Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
A randomized, blinded study to evaluate the efficacy and safety of umeclidinium 62.5 µg compared with tiotropium 18 µg in patients with COPD
Authors Feldman G, Maltais F ![]() , Khindri S, Vahdati-Bolouri M, Church A, Fahy WA, Trivedi R
, Khindri S, Vahdati-Bolouri M, Church A, Fahy WA, Trivedi R
Received 15 December 2015
Accepted for publication 4 February 2016
Published 7 April 2016 Volume 2016:11(1) Pages 719—730
DOI https://doi.org/10.2147/COPD.S102494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Gregory Feldman,1 François Maltais,2 Sanjeev Khindri,3 Mitra Vahdati-Bolouri,3 Alison Church,4 William A Fahy,3 Roopa Trivedi4,5
1S. Carolina Pharmaceutical Research, Spartanburg, SC, USA; 2Institut universitaire de cardiologie et de pneumologie de Québec, Quebec, QC, Canada; 3GSK, Respiratory Research and Development, Middlesex, UK; 4GSK, Respiratory and Immuno-Inflammation Research, Triangle Park, NC, USA; 5Pearl Therapeutics Inc., Durham, NC, USA
Background: The long-acting muscarinic antagonists umeclidinium (UMEC) and tiotropium (TIO) are approved once-daily maintenance therapies for COPD. This study investigated the efficacy and safety of UMEC versus TIO in COPD.
Methods: This was a 12-week, multicenter, randomized, blinded, double-dummy, parallel-group, non-inferiority study. Patients were randomized 1:1 to UMEC 62.5 µg plus placebo or TIO 18 µg plus placebo. The primary end point was trough forced expiratory volume in 1 second (FEV1) at day 85 (non-inferiority margin -50 mL; per-protocol [PP] population). Other end points included weighted mean FEV1 over 0–24 and 12–24 hours post-dose. Patient-reported outcomes comprised Transition Dyspnea Index score, St George’s Respiratory Questionnaire total score, and COPD Assessment Test score. Adverse events were also assessed.
Results: In total, 1,017 patients were randomized to treatment. In the PP population, 489 and 487 patients received UMEC and TIO, respectively. In the PP population, change from baseline in trough FEV1 was greater with UMEC versus TIO at day 85, meeting non-inferiority and superiority margins (difference: 59 mL; 95% confidence interval [CI]: 29–88; P<0.001). Similar results were observed in the intent-to-treat analysis of trough FEV1 at day 85 (53 mL, 95% CI: 25–81; P<0.001). Improvements in weighted mean FEV1 over 0–24 hours post-dose at day 84 were similar with UMEC and TIO but significantly greater with UMEC versus TIO over 12–24 hours post-dose (70 mL; P=0.015). Clinically meaningful improvements in Transition Dyspnea Index and St George’s Respiratory Questionnaire were observed with both treatments at all time points. No differences were observed between UMEC and TIO in patient-reported outcomes. Overall incidences of adverse events were similar for UMEC and TIO.
Conclusion: UMEC 62.5 µg demonstrated superior efficacy to TIO 18 µg on the primary end point of trough FEV1 at day 85. Safety profiles were similar for both treatments.
Keywords: tiotropium, umeclidinium, COPD, non-inferiority, long-acting muscarinic antagonist
Introduction
COPD contributes significantly to morbidity and mortality, and is predicted to be the third leading cause of death worldwide by 2030.1,2 COPD is characterized by chronic and progressive breathlessness, cough, and sputum production, which can all be a major cause of disability.1
Bronchodilators are central to the pharmacological management of COPD.1 Long-acting muscarinic antagonists (LAMAs) are an integral part of management in stable COPD and have been shown to provide improvements in lung function, hyperinflation, symptoms, and health status, while reducing exacerbations.3–5 However, until recently, tiotropium (TIO) was the only LAMA available for patients with COPD.6 Three other LAMAs, aclidinium bromide (twice daily),7 glycopyrronium bromide (once daily),8 and umeclidinium (UMEC; once daily),9,10 have now been approved for the treatment of COPD. The results of two randomized active comparator clinical trials of twice-daily aclidinium bromide 400 μg and once-daily glycopyrronium 50 μg showed that both these drugs had similar efficacy to TIO 18 μg, as assessed by trough forced expiratory volume in 1 second (FEV1).11,12 However, limited data are available comparing the efficacy and safety of UMEC with other LAMAs.
The main aim of this study was to investigate whether treatment with once-daily UMEC 62.5 μg was non-inferior to TIO 18 μg, as assessed by trough FEV1 on treatment day 85 in symptomatic patients with moderate-to-severe COPD. Additionally, efficacy, as assessed by other lung function and patient-reported outcomes (PROs), and safety of UMEC and TIO were also evaluated.
Methods
Study design
This was a 12-week, multicenter, randomized, blinded, double-dummy, parallel-group study, conducted in patients with COPD between September 2014 and June 2015 (GSK study number: 201316; Clinicaltrials.gov identifier: NCT02207829) in Canada, Chile, Denmark, France, Germany, Italy, Romania, Korea, South Africa, the Russian Federation, the Ukraine, and the USA.
Patients meeting the eligibility criteria at screening (Visit 1) completed a 7–14-day run-in period prior to randomization. Eligible patients were then randomized at clinic Visit 2 (day 1) to 12 weeks of treatment. Clinic visits during the 12-week treatment period were on days 2, 28, 56, 84, and 85. In a subset of patients (n=250), 24-hour serial spirometry was conducted at days 1 and 84. Inhaler assessments were conducted to evaluate errors, ease of use, and inhaler preference.
The study protocol and written informed consent were reviewed and approved by the Chesapeake Institutional Review Board, as well as each relevant national, regional, or independent ethics committee or institutional review board, in accordance with Good Clinical Practice. This study was conducted in accordance with Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki (2008).13
Patients
Patients eligible for inclusion were ≥40 years of age with a diagnosis of COPD in accordance with the American Thoracic Society/European Respiratory Society definition,14 current or former cigarette smokers with ten or more pack-years cigarette smoking history, had a pre- and post-albuterol/salbutamol FEV1/forced vital capacity (FVC) ratio of <0.70, and a post-albuterol/salbutamol FEV1 of 30%–70% of predicted normal values. Patients also had a dyspnea score of ≥2 on the modified Medical Research Council Dyspnea Scale at Visit 1.
