Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 6
A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban
Authors Frost C, Song Y, Barrett YC, Wang J, Pursley J, Boyd RA, LaCreta F ![]()
Received 22 January 2014
Accepted for publication 17 September 2014
Published 13 November 2014 Volume 2014:6 Pages 179—187
DOI https://doi.org/10.2147/CPAA.S61131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Arthur E. Frankel
Charles Frost,1 Yan Song,1 Yu Chen Barrett,1 Jessie Wang,2 Janice Pursley,3 Rebecca A Boyd,4 Frank LaCreta1
1Exploratory Clinical and Translational Research, 2Exploratory Development Global Biometric Sciences, 3Analytical and Bioanalytical Development, Bristol-Myers Squibb Company, Princeton, NJ, USA; 4Global Innovative Pharma Business Clinical Pharmacology, Pfizer Inc., Groton, CT, USA
Background: Currently, there are no direct comparisons of apixaban and rivaroxaban, two new oral direct factor Xa inhibitors approved for management of thromboembolic disorders.
Objective: Compare the pharmacokinetics and anti-factor Xa activity (AXA) of apixaban and rivaroxaban.
Methods: In this randomized, open-label, two-period, two-treatment crossover study, healthy subjects (N=14) received apixaban 2.5 mg twice daily (BID) and rivaroxaban 10 mg once daily (QD) for 4 days with a ≥4.5-day washout. Plasma samples were obtained for pharmacokinetic and AXA assessments; parameters were calculated using noncompartmental methods.
Results: Median time-to-maximum concentration was 2 hours for both compounds, and the mean half-life was 8.7 and 7.9 hours for apixaban and rivaroxaban, respectively. Daily exposure, the area under the curve (AUC(0–24)), appeared similar for rivaroxaban (1,094 ng · h/mL) and apixaban (935 ng · h/mL), whereas mean peak-to-trough plasma concentration ratio was 3.6-fold greater for rivaroxaban (16.9) than apixaban (4.7). Coefficient of variation for exposure parameters (AUC0–24, Cmax, Cmin) was 20%–24% for apixaban versus 29%–46% for rivaroxaban. Peak AXA, AXA AUC(0–24), and AXA fluctuation were ~2.5-, 1.3-, and 3.5-fold higher for rivaroxaban than apixaban, respectively. Trough concentrations and AXA were lower for rivaroxaban (10 ng/mL and 0.17 IU/mL vs 17 ng/mL and 0.24 IU/mL for apixaban, respectively). Rivaroxaban exhibited a steeper concentration–AXA response (slope: 0.0172 IU/ng vs 0.0134 IU/ng for apixaban, P<0.0001).
Conclusion: Apixaban 2.5 mg BID demonstrated less intersubject variability in exposure, lower AXA AUC, and higher trough and smaller peak-to-trough fluctuations in plasma concentration and AXA, suggesting more constant anticoagulation compared with rivaroxaban 10 mg QD. However, the clinical impact of these differences on the relative efficacy and safety of apixaban and rivaroxaban remains to be determined.
Keywords: apixaban, pharmacodynamics, pharmacokinetics, rivaroxaban, safety
A letter to the Editor has been received and published for this article.
Corrigendum for this paper has been published.
