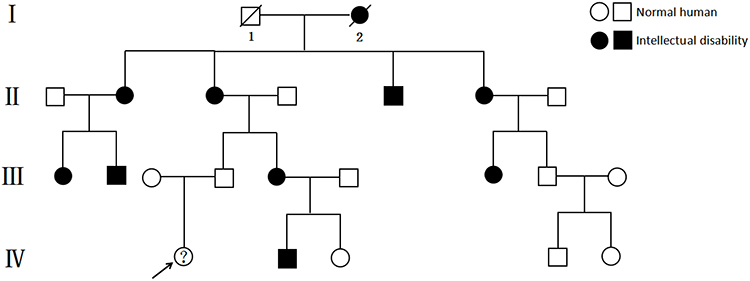 |
Figure 3 This is the familial intellectual disability genetic genealogy. |
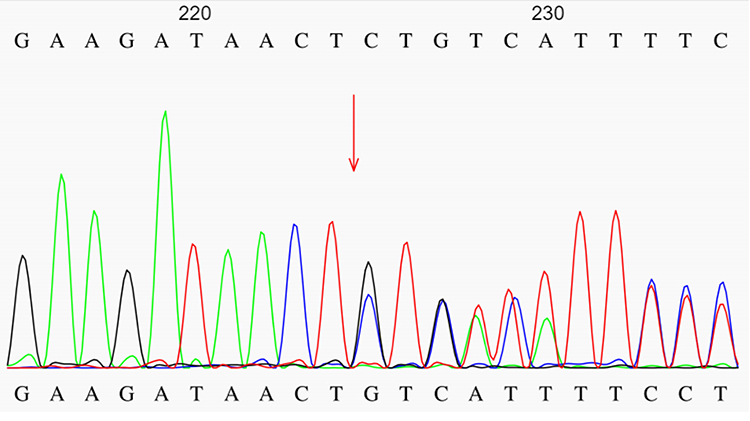 |
Figure 4 Sanger sequencing confirmed that the patient’s NFl gene underwent mutations. The red arrow indicated the NF1 gene mutation site. |
Discussion
NHIE, also known as perinatal asphyxia, is a disease characterized by clinical and laboratory evidence of acute or subacute brain injury related to perinatal hypoxic-ischemic events.4 NHIE is the leading cause of perinatal mortality and cerebral palsy worldwide, with two to three cases occurring in every 1000 newborns.4–6 Brain damage caused by NHIE is mainly caused by acute or chronic hypoxic-ischemic events, which lead to metabolic brain dysfunction and subsequent cell death.4 The diagnostic rates of neonatal brain injury using CT, magnetic resonance imaging, and transcranial Doppler ultrasound are high; therefore, they can effectively ascertain early diagnosis, assess severity, and determine the prognosis of neonatal brain injury.3,7 If not diagnosed and treated in a timely manner, then NHIE can lead to neurological sequelae, including behavioral abnormalities, cognitive impairment and intellectual impairment. Severe cases are associated with the risks of cerebral palsy and death. Neurological sequelae cause heavy economic and mental burdens on families and society; therefore, the diagnosis and treatment of NHIE should be expanded to improve the prognosis and reduce mortality.