Key exclusion criteria included pregnancy, a current diagnosis of asthma or other significant respiratory disorder or other condition that may affect respiratory function (eg, unstable or life-threatening cardiac disease, a neurological condition), lung volume reduction surgery, or hospitalization for COPD/pneumonia within 12 weeks prior to Visit 1. Patients were also excluded for the use of long-term oxygen therapy (prescribed for >12 hours per day) and use of COPD maintenance medications other than study medication, with the exception of inhaled corticosteroids (ICSs). The use of prohibited medications (Table S1) within the specified time periods also excluded patients from the study.
Treatments
After the run-in period, patients were randomized 1:1 to once-daily UMEC 62.5 μg (delivering 55 μg) administered via the ELLIPTA™ dry powder inhaler (DPI) plus placebo (PBO) administered via the HandiHaler®, or once-daily TIO 18 μg (delivering 10 μg) administered via the HandiHaler® plus PBO administered via the ELLIPTA™ DPI. Patients were provided albuterol/salbutamol for use as a rescue medication. Active and PBO inhalers were identical in appearance, and all patients and physicians were masked to study treatment as described previously.15,16 Further details on study blinding are provided in the Supplementary materials section.
The randomization code was generated by GSK using a validated computerized system RandAll version NG. Patients were randomized using RAMOS interactive voice technology. Randomization was further stratified according to whether or not the patient participated in 24-hour serial FEV1 assessments.
Outcomes and assessments
Primary end point
Trough FEV1 on day 85 (defined as the mean of the FEV1 values obtained 23 and 24 hours after dosing on day 84) in the per-protocol (PP) population.
Other lung function outcomes
Other lung function end points included the following: trough FEV1 on day 85 in the intent-to-treat (ITT) population; trough FEV1 on days 2, 28, 56, and 84 (ITT population); trough FVC on days 2, 28, 56, 84, and 85 (ITT population); weighted mean (WM) FEV1 over 0–12 hours post-dose, 12–24 hours post-dose, and 0–24 hours post-dose, each on days 1 and 84 (subset analysis); and serial FEV1 on days 1 and 84 (subset analysis).
Patient-reported outcomes
PROs included Transition Dyspnea Index (TDI) focal score and the proportion of TDI responders on days 28, 56, and 84. TDI responders were defined as patients with a ≥1 unit TDI focal score.17 St George’s Respiratory Questionnaire (SGRQ) total score and the proportion of SGRQ responders on days 28 and 84 were also assessed. SGRQ responders were defined by a reduction from baseline of ≥4 units in SGRQ total score.18 Finally, COPD Assessment Test (CAT) score and the proportion of CAT responders (defined as a reduction from baseline of ≥2 units in CAT score19) were assessed on days 28 and 84. Rescue medication was also an end point and was assessed by the mean number of puffs/day of rescue medication and percentage of rescue-free days over the study duration (weeks 1–12).
Inhaler assessments
Patient preference for the ELLIPTA™ DPI compared with the HandiHaler® was assessed at the end of the patient’s treatment phase, and “ease of use” rating for the ELLIPTA™ DPI compared with the HandiHaler® was assessed on days 28 and 84. Inhaler errors (IEs) were assessed in a subset of patients on days 1, 28, and 84 using the IE checklists, based on steps in the patient information leaflets for each inhaler.20,21 A critical error was predefined as an error that was most likely to result in no or only minimal medication being inhaled. An overall error included a critical and noncritical error.
Post hoc analyses
Post hoc analyses were performed on trough FEV1 in the ITT population. These included a subgroup analysis by Global initiative for chronic Obstructive Lung Disease (GOLD) Grade 1/2 and Grade 3/4, and GOLD Groups B and D. Additionally, a subgroup analysis by ICS use at screening was performed. An analysis by GOLD Grade 1/2 and Grade 3/4, each split by ICS use, was also performed.
A post hoc FEV1 responder analysis (by GOLD grade) was performed, whereby a response was defined as an increase of ≥100 mL above baseline in trough FEV1 (minimum clinically important difference [MCID]22).
Safety end points
Safety assessments included the incidence of adverse events (AEs) and vital sign measurements. The incidence of COPD exacerbations was also assessed. A COPD exacerbation was defined as an acute worsening of symptoms of COPD requiring the use of any treatment beyond study medication or rescue albuterol/salbutamol. This included the use of systemic corticosteroids, antibiotics, and/or emergency treatment or hospitalization. Patients experiencing an exacerbation remained in the study at the discretion of the study investigator if they received ≤14 days of treatment with systemic corticosteroids or antibiotics and did not require hospitalization.
Statistical analysis
Non-inferiority and superiority criteria
The non-inferiority margin was set at −50 mL, which is half the generally accepted MCID for trough FEV1.22 UMEC was therefore considered non-inferior to TIO if the lower boundary of the 95% confidence interval (CI) around the UMEC versus TIO treatment difference was greater than −50 mL, and superior if it was greater than 0 mL.
Testing hierarchy
Inferences drawn from the P-values presented in this study were as follows: If non-inferiority of UMEC versus TIO was demonstrated for the primary end point, inference was drawn from P-values for treatment comparisons that were statistically significant (P<0.05). If superiority of UMEC versus TIO was demonstrated for the primary end point, P-values were used to indicate the strength of that superiority, and inference was drawn from P-values for treatment comparisons (statistically significant if P<0.05). If non-inferiority of UMEC versus TIO was not demonstrated for the primary end point, no inference was drawn from P-values for treatment comparisons on other end points.
Analysis populations
The ITT population comprised all patients randomized to treatment who received at least one dose of study medication.
The PP population comprised all patients in the ITT population, including those who did not complete the study, who did not have a protocol deviation considered to impact efficacy. The PP population was used for analysis of the primary comparison of the primary end point only, to avoid bias of the results toward equivalence, which could make a truly inferior treatment appear to be non-inferior.23 This approach was intended to maximize any true differences between treatments and has been employed in a previous non-inferiority study.11 As the study was not designed to detect non-inferiority on the other end points, the ITT population (or a subset thereof) has been used for other end points because it adheres to the randomization procedure and is generally conservative.24 Patients experiencing a COPD exacerbation were also excluded from PP analyses from the onset of the exacerbation, owing to the potential impact that the exacerbation or medications used to treat it may have had on efficacy data.
The 24-hour (TFH) population comprised all patients in the ITT population for whom 24-hour spirometry was performed. The TFH population was used for the analysis of serial FEV1 and 0–12-, 12–24-, and 0–24-hour WM FEV1 end points.
The IE population comprised all patients in the ITT population who completed the IE checklist. The IE population was used for the analysis of critical and overall IE end points.
Sample size
Sample size calculations used a one-sided 2.5% significance level and an estimate of residual standard deviation for trough FEV1 of 220 mL.