Introduction
Apixaban and rivaroxaban are oral direct reversible factor Xa (FXa) inhibitors approved for the prevention of several thromboembolic disorders. Based on the current literature, the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of these two FXa inhibitors share some general features.1,2 The bioavailability of apixaban is ~50% and is not significantly influenced by dose or administration with meals.3 Rivaroxaban bioavailability ranges from 66%–100%, depending on the dose and whether or not the dose is administered with a meal. Rivaroxaban requires administration with food to achieve similar bioavailability between a 10 mg dose and doses ≥15 mg.4 Both agents are readily absorbed, achieving maximal plasma concentrations ~3 hours after administration. Protein binding is similar for the two compounds (apixaban ~87%, rivaroxaban ~93%). Apixaban and rivaroxaban also share similar pathways of elimination including metabolism, mainly by cytochrome P450 3A4 (CYP3A4), as well as biliary and renal elimination, and have relatively similar terminal half-lives (apixaban ~12 hours, rivaroxaban ~10 hours). Despite these similar PK properties, these agents are administered differently. Rivaroxaban is administered once daily (QD), twice daily (BID), or a combination of QD and BID, depending on the indication, whereas apixaban is administered BID for all indications. Although individual literature reports are helpful to form a general understanding of their relative PK and PD profiles, no direct comparisons of these agents have been performed in humans. Such a comparison could help elucidate how their PK and PD properties, along with their posology, relate to observed clinical outcomes, and may aid in the transition between therapies. The purpose of this study was to compare the PK and PD (anti-factor Xa activity [AXA]) profiles of apixaban (2.5 mg BID) and rivaroxaban (10 mg QD) in healthy subjects at the doses and schedules approved for prevention of venous thromboembolism (VTE) following elective knee or hip replacement surgery.5,6
Methods
Study design
This study (EudraCT 2009-017278-19) was a randomized, open-label, two-period, two-treatment crossover study in 14 healthy subjects. Primary objectives were to assess the multiple-dose, steady-state PK profiles and compare plasma concentration peak-to-trough ratios (Cmax/Cmin) of rivaroxaban 10 mg QD with those of apixaban 2.5 mg BID following oral administration in healthy subjects. Secondary objectives included assessment of the multiple-dose, steady-state AXA profiles and safety and tolerability of these agents. The study was carried out in compliance with the International Conference on Harmonisation guidelines for Good Clinical Practice and in accordance with the principles of the Declaration of Helsinki, including all amendments in effect up to the time the study was conducted. Study protocol and informed consent forms were reviewed and approved by an independent ethical committee (Stichting Beoordeling Biomedisch Ethiek Onderzoek, Assen, the Netherlands), and all parts of the study were in compliance with the EU Clinical Trial Directive (EU CTD) 2001/20/EC.
Screening was carried out within 21 days prior to study drug administration and all subjects provided written informed consent prior to the start of any study-related procedures.
Eligible subjects were admitted to the clinical facility (PRA International, Zuidlaren, the Netherlands) the day before the first treatment period and remained in the clinical facility for the duration of the study. Subjects were randomized 1:1, according to a computer-generated randomization scheme, to receive either apixaban 2.5 mg BID (every 12 hours) or rivaroxaban 10 mg QD (every 24 hours) for 4 days followed by a ≥4.5-day washout before receiving the alternate treatment (Figure 1). Four days of dosing was selected to ensure that both agents achieved steady state; apixaban achieves steady state within 2–3 days of administration with modest accumulation (<2-fold) following BID dosing, and rivaroxaban does not show appreciable accumulation following QD administration.7,8
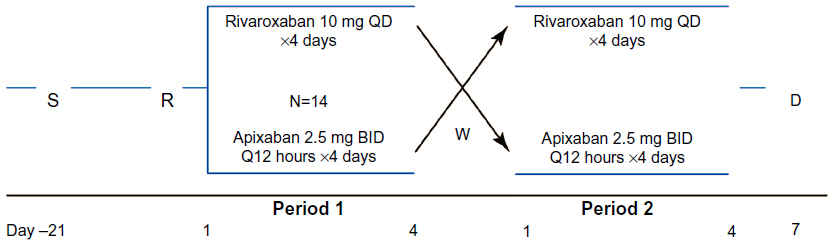 | Figure 1 Study design schematic. |
Inclusion and exclusion criteria
Eligibility criteria included age 18–45 years, body mass index of 18–30 kg · m−2, inclusive, and a healthy condition according to physical examination, vital sign assessment, clinical laboratory assessment, and medical history. Reasons for exclusion from the study included any significant or acute medical illness or relevant trauma; gastrointestinal disease or surgery in the previous 3 months; a history of abnormal bleeding or coagulation disorder; any significant head injury within the previous 2 years; any major surgery within 4 weeks prior to dosing or planned within 2 weeks after completion of the study; or any history of acute or chronic pancreatitis. Female subjects were excluded if pregnant or breastfeeding and were required to have a negative serum or urine pregnancy test within 24 hours prior to dosing. All subjects were required to use an acceptable method of contraception for the duration of the study. Subjects were also excluded for a recent history of smoking or alcohol abuse.