NF1 is an autosomal dominant inherited disease of the nervous system caused by mutations of the NF1 gene, and its global incidence rate is approximately 1 in 3000.8 Approximately 50% of patients have familial genetic mutations, and the others have sporadic mutations.9 NF1 is also known as von Recklinghausen disease. Typical clinical symptoms of NF1 include CALMs, multiple neurofibromas, and axillary or inguinal freckles; among these symptoms, neurofibromas are the most common and characteristic of NF1.8 The clinical symptoms of this disease vary greatly and can cause external damage and dysfunction, increase the risk of tumor malignancy, seriously affect the quality of life of patients, and cause psychological and economic burdens.10 In 2021, the International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC) proposed the following revisions to the 1987 diagnostic standard for NF1 that incorporated the genetic diagnosis: six or more CALMs with diameters larger than 5 mm before puberty or 15 mm after puberty; two or more neurofibromas of any type or one plexiform neurofibroma (pNF); freckles in the armpit or groin area; optic pathway glioma; slit-lamp examination revealing two or more Lisch nodules (iris hamartomas); optical coherence tomography/near-infrared imaging revealing two or more choroidal abnormalities; characteristic bone lesions, including developmental dysplasia of the sphenoid bone, anterior-lateral curvature of the tibia, or pseudarthrosis of the long bone; and pathogenic heterozygous NF1 variants with an allele variant score of up to 50% in normal tissues (such as white blood cells).11,12 For individuals without a history of parental illness, the presence of two or more clinical characteristics can lead to a diagnosis of NF1. Individuals with a history of parental illness and one or more clinical features can be diagnosed with NF1. If the patient only has CALMs and freckles in the axillary or inguinal area, then the possibility of Legius syndrome should be considered simultaneously with NF1, especially for patients with bilateral freckles.
This consensus standard has high specificity and sensitivity for adult NF1, and only approximately half of the pediatric patients who develop NF1 before age 1 year and do not have a family history that meets this standard.13,14 Therefore, the American Academy of Pediatrics has summarized several suggestions regarding genetic testing for NF1 in children.15 Genetic testing can confirm a suspected diagnosis before the clinical diagnosis is confirmed (or before the appearance of the second clinical feature). Gene detection can distinguish between NF1 and Legius syndrome which is associated with SPRED1. Genetic testing may help screen for atypical features such as isolated pNF, optic pathway glioma, and tibial dysplasia in children. For pediatric patients, the current clinical diagnostic standards have low sensitivity, and physical signs gradually appear with age. Genetic testing can be performed to diagnose NF1 as early as possible. Currently, there is no direct treatment for NF1; the most important intervention measures are early diagnosis and symptomatic treatment.
First-line treatments include surgical resection and carbon dioxide laser ablation, which is especially effective for small neurofibromas.16 Other methods include laser photocoagulation and radiofrequency ablation. Before surgical intervention is performed, the patient’s condition, potential postoperative pain, secondary psychological disorders that can be caused by postoperative changes in appearance, progressive neurological symptoms, and risk of permanent defects should be considered. Currently, multiple clinical drug trials related to NF1 are being conducted globally, and their results regarding safety and efficacy will require further investigation. A Phase II clinical trial of selumetinib conducted in 2020 showed that 35 (70%) children with pNF who were not suitable for surgical resection experienced a sustained tumor reduction of 20% or more.17 As a result, in April 2020, the Food and Drug Administration approved this drug to treat symptomatic and/or progressive pNF in patients 3 years or older who cannot undergo surgery.
NF1 involves multidisciplinary clinical manifestations and requires management as well as social and psychological interventions. Furthermore, it is necessary to strengthen the understanding of the disease among physicians. Primary healthcare is particularly important for tracking and observing disease development in children with NF1. For pediatric patients with NF1, regular health examinations and observations should be conducted at all stages of growth to assess the impact of disease on their health, personal growth, and behavior. Reports of NF1 cases complicated by NHIE are rare; however, we believe that such cases are inevitable. For example, prenatal ultrasound examinations can clarify the placental position, umbilical cord condition, fetal position, amniotic fluid condition, amniotic fluid contamination or reduction, umbilical cord entanglement around the neck or trunk, and fetal distress, which may cause hypoxia and ischemia. The importance of prenatal examinations should be emphasized because the results can be used to plan the delivery process, estimate the possible situations that could occur during the delivery process, and prepare for and manage the outcomes accordingly, thereby minimizing the impact on the fetus and newborn. The application of noninvasive prenatal genetic testing and other technologies provide a new diagnostic basis for the screening of fetal anomalies, which can help strengthen the physicians’ understanding of the patients’ conditions and allow for comprehensive treatment.