With a non-inferiority margin of −50 mL, and assuming that the true mean treatment difference was 0 mL, it was calculated that a study with 816 evaluable patients would have 90% power to determine the non-inferiority of UMEC compared with TIO based on trough FEV1 at day 85 (PP population). A total of 1,006 patients were planned for randomization, based on an estimated 10% of patients providing a day 85 assessment being excluded from the PP population and a 10% withdrawal rate. The sample sizes for the TFH and IE populations were chosen to provide an adequate number of data points to characterize the lung function response over 24 hours and the IEs, respectively, and were not based on providing an adequate number of patients for statistical analysis purposes.
Results
Patient disposition and characteristics
Of the 1,259 patients enrolled, 1,017 were randomized to treatment and comprised the ITT population, of which 941 patients completed the study (UMEC, n=467; TIO, n=474; Figure 1). The PP population comprised 976 patients (UMEC, n=489; TIO, n=487; Figure 1). There were 295 patients in the TFH population (UMEC, n=148; TIO, n=147).
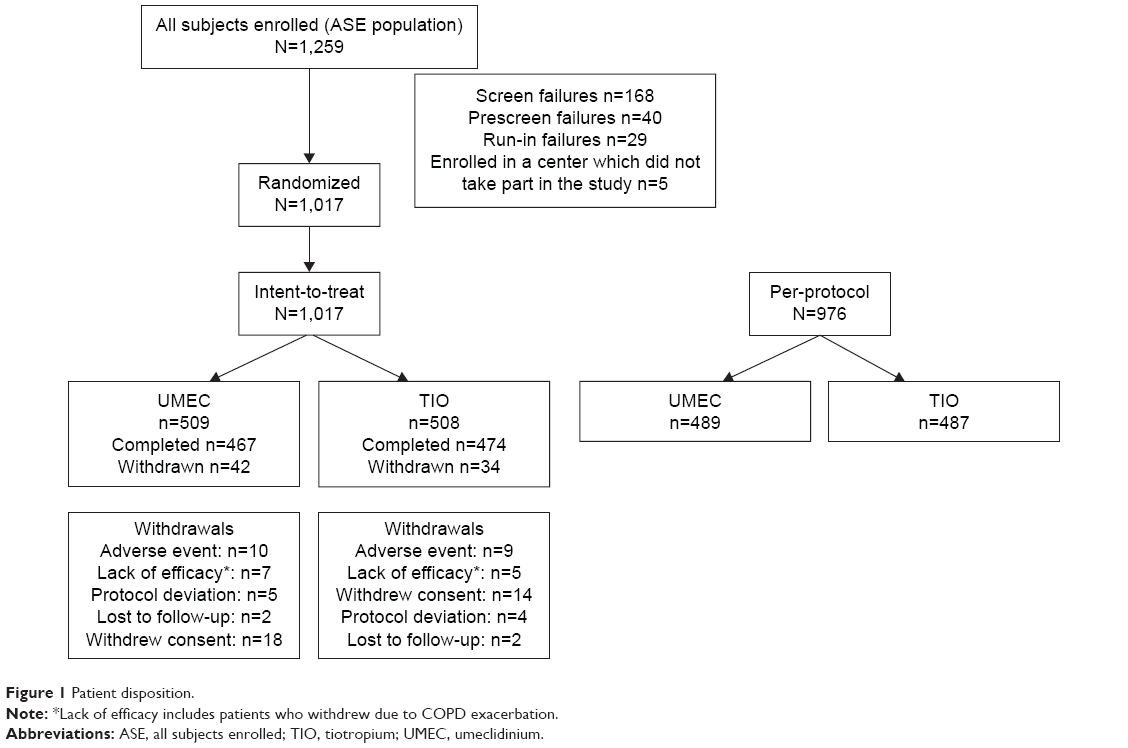 | Figure 1 Patient disposition. |
Patients had symptomatic moderate-to-severe COPD (GOLD Grade 2–3 and GOLD Groups B and D), and the majority of patients were male (Table 1). Baseline demographics were similar between UMEC and TIO treatment groups (Table 1).
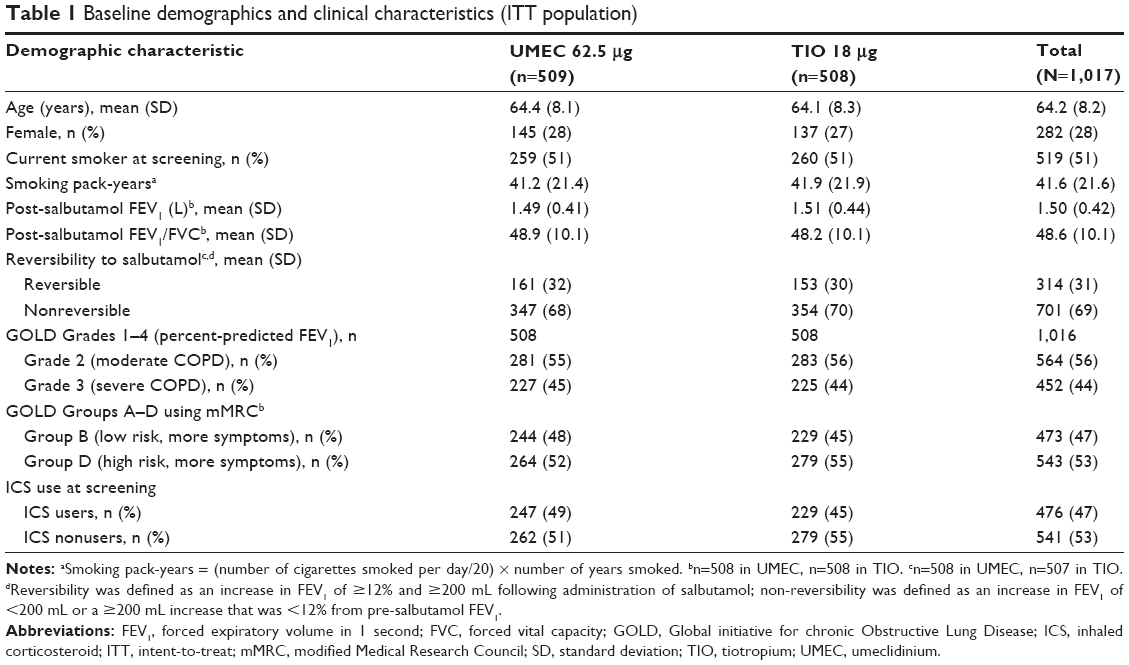 | Table 1 Baseline demographics and clinical characteristics (ITT population) |
Lung function end points
Primary efficacy end point
The least squares (LS) mean change from baseline in trough FEV1 was greater with UMEC than with TIO at day 85 in the PP population (difference: 59 mL, 95% CI: 29–88; P<0.001; Figure 2). Similar results were observed in the analysis of trough FEV1 at day 85 for the ITT population (53 mL, 95% CI: 25–81; P<0.001; Table 2).