Assessments
Blood sampling
Blood samples (4 mL) for measurement of rivaroxaban plasma concentrations were collected in potassium EDTA tubes predose on days 1 and 4 of the treatment period and at 0.5, 1, 2, 3, 4, 5, 6, 8, 12, 13, 14, 15, 16, 17, 18, 20, 24, 36, 48, and 72 hours after the morning dose on day 4 of the treatment period. Blood samples (2.7 mL) for measurement of apixaban plasma concentrations were collected in 3.2% sodium citrate tubes predose on days 1 and 4 of each treatment period and at 0.5, 1, 2, 3, 4, 5, 6, and 8 hours after the morning dose, and at predose (12 hours) and 0.5, 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, and 60 hours after the evening dose on day 4 of the treatment period. Samples were stored on ice immediately after collection and processed within 30 minutes by centrifugation for 15 minutes at 1,500× g, 4°C. Plasma from each sample was then transferred to a separate vial and stored at ≤−20°C until shipped on dry ice for analysis.
Apixaban and rivaroxaban samples were assayed by validated liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) methods at Intertek Pharmaceutical Services (formerly known as Alta Analytical Laboratory, El Dorado Hills, CA, USA) and at PPD Inc. (Middleton, WI, USA), respectively. For apixaban, sample extraction for plasma utilized protein precipitation. Apixaban-M4 (a hydroxylated metabolite of apixaban) was used as the internal standard for the assay. The lower limit of quantification (LLOQ) for apixaban was 1.00 ng/mL. The between-run and within-run variability (coefficient of variation [CV]) for apixaban in quality control samples were ≤6.7% and ≤4.1%, respectively, with deviations from the nominal concentration of no more than ±3.7%. All samples were analyzed within the period of analyte stability. For rivaroxaban, sample extraction for plasma utilized liquid–liquid extraction. Rivaroxaban-d4 (deuterium-radiolabeled rivaroxaban) was used as the internal standard for the assay. The LLOQ for rivaroxaban was 0.5 ng/mL. The between-run and within-run variability (CV) for rivaroxaban in quality control samples were ≤5.1% and ≤8.8%, respectively, with deviations from the nominal concentration of no more than ±5.6%. All samples were analyzed within the period of analyte stability.
Pharmacokinetics
PK parameters for apixaban and rivaroxaban were derived from the plasma concentration–time data on day 4 and included maximum observed plasma concentration within the dosing interval (Cmax), minimum observed plasma concentration within the dosing interval (Cmin), time of Cmax (Tmax), and area under the plasma concentration–time curve within the dosing interval (AUC(0–12) and AUC(0–24), respectively). Apixaban daily AUC (AUC(0–24)) was calculated as the sum of the AUC(0–12) and AUC(12–24) observed following administration of the morning and evening dose, respectively. Terminal half-life (T1/2) was calculated following the last dose (ie, following the day 4 evening dose for apixaban and following the day 4 morning dose for rivaroxaban). Plasma concentration peak-to-trough ratios (Cmax/Cmin) were calculated.
Anti-factor Xa activity
Blood samples (2.7 mL) for the determination of AXA for both drugs were collected in 3.2% sodium citrate tubes at the same time points as the PK samples, and kept at room temperature for no more than 30 minutes before centrifuging at 18°C and 1,500× g for 15 minutes. Plasma was collected from each sample and stored in cryovials at ≤−70°C. Samples were shipped on dry ice to Esoterix Coagulation Laboratory (Englewood, CO, USA) for analysis of AXA using a validated chromogenic anti-FXa assay (Rotachrom®; Diagnostica Stago Inc., Parsippany, NJ, USA)9 with a reportable range of 0.10 IU/mL to 18.5 IU/mL for apixaban and rivaroxaban.9,10
AXA parameters for both compounds were derived from plasma AXA time data obtained on day 4 over the dosing interval as described above for PK. Parameters assessed included peak and trough plasma AXA, time to peak AXA (Tpeak), area under the plasma AXA–time curve, and AXA half-life.
Safety
Physical examination, measurement of vital signs, 12-lead electrocardiogram (ECG), and blood/urine clinical laboratory testing were performed at screening, prior to each treatment period, and at study discharge, with close monitoring for adverse events (AEs) throughout the study. All AEs were coded by the investigator using the Medical Dictionary for Regulatory Activities (MedDRA; v12.1) and recorded along with severity, timing of onset, relationship to study drug, and outcome.