Conclusion
NF1 involves multidisciplinary clinical manifestations, seriously affecting the quality of life of patients. Its treatment is the main difficulty currently. NHlE is a common and serious illness during the perinatal period. The patients in this report was given observation therapy and received short-term follow-up. This article aims to raise the awareness of clinical physicians among two diseases and provide patients and guardians more disease education. Early diagnosis and intervention can effectively prevent disease progression and reduce economic burden.
Ethics Approval and Informed Consent
The patient’s parents have signed the informed consent and agreed to publish it. They gave their consent for the publication of identifiable details, which can include photographs and/or case history and/or details within the text to be published in the article. They confirmed that they had seen and been given the opportunity to read the article.
Consent for Publication
Approval for the publication of the patient’s case details was obtained from Shandong Provincial Hospital.
Acknowledgments
We would like to acknowledge the patient and her parents who participated in the test and we are grateful for their kind contributions.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nature Rev Dis Primers. 2017;3(1):1–7.
2. Garcia B, Catasus N, Ros A, et al. Neurofibromatosis type 1 families with first-degree relatives harbouring distinct NF1 pathogenic variants. Genetic counselling and familial diagnosis: what should be offered? J Med Genet. 2022;59(10):1017–1023. doi:10.1136/jmedgenet-2021-108301
3. Krishnan P, Shroff M. Neuroimaging in Neonatal Hypoxic Ischemic Encephalopathy. Indian J Pediatr. 2016;83(9):995–1002. doi:10.1007/s12098-016-2042-1
4. Paul SP, Abdelrhim H, Heep A. Management of Hypoxic-ischemic Encephalopathy. Indian J Pediatr. 2015;82(6):493–496. doi:10.1007/s12098-014-1592-3
5. Yıldız EP, Ekici B, Tatlı B. Neonatal hypoxic ischemic encephalopathy: an update on disease pathogenesis and treatment. Expert Rev Neurother. 2017;17(5):449–459. doi:10.1080/14737175.2017.1259567
6. Cerio FG, Lara-Celador I, Alvarez A, Hilario E. Neuroprotective therapies after perinatal hypoxic-ischemic brain injury. Brain Sci. 2013;3(1):191–214. doi:10.3390/brainsci3010191
7. Liu JX, Fang CL, Zhang K, et al. Transcranial Doppler Ultrasonography detection on cerebrovascular flow for evaluating neonatal hypoxic-ischemic encephalopathy modeling. Front Neurosci. 2023;17:962001. doi:10.3389/fnins.2023.962001
8. Liu Y, Zhang Z, Liang M, Liu Y, Zhang N, Xu K. Comprehensive imaging analysis of a patient with neurofibromatosis type 1 combined with hypophosphatemic osteomalacia: a case description. Quant Imaging Med Surg. 2023;13(7):4777–4784. doi:10.21037/qims-22-1217
9. Farschtschi S, Mautner VF, McLean ACL, Schulz A, Friedrich RE, Rosahl SK. The Neurofibromatoses. Dtsch Arztebl Int. 2020;117(20):354–360. doi:10.3238/arztebl.2020.0354
10. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–843. doi:10.1016/S1474-4422(14)70063-8
11. Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021;23(8):1506–1513. doi:10.1038/s41436-021-01170-5
12. NIH. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172–178.
13. Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol. 2013;115:939–955. doi:10.1016/B978-0-444-52902-2.00053-9
14. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 Pt 1):608–614. doi:10.1542/peds.105.3.608
15. Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR. Health Supervision for Children with Neurofibromatosis Type 1. Pediatrics. 2019;143(5):e20190660. doi:10.1542/peds.2019-0660
16. Bergqvist C, Servy A, Valeyrie-Allanore L, et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020;15(1):37. doi:10.1186/s13023-020-1310-3
17. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020;382(15):1430–1442. doi:10.1056/NEJMoa1912735
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.