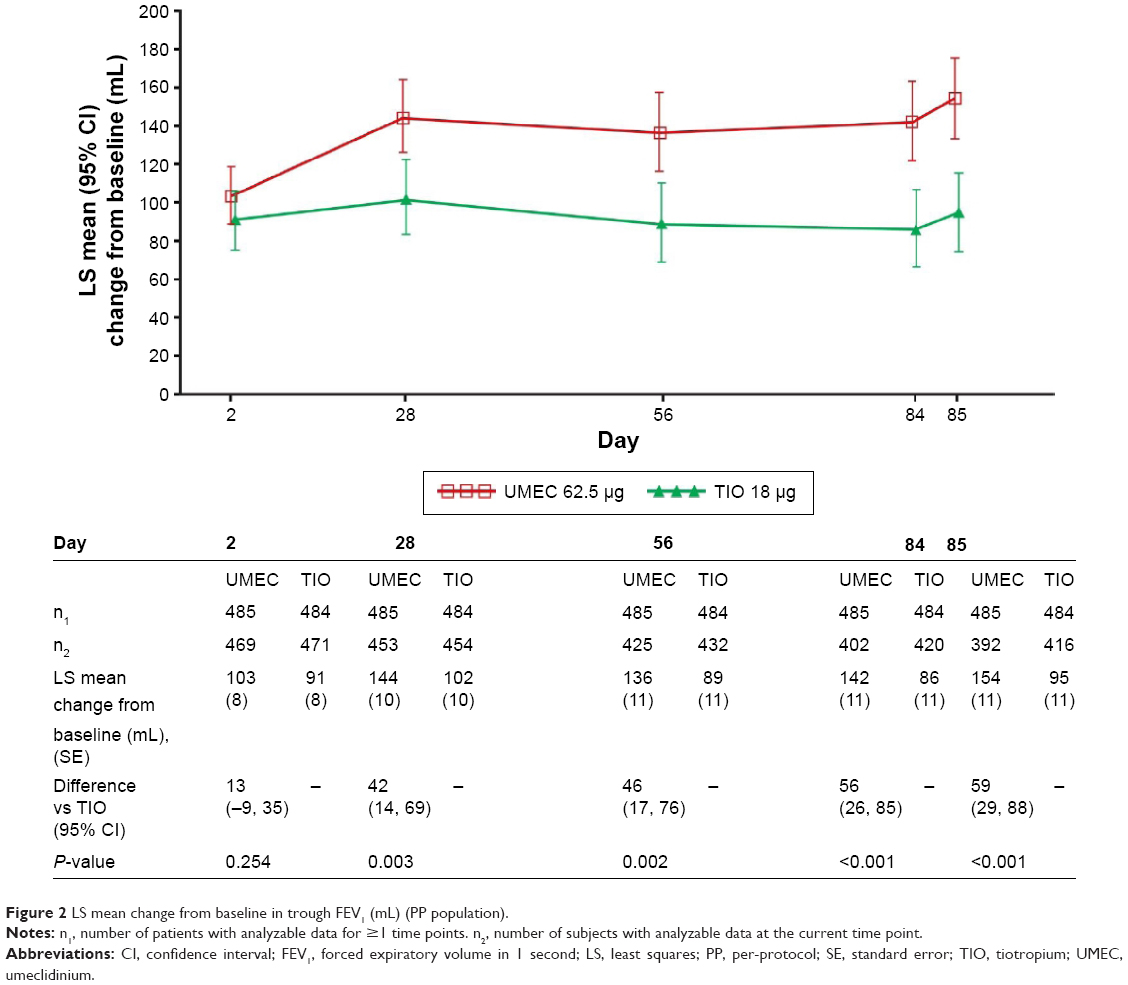 | Figure 2 LS mean change from baseline in trough FEV1 (mL) (PP population). |
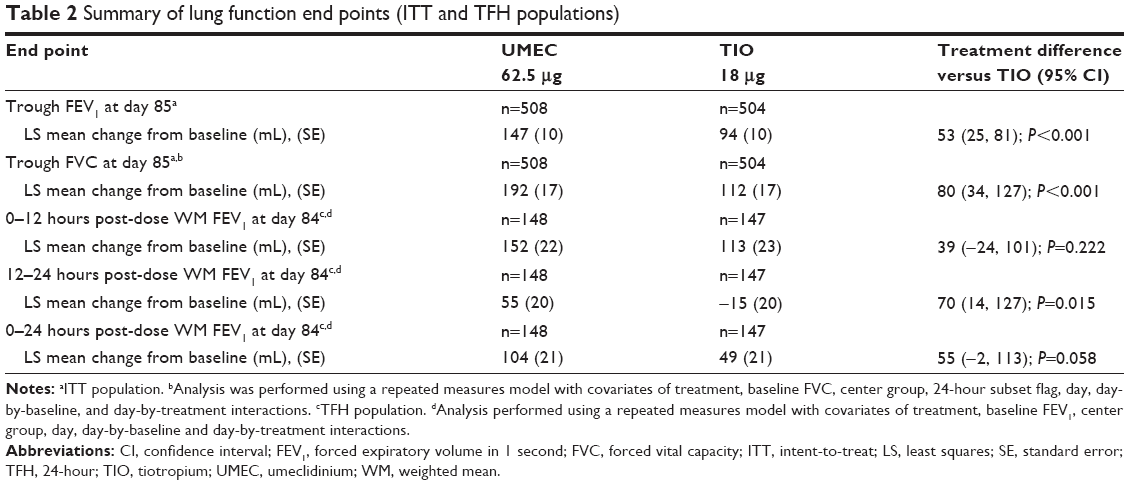 | Table 2 Summary of lung function end points (ITT and TFH populations) |
Other lung function end points
UMEC resulted in a statistically significant difference in LS mean change from baseline trough FEV1 versus TIO at days 28, 56, and 84 (all P≤0.003) but not at day 2 (Figure 2). UMEC also demonstrated a statistically significant difference in LS mean change from baseline trough FVC versus TIO at days 28, 56, 84, and 85 (all P≤0.016) but not at day 2 (Table S2).
UMEC was similar to TIO in 0–12 and 0–24 hours post-dose WM FEV1 at day 84 (0–12 hours post-dose: 39 mL, 95% CI: −24 to 101; P=0.222; and 0–24 hours post-dose: 55 mL, 95% CI: −2 to 113; P=0.058; Table 2). Treatment with UMEC resulted in statistically significant improvements in 12–24 hours post-dose WM FEV1 at day 84 compared with TIO (70 mL, 95% CI: 14–127; P=0.015; Table 2).