Statistical methods
Data from 12 subjects were expected to provide ≥90% probability that the lower limit of the 90% confidence interval (CI) for the geometric mean ratio of Cmax/Cmin values (rivaroxaban/apixaban) would be >1. An additional two subjects were enrolled to allow for early withdrawals. This estimate was based on the assumptions that the expected Cmax/Cmin ratio was ≥30% greater for rivaroxaban than apixaban,11,12 Cmax/Cmin would be log-normally distributed, and intersubject standard deviation would not be greater than 0.22.13
Mean and individual steady-state concentration–time and AXA–time profiles were plotted for both apixaban and rivaroxaban. Scatter plots of AXA versus plasma concentration were plotted for both compounds and analyzed by linear regression. Individual PK and PD parameters were estimated using noncompartmental methods with WinNonlin® Professional (v5.0.1; Pharsight Corporation, Sunnyvale, CA, USA). Terminal elimination rate constants were estimated using the WinNonlin algorithm and AUC parameters were calculated using the log-linear trapezoidal rule (WinNonlin Method 1). Actual sampling times were used for all parameter calculations. Descriptive statistics for PK and PD parameters were tabulated.
Results
Demographics and disposition
The study was conducted between February and March 2010. A total of 14 healthy subjects (eleven male, three female) were enrolled and received study medication. All 14 subjects completed the study. A summary of subject demographics is shown in Table 1.
 | Table 1 Population demographics |
Pharmacokinetics
Mean and individual plasma concentration–time profiles from day 4 of treatment for both compounds are shown in Figure 2, and summary statistics of the PK data are provided in Table 2. Median time to peak plasma concentration (Tmax) was 2 hours for both compounds. Peak concentrations were followed by a multiphasic decline with a mean terminal half-life of 8.7 hours for apixaban versus 7.9 hours for rivaroxaban. Evaluation of mean trough concentrations on days 4 and 5 indicated that steady state for both apixaban and rivaroxaban had been reached by the fourth day of dosing. On day 4, rivaroxaban Cmax was 171 ng/mL, ~2-fold higher than for apixaban (81 ng/mL). Conversely, the Cmin for rivaroxaban (10 ng/mL) was ~40% lower than for apixaban (17 ng/mL). The greater fluctuation in rivaroxaban plasma concentration over the dosing interval was reflected in the differences in the Cmax/Cmin ratio. The geometric mean of the Cmax/Cmin ratio for rivaroxaban (16.9) was 3.6-fold higher (90% CI: 2.82–4.61) than that for apixaban (4.7). Despite the difference in Cmax/Cmin ratio over the dosing interval, the daily AUC at steady state (AUC0–24) was only ~17% higher for rivaroxaban (mean [%CV] AUC(0–24): rivaroxaban =1,094 [29] ng · h/mL; apixaban =935 [24] ng · h/mL). Intersubject variability, expressed as %CV, was lower for apixaban exposure parameters (20%–24%) than for rivaroxaban exposure parameters (29%–46%).