On day 1, the difference between treatment groups in LS mean change from baseline in FEV1 was similar at all time points through 24 hours post-dose (Figure 3A), but on day 84, the difference between UMEC and TIO in LS mean change from baseline in FEV1 favored UMEC through 24 hours post-dose, with statistically significant differences at pre-dose (P=0.005) and from 21 hours onward (21 hours post-dose: 76 mL, 95% CI: 11–141, P=0.022; and 24 hours post-dose: 67 mL, 95% CI: 8–126; P=0.026; Figure 3B).
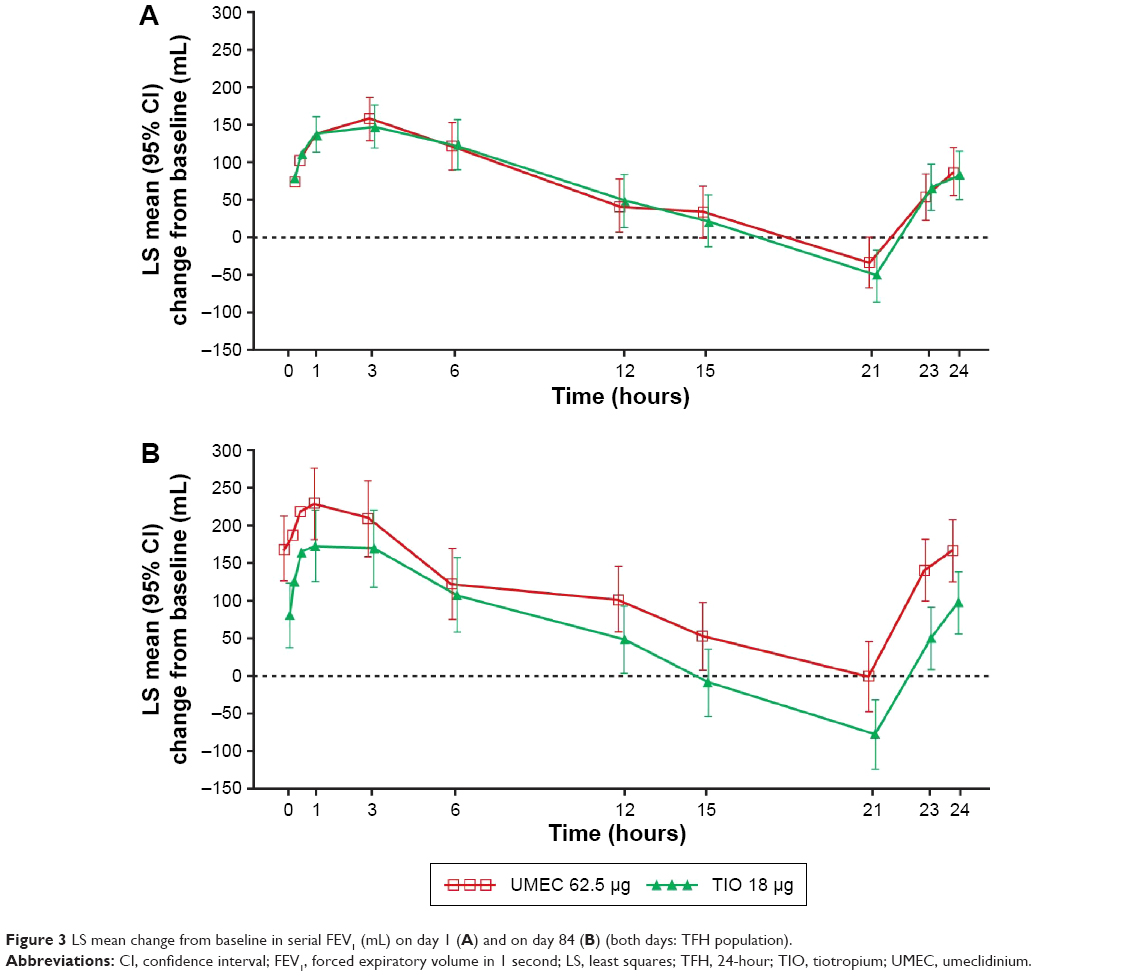 | Figure 3 LS mean change from baseline in serial FEV1 (mL) on day 1 (A) and on day 84 (B) (both days: TFH population). |
Patient-reported outcomes
TDI focal score, SGRQ total score, and CAT score
Similar improvements were observed in TDI, SGRQ, and CAT score for UMEC and TIO (Table 3). Both treatments demonstrated improvements in the LS mean change from baseline in SGRQ total score that met the MCID of ≥4 units at day 84 (Table 3). These trends were also observed on days 28 and 56 (Table S3). No differences were observed in SGRQ or CAT score between UMEC and TIO at day 28 (Table S3) or day 84 (Table 3). UMEC and TIO resulted in TDI focal scores that exceeded the MCID of 1 unit at day 84 (Table 3). However, no treatment difference between UMEC and TIO was observed for TDI at days 28 and 56 (Table S3) and day 84 (Table 3). Approximately half of patients in each treatment group were considered to be responders to TDI, SGRQ, and CAT on day 84 with no difference in the odds of being a responder versus being a nonresponder for each end point (Table 3).
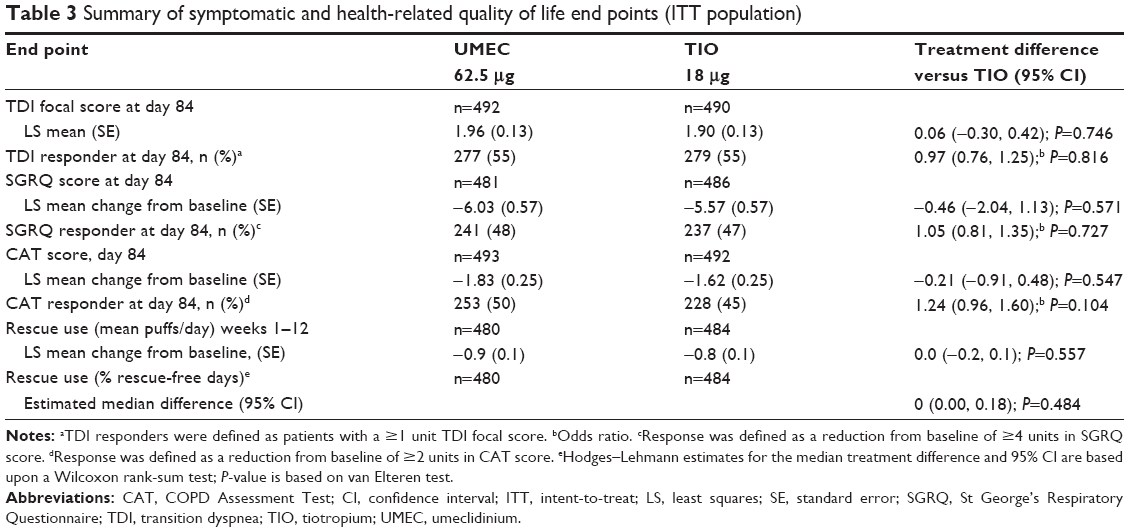 | Table 3 Summary of symptomatic and health-related quality of life end points (ITT population) |
Rescue medication use
There were no differences between treatment groups in the LS mean change from baseline in rescue medication use over weeks 1–12 (0.0 puffs/day, 95% CI: −0.2 to 0.1; Table 3), or in the median percentage of rescue-free days (0.0, 95% CI: 0.00 to 0.18; Table 3).
Inhaler assessments
At the end of the treatment period, a larger proportion of patients indicated an overall device preference for the ELLIPTA™ inhaler compared with the HandiHaler® (57% and 19%, respectively; Table 4). At day 84, 95% of patients rated the ease of use of the ELLIPTA™ inhaler as “Very easy” or “Easy”, while 78% rated the ease of use of the HandiHaler® as “Very easy” or “Easy” (Table 4). The proportion of patients reporting “Neutral” in the ease-of-use assessment at day 84 was 16% for the HandiHaler® and 5% for the ELLIPTA™ inhaler (Table 4). On days 1, 28, and 84, the proportion of patients with at least one overall error ranged between 8% and 13% and was similar between both treatment groups. The proportion of patients with critical errors with the ELLIPTA™ inhaler and with the HandiHaler® was very low at all clinic visits for both inhaler types and ranged between 1% and 4%.
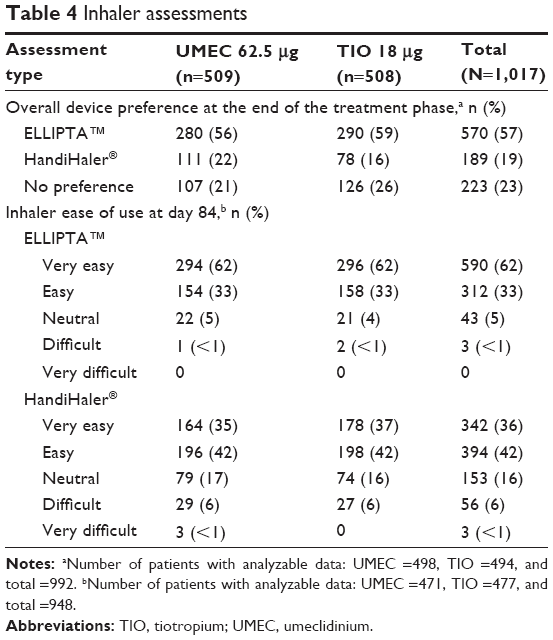 | Table 4 Inhaler assessments |
Post hoc analyses
The LS mean change from baseline in trough FEV1 for UMEC at day 85 was greater for patients with GOLD Grade 2 COPD than those with GOLD Grade 3 COPD with both UMEC and TIO (Table S4). A greater difference in the LS mean change from baseline in trough FEV1 in favor of UMEC versus TIO was observed for patients with GOLD Grade 2 COPD (63 mL, 95% CI: 25–100; P=0.001; Table S4) but not for patients with GOLD Grade 3 COPD at day 85 (39 mL, 95% CI: −4 to 82; P=0.074; Table S4). Patients in GOLD Group B and Group D had significant differences in the LS mean change from baseline in trough FEV1 in favor of UMEC versus TIO (57 mL, 95% CI: 16–98; P=0.006; and 46 mL, 95% CI: 7–85; P=0.020, respectively; Table S4). The LS mean change from baseline in trough FEV1 was greater for UMEC compared with TIO for both ICS users and ICS nonusers (Table S4). For patients with GOLD Grade 3 COPD, a greater difference in the LS mean change from baseline in trough FEV1 in favor of UMEC was observed for ICS users (65 mL, 95% CI: 7–123; P=0.028) but not for ICS nonusers (9 mL, 95% CI: −55 to 72; P=0.784). UMEC was associated with a greater chance of having an increase in trough FEV1 from baseline of ≥100 mL above baseline at day 85 compared with TIO (UMEC 53%; TIO 45%; odds ratio: 1.35, 95% CI: 1.06–1.74; P=0.017; Table S4), and at days 28, 56, and 84 (Figure 4).
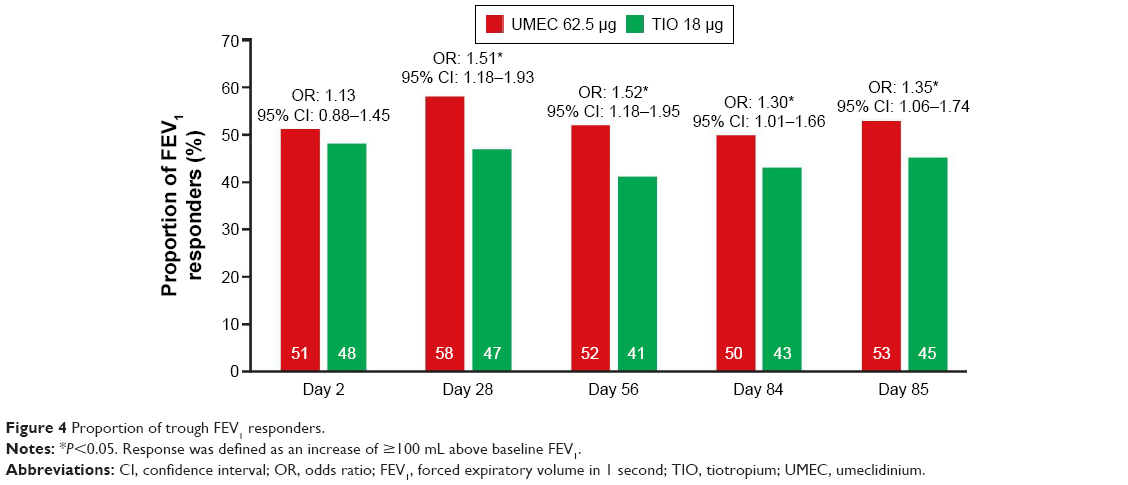 | Figure 4 Proportion of trough FEV1 responders. |
Safety
The overall incidence of AEs during treatment was similar between both treatment groups (32% in the UMEC group and 30% in the TIO group; Table 5). The most common AEs were headache (6% for both treatments) and nasopharyngitis (5% for both treatments).