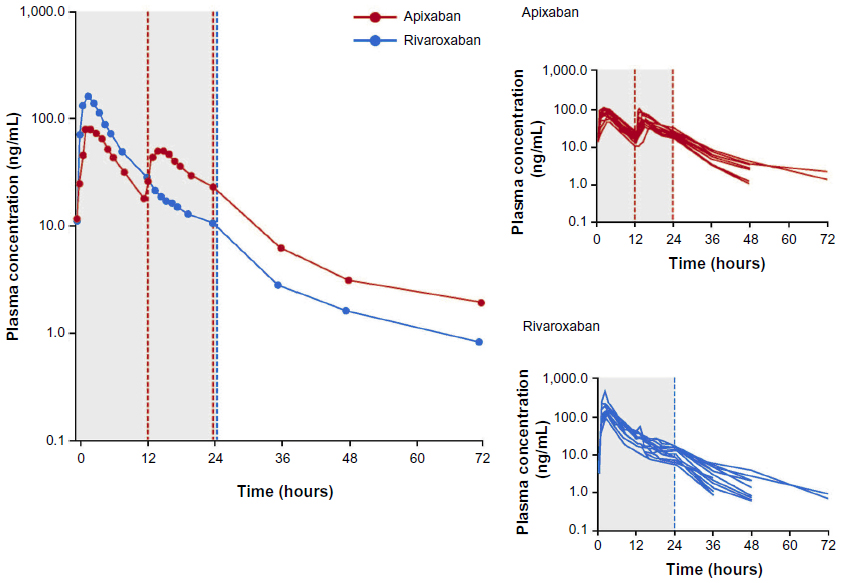 | Figure 2 Arithmetic mean plasma concentration over time at steady state after treatment with rivaroxaban or apixaban. |
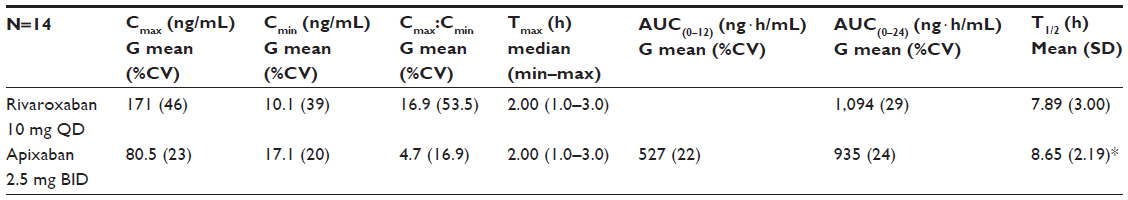 | Table 2 Summary statistics: steady-state pharmacokinetics of rivaroxaban and apixaban |
Pharmacodynamics
The results of the PD analyses were similar to those of the PK analyses for apixaban and rivaroxaban. Plots of mean and individual AXA over time are shown in Figure 3, and summary statistics of the PD data are provided in Table 3. A close temporal relationship between plasma concentration and AXA was seen for both compounds across the dosing interval (compare Figures 2 and 3). Plotting AXA against plasma concentration of each drug (Figure 4) confirmed a direct linear relationship for both compounds; the slope of the regression lines appeared to be different (slope: rivaroxaban 0.0172 IU/ng vs apixaban 0.0134 IU/ng; P<0.0001). Consistent with the PK findings, AXA fluctuated less over the dosing interval for apixaban than for rivaroxaban. The geometric mean peak-to-trough AXA ratio for rivaroxaban was 3.5-fold higher than for apixaban (16.5 vs 4.7, respectively): peak AXA was ~2.5-fold higher for rivaroxaban than for apixaban (2.82 IU/mL [%CV 51] vs 1.12 IU/mL [%CV 21]), whereas trough AXA was ~30% lower for rivaroxaban than for apixaban (Table 3). The AXA geometric mean AUC(0–24) was ~34% higher for rivaroxaban than for apixaban (17.8 IU · h/mL vs 13.3 IU · h/mL). Apixaban mean AXA half-life (8.9 hours) was similar to the half-life observed for plasma concentration (8.7 hours). Rivaroxaban mean AXA half-life could not be estimated over a similar time interval as for plasma concentration since AXA results for all but two subjects were below the assay’s LLOQ 24 hours after administration.
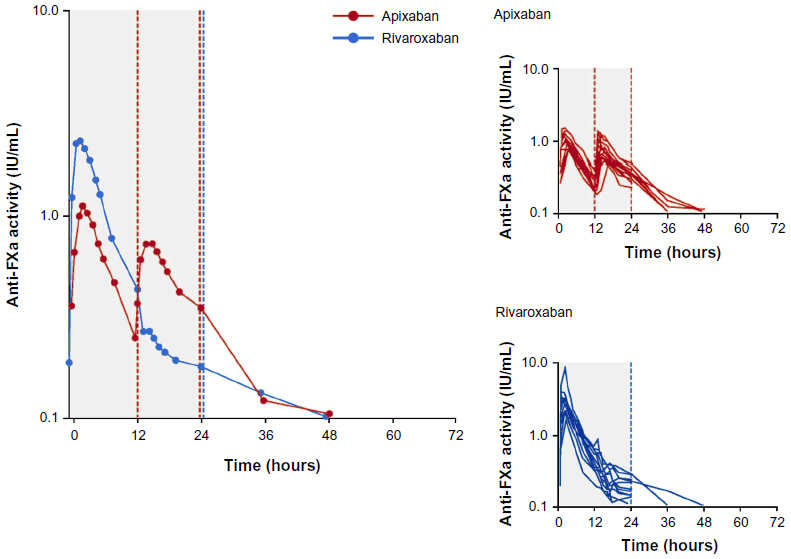 | Figure 3 Arithmetic mean anti-FXa activity* over time at steady state on day 4 of treatment with rivaroxaban or apixaban. |
 | Figure 4 Plot of anti-FXa activity versus apixaban and rivaroxaban plasma concentration. |
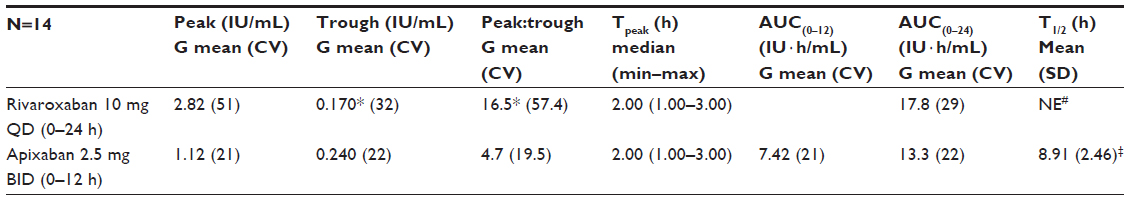 | Table 3 Summary statistics: steady state anti-FXa activity of rivaroxaban and apixaban |
Safety