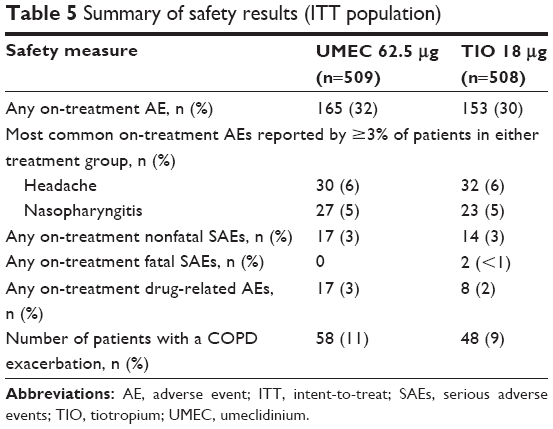 | Table 5 Summary of safety results (ITT population) |
Two deaths occurred on-treatment during the study (TIO, <1%). Neither of the two events (alcohol poisoning, seizure) were considered related to study drug by the reporting investigator.
A total of 58 patients (11%) in the UMEC group and 48 patients (9%) in the TIO group experienced an on-treatment COPD exacerbation. There were minimal changes from baseline in vital signs over the treatment period for either treatment group (Table S5). The incidence of AEs of special interest was similar between the two treatment groups (≤2% in both groups; Table S6).
Discussion
This study aimed to determine whether once-daily UMEC 62.5 μg was non-inferior to TIO 18 μg by trough FEV1 in patients with symptomatic moderate-to-severe COPD. In this randomized trial, UMEC 62.5 μg was found to be both non-inferior and superior to TIO 18 μg, when assessed by trough FEV1 at day 85 in the PP population (treatment difference: 59 mL, 95% CI: 29–88), as the lower bound of the 95% CI around the treatment difference exceeded 0 mL. Similar differences in this primary end point were observed in the ITT population.
As with the primary end point, UMEC demonstrated statistically significant improvements in trough FVC at days 28, 56, 84, and 85 compared with TIO. Interestingly, the treatment difference for trough FEV1 and FVC did not reach statistical significance after the first dose (day 2) but was apparent at day 28, the first assessment after repeat dosing. The 12- to 24-hour WM FEV1 at day 84 was statistically significantly greater for UMEC versus TIO, whereas there were no differences between the treatments for WM FEV1 over 0–12 and 0–24 hours. This result was further supported by serial FEV1 measurements and demonstrates the 24-hour duration of bronchodilation action with UMEC, possibly suggesting that UMEC has greater efficacy in the second half of a 24-hour dosing interval than TIO. Analyses performed post hoc highlighted that more patients receiving UMEC than patients receiving TIO achieved a clinically important (≥100 mL) improvement in trough FEV1. Patients also generally favored using the ELLIPTA™ inhaler over the HandiHaler®, as shown by the larger proportion of patients reporting a device preference and greater ease of use for the ELLIPTA™ inhaler compared with the HandiHaler®.
A possible explanation for the difference in bronchodilation between UMEC and TIO is the dose–response relationships of the two drugs. Dose–response evaluations for UMEC and TIO in COPD have shown that both drugs have relatively flat dose–response curves.25–27 Nevertheless, these evaluations showed dose differentiation for both drugs, with maximal improvements in lung function generally observed at doses above the approved doses evaluated in the current study.25–27 Therefore, a potential explanation for the difference in bronchodilation between UMEC and TIO is that the approved dose of UMEC (62.5 μg)9,10 is more optimally positioned near the top of its dose–response curve compared with the approved dose of TIO (18 μg).28 Additionally, while both compounds are very potent muscarinic antagonists, they are chemically distinct, providing a different array of receptor–ligand interactions.29 Although quite speculative, differences in interactions with muscarinic receptors may also contribute to differences in functional activity. However, to understand differences in pharmacology between these two drugs, they would have to be tested at equi-effective doses, preferably in the same subjects.
Finally, because of the differences in administration between the two medications and the different device used for each, it is not possible to determine whether the differences in bronchodilator effect observed are due to pharmacologic effects of the drugs themselves or to differences in the devices that might have influenced inhalation technique and/or drug absorption and lung deposition. There was a higher patient preference for the ELLIPTA™ device compared with the HandiHaler® device, particularly for ease of use. The preferred device may have been used more effectively, thus leading to improved administration of the drug. This in turn could have contributed to the greater improvement in lung function results for UMEC compared with TIO. However, further studies would be necessary to confirm or differentiate these effects.
The incidence of any AEs was similar between both treatment groups, and the most common AEs were headache and nasopharyngitis, in agreement with previous studies of UMEC 62.5 μg.6
Improvements from baseline in TDI, SGRQ score, and CAT score were demonstrated with both UMEC and TIO. No differences in rescue medication use were observed between UMEC and TIO, as assessed by mean number of puffs per day of rescue medication and percentage of rescue-free days over weeks 1–12. There were no statistically significant between-group differences for PROs.
The reason for the difference between objective lung function assessments and the subjective PRO assessments is likely to be multifaceted. It is possible that the extra ≈60 mL in trough FEV1 at day 85 observed with UMEC versus TIO in this study is the result of improved bronchodilation during the second part of the day, when patients’ levels of activity are not sufficient to provoke symptoms.30 Furthermore, patients with COPD have reported that the early morning involves the greatest burden on them in terms of symptoms.30 Therefore, PROs which focus on the early morning period may be required to detect a treatment difference in PRO assessments between UMEC and TIO. In support of this hypothesis, it is notable that significant reductions in the limitation of morning activities were documented in favor of aclidinium versus TIO.12 Additional studies are required to explore this further. In addition, because PROs are subjective outcomes and marked clinically important benefits on TDI and SGRQ were seen from baseline with both LAMAs, alternative study designs may be needed to increase sensitivity to detect differences in PROs between the two active LAMAs.