Apixaban and rivaroxaban were well-tolerated in this study. A total of 22 AEs were reported by ten subjects after the start of study medication. Twelve AEs were reported by seven subjects following rivaroxaban administration, consisting of contusion, pharyngitis, and rhinitis, each reported twice, and single reports of epistaxis, abdominal discomfort, abdominal distension, diarrhea, paresthesia, and procedural pain. Ten AEs were reported by eight subjects following administration of apixaban, consisting of three reports of dizziness, two reports of contusion, and single reports of epistaxis, catheter site hematoma, catheter site-related reaction, cough, and musculoskeletal chest pain. All AEs reported during the study were mild and resolved without treatment. No deaths or serious AEs were reported for either compound during the study, and no subjects discontinued treatment because of AEs or for other reasons. There were no findings of clinical relevance with respect to clinical laboratory tests, vital signs, ECGs, or physical examinations.
Discussion
This study provides the first head-to-head comparison of the PK and PD profiles for apixaban administered BID and rivaroxaban administered QD. Both apixaban 2.5 mg BID and rivaroxaban 10 mg QD were readily absorbed, reaching Tmax in 2 hours, and both agents had multiphasic elimination profiles with terminal half-lives of ~8 hours. For apixaban 2.5 mg BID, there is significantly less daily fluctuation in plasma concentrations and AXA (ie, lower peak-to-trough ratio), as well as less variability in PK parameters (ie, Cmax and AUC), than for rivaroxaban 10 mg QD. The PK results for apixaban in this study were generally consistent with previously published multiple-dose data on apixaban7,14 in healthy subjects. The fluctuations in mean apixaban concentrations following administration of apixaban 2.5 mg BID in multiple ascending dose studies in Western and Japanese subject populations were ~3.0 and ~4.2, respectively, and the mean terminal half-lives were 8.1 and 9.7 hours, respectively.7,14 To our knowledge, the multiple-dose steady-state PK of rivaroxaban 10 mg QD in healthy subjects have not previously been reported, and the limited available data for 5 mg QD8 do not permit estimation of the concentration fluctuation, although the half-life (8.4 hours) was similar to that in the current study.
Although the PK and PD profiles of rivaroxaban and apixaban in VTEp patients differ somewhat from that in healthy subjects due to differences in covariates impacting the PK, the observations in healthy subjects are generally relevant to the patient population. Specifically, for rivaroxaban, it appears that in VTEp patients, the Cmax value is somewhat lower and the Cmin value is slightly higher than that observed in our study,5 with a resulting Cmax/Cmin ratio of ~7. For apixaban, the Cmax value in VTEp patients is similar to that in our study, but the Cmin value is higher,6 resulting in a Cmax/Cmin ratio of ~1.5. Although the fluctuation values are lower, the difference between the two drugs is similar to that observed in healthy subjects.
Given that direct linear relationships between plasma concentration and AXA were evident for both apixaban and rivaroxaban, AXA findings are largely consistent with the PK of these agents. Peak AXA was observed at 2 hours postdose, coinciding with the Cmax for the respective compound, and apixaban maintained a lower AXA peak-to-trough ratio over the dosing interval. The difference between apixaban 2.5 mg BID and rivaroxaban 10 mg QD total daily AXA (AXA AUC(0–24)) was somewhat greater than the difference observed for exposure, with AXA AUC(0–24) being ~34% higher for rivaroxaban. This finding is consistent with the difference observed in the slope of the AXA concentration regression lines for rivaroxaban and apixaban. Although the regression for rivaroxaban was determined over a wider range of concentrations than that for apixaban, this is unlikely to provide an explanation for the difference in slope. Rivaroxaban has also been shown to have a greater effect than apixaban in the prothrombin time assay, at a concentration of 1,000 ng/mL, that cannot be attributed to the ~5% difference in molecular weight.9
Despite a slightly lower daily AXA(0–24), apixaban AXA activity persisted well beyond the time at which the next scheduled BID dose was to be administered, whereas rivaroxaban AXA was near or below the lower limit of detection of the assay at the time of the next scheduled QD dose. Although QD administration may be favored based on convenience, it clearly results in higher peaks and lower troughs.12,15 The impact of greater fluctuation in levels of anticoagulation on clinical outcomes with oral FXa inhibitors is not known. Thus, the clinical importance of differences in rivaroxaban and apixaban AXA observed over their respective dosing intervals in this study remains to be determined.
The safety and efficacy of apixaban and rivaroxaban dosed QD and BID have been studied in Phase II clinical trials of VTE prevention. A Phase II dose-ranging clinical trial in patients undergoing total knee replacement was conducted to evaluate the efficacy and safety of apixaban total daily doses of 5, 10, and 20 mg administered in QD and BID regimens.16 Numerically lower rates of VTE and death were observed with the apixaban 2.5-, 5-, and 10-mg BID regimens compared with the same total daily doses administered QD, whereas bleeding was similar for both BID and QD administration. A pharmacometric analysis performed with the apixaban Phase II VTE prevention data further supported a trend for a more favorable risk–benefit profile for the apixaban BID regimen.15 Thus, a BID regimen was selected for further study.
QD and BID regimens of rivaroxaban were evaluated in four separate Phase II studies of VTE prevention.17–20 No clear dose–response relationship was seen for efficacy across these trials, whereas bleeding increased with increasing doses. Comparisons of rivaroxaban QD and BID regimens have mainly focused on cross-study assessments, with the exception of one study20 that included a single QD regimen among several BID regimens. Overall, the safety and efficacy profiles of the rivaroxaban QD and BID regimens appeared to be similar,20 and a QD regimen was chosen for further study. While the outcomes of the Phase III clinical trials in VTE prevention have been favorable for both apixaban and rivaroxaban relative to low-molecular-weight heparin,21–27 no comparisons of dosing frequency were performed in these trials.
This study was performed in a small number of healthy subjects receiving apixaban or rivaroxaban under supervision. As already mentioned, PK/PD differences in specific patient populations may alter the risk–benefit profiles of these agents in real-world clinical use. Additionally, factors such as compliance may impact outcomes, especially over the longer-term outpatient settings where dosing cannot be monitored. While compliance can be affected by dosing regimen, it can also be affected by the side-effect profile of the drug in question, as well as other factors. The compliance of both agents during real-world clinical use and its impact on clinical outcomes remains to be determined.
In summary, this study reports the first head-to-head comparison of apixaban and rivaroxaban PK and PD. Apixaban 2.5 mg BID demonstrated lower fluctuations in peak-to-trough plasma concentration and AXA throughout the dosing interval, lower interindividual variability in concentration and AXA, and a lower AXA AUC compared with rivaroxaban 10 mg QD. Although this study provides an informative comparison of apixaban and rivaroxaban PK and PD at dose regimens used for the prevention of VTE, it is important to note that it is difficult to extrapolate data from this healthy subject study to patients. It is possible that other factors encountered in clinical practice, such as drug interactions, varying degrees of renal function, and other patient characteristics may influence both the relative PK and PD profiles observed in this study and their relationships with real-world efficacy and safety outcomes. Although efficacy and safety exposure–response relationships in the prevention of VTE have been reported for apixaban, using AUC as the measure of exposure,15 these relationships have not been reported for rivaroxaban. Therefore, the impact of greater or lesser fluctuation in plasma concentration or AXA resulting from their respective dosing regimens on the relative efficacy and safety profiles of apixaban and rivaroxaban in clinical practice remains to be determined.
Acknowledgments
Study conduct, analysis, and reporting were supported by Dr Maria Velinova and the staff of PRA International, the Netherlands. The authors would like to thank Dr John Alexander for his helpful review of the manuscript. Professional editorial assistance was provided by Andy Shepherd and Dana Fox at Caudex Medical, and funded by Bristol-Myers Squibb Company and Pfizer Inc.
Author contributions
All of the authors (C Frost, Y Song, YC Barrett, J Wang, J Pursley, RA Boyd, and F LaCreta) were: involved in the conception and design (protocol development and/or design advice) and/or acquisition of data (study investigator), or data analysis and interpretation; drafting/revising the publication for content; and approval of the final version to be published.
Disclosure
At the time of research, all authors of this study were employees of Bristol-Myers Squibb Company or Pfizer Inc. This study was sponsored by Bristol-Myers Squibb and Pfizer.
References
Xarelto® (rivaroxaban) [prescribing information]. Leverkusen, Germany: Bayer Healthcare AG; 2011. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202439s001lbl.pdf. Accessed February 6, 2013. | |
Eliquis® (apixaban) tablets [prescribing information]. New York: Bristol-Myers Squibb Company; 2012. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202155s000lbl.pdf. Accessed January 24, 2013. | |
Frost C, Wang J, Nepal S, et al. Apixaban, an oral, direct factor Xa inhibitor: single-dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75(2):476–487. | |
Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51(7):549–561. | |
Xarelto® (rivaroxaban). Summary of product characteristics. Leverkusen, Germany: Bayer Pharma AG. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000944/WC500057108.pdf. Accessed May 24, 2012. | |
Eliquis® (apixaban) tablets. Summary of product characteristics. New York: Bristol-Myers Squibb Pfizer EEIG. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf. Accessed June 2, 2013. | |
Frost C, Nepal S, Wang J, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76(5):776–786. | |
Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939 – an oral, direct Factor Xa inhibitor – after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61(12):873–880. | |
Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263–1271. | |
Upreti VV, Wang J, Barrett YC, et al. Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br J Clin Pharmacol. 2013;76(6):908–916. | |
Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct factor Xa inhibitor, in healthy subjects. J Clin Pharmacol. 2006;46(5):549–558. | |
Mueck W, Borris LC, Dahl OE, et al. Population pharmacokinetics and pharmacodynamics of once- and twice-daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Haemost. 2008;100(3):453–461. | |
Cui Y, Song Y, Wang J, et al. Single- and multiple-dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin Pharmacol. 2013;5:177–184. | |
Yamahira N, Frost C, Fukase H, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int J Clin Pharmacol Ther. 2014;52(7):564–573. | |
Leil TA, Feng Y, Zhang L, Paccaly A, Mohan P, Pfister M. Quantification of apixaban’s therapeutic utility in prevention of venous thromboembolism: selection of phase III trial dose. Clin Pharmacol Ther. 2010;88(3):375–382. | |
Lassen MR, Davidson BL, Gallus A, Pineo G, Ansell J, Deitchman D. The efficacy and safety of apixaban, an oral, direct factor Xa inhibitor, as thromboprophylaxis in patients following total knee replacement. J Thromb Haemost. 2007;5(12):2368–2375. | |
Eriksson BI, Borris LC, Dahl OE, et al; ODIXa-HIP Study Investigators. A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement. Circulation. 2006;114(22):2374–2381. | |
Eriksson BI, Borris L, Dahl OE, et al; ODIXa-HIP Study Investigators. Oral, direct Factor Xa inhibition with BAY 59-7939 for the prevention of venous thromboembolism after total hip replacement. J Thromb Haemost. 2006;4(1):121–128. | |
Turpie AG, Fisher WD, Bauer KA, et al; OdiXa-Knee Study Group. BAY 59-7939: an oral, direct factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost. 2005;3(11):2479–2486. | |
Eriksson BI, Borris LC, Dahl OE, et al. Dose-escalation study of rivaroxaban (BAY 59-7939) – an oral, direct Factor Xa inhibitor – for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Res. 2007;120(5):685–693. | |
Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358(26):2765–2775. | |
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361(6):594–604. | |
Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet. 2008;372(9632):31–39. | |
Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358(26):2776–2786. | |
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P; ADVANCE-2 investigators. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375(9717):807–815. | |
Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM; ADVANCE-3 Investigators. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med. 2010;363(26):2487–2498. | |
Turpie AG, Lassen MR, Davidson BL, et al; RECORD4 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373(9676):1673–1680. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.