Until this investigation, studies specifically designed and powered to compare the efficacy and safety of UMEC with blinded TIO were lacking. A Phase II 14-day study, which included an open-label TIO 18 μg arm, demonstrated that short-term efficacy was achievable with UMEC 62.5 μg and TIO 18 μg.26 However, previous studies comparing twice-daily aclidinium and glycopyrronium with TIO failed to show similar findings.11,12 In a study which compared aclidinium with TIO, aclidinium showed comparable efficacy to TIO by change from baseline morning trough FEV1 at week 6 (treatment difference: 38 mL, not significant).12 Similarly, glycopyrronium was shown to be non-inferior to TIO by trough FEV1 at week 12 (difference: 0 mL, P<0.001).11
There are potential limitations to the interpretation of the data in this study. First, there were differences in the markings between the TIO and PBO capsules which may be seen as influencing the blinding of study treatment. However, a similar methodology to account for this has been used in previous studies that have detected no differences in trough FEV1 between the alternative LAMA therapy glycopyrronium and TIO (eg, the GLOW5 comparator trials of glycopyrronium versus TIO).11 Second, this was a controlled, short-term study in which patients were supervised while administering their study medication. Therefore, patients were expected to have minimal critical errors in device handling. Additionally, critical errors were measured in a subset of patients only, and so these results are less likely to be definitive. Longer-term studies in a real-world population are required to further characterize the frequency of critical errors with these inhalers. Finally, the duration of this study was too short to evaluate the comparative efficacy of UMEC and TIO on exacerbation rate.
Conclusion
The results of this randomized trial indicate that treatment with UMEC provides superior efficacy when assessed by trough FEV1 at day 85 versus TIO in patients with symptomatic moderate-to-severe COPD. Both treatments showed clinically meaningful improvements in PRO assessments, with no significant differences between treatment groups for TDI, SGRQ, and CAT scores. UMEC and TIO showed similar tolerability and safety profiles.
Acknowledgments
This study was funded and conducted by GSK (GSK study number: 201316; Clinicaltrials.gov identifier: NCT02207829). Palvi Shah (GSK Clinical Statistics, Stockley Park, UK) provided substantial contributions to the study data analysis, data interpretation, and preparation of the manuscript. Medical writing assistance was provided by Stuart Wakelin and Matthew Robinson of Fishawack Indicia Ltd (UK), funded by GSK.
Disclosure
GF has no conflicts of interest in this work. FM holds a CIHR/GSK research chair on COPD and has received fees for speaking at conferences sponsored by Boehringer Ingelheim (BI), Novartis, and Grifols, and has served on advisory boards for GSK and BI. He has also received research grants from GSK, BI, AstraZeneca, Nycomed, and Novartis, and has received an unrestricted research grant from BI and GSK. SK, MVB, AC, and WAF are employees of GSK and hold stocks/shares in GSK. RT was an employee of GSK and holder of stock/shares in GSK at the time of the study but is now employed by Pearl Therapeutics. The authors report no other conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease (GOLD) [updated 2015]. Available from: http://www.goldcopd.org/. Accessed August 12, 2015. | ||
World Health Organization – burden of COPD; 2015. Available from: http://www.who.int/respiratory/copd/burden/en/. Accessed August 19, 2015. | ||
Barr RG, Bourbeau J, Camargo CA, Ram FS. Inhaled tiotropium for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;(2):CD002876. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. | ||
O’Donnell DE, Fluge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnoea and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832–840. | ||
Trivedi R, Richard N, Mehta R, Church A. Umeclidinium in patients with COPD: a randomised, placebo-controlled study. Eur Respir J. 2014;43(1):72–81. | ||
Eklira Genuair EMA assessment report; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002211/WC500132663.pdf. Accessed August 12, 2015. | ||
Seebri Breezhaler EMA assessment report; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002430/WC500133771.pdf. Accessed September 23, 2015. | ||
INCRUSE ELLIPTA: highlights of prescribing information; 2013. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Incruse_Ellipta/pdf/INCRUSE-ELLIPTA-PI-PIL.PDF. Accessed August 12, 2015. | ||
Summary of product characteristics: Incruse 55 micrograms inhalation powder; 2015. Available from: https://www.medicines.org.uk/emc/medicine/29394. Accessed August 12, 2015. | ||
Chapman KR, Beeh KM, Beier J, et al. A blinded evaluation of the efficacy and safety of glycopyrronium, a once-daily long-acting muscarinic antagonist, versus tiotropium, in patients with COPD: the GLOW5 study. BMC Pulm Med. 2014;14:4. | ||
Beier J, Kirsten AM, Mroz R, et al. Efficacy and safety of aclidinium bromide compared with placebo and tiotropium in patients with moderate-to-severe chronic obstructive pulmonary disease: results from a 6-week, randomized, controlled Phase IIIb study. COPD. 2013;10(4):511–522. | ||
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects; 2008. Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdf. Accessed September 29, 2015. | ||
Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2(6):472–486. | ||
Maleki-Yazdi MR, Kaelin T, Richard N, Zvarich M, Church A. Efficacy and safety of umeclidinium/vilanterol 62.5/25 mcg and tiotropium 18 mcg in chronic obstructive pulmonary disease: results of a 24-week, randomized, controlled trial. Respir Med. 2014;108(12):1752–1760. | ||
Witek TJ Jr, Mahler DA. Minimal important difference of the transition dyspnoea index in a multinational clinical trial. Eur Respir J. 2003;21(2):267–272. | ||
Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD. 2005;2(1):75–79. | ||
Kon SS, Canavan JL, Jones SE, et al. Minimum clinically important difference for the COPD Assessment Test: a prospective analysis. Lancet Respir Med. 2014;2(3):195–203. | ||
Ingelheim B. Spiriva® 18 microgram, inhalation powder, hard capsule – package leaflet: information for the user; 2014. Available from: https://www.medicines.org.uk/emc/PIL.16286.latest.pdf. Accessed October 20, 2015. | ||
GSK. Incruse® 55 micrograms inhalation powder, pre-dispensed – package leaflet: information for the user; 2015. Available from: https://www.medicines.org.uk/emc/PIL.29387.latest.pdf. Accessed October 20, 2015. | ||
Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2(1):111–124. | ||
Snapinn SM. Noninferiority trials. Curr Control Trials Cardiovasc Med. 2000;1(1):19–21. | ||
Lewis JA, Machin D. Intention to treat – who should use ITT? Br J Cancer. 1993;68(4):647–650. | ||
Church A, Beerahee M, Brooks J, Mehta R, Shah P. Dose response of umeclidinium administered once or twice daily in patients with COPD: a randomised cross-over study. BMC Pulm Med. 2014;14:2. | ||
Donohue JF, Anzueto A, Brooks J, Mehta R, Kalberg C, Crater G. A randomized, double-blind dose-ranging study of the novel LAMA GSK573719 in patients with COPD. Respir Med. 2012;106(7):970–979. | ||
Littner MR, Ilowite JS, Tashkin DP, et al. Long-acting bronchodilation with once-daily dosing of tiotropium (Spiriva) in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1136–1142. | ||
Summary of Product Characteristics: Spiriva 18 microgram inhalation powder, hard capsule. Available from: https://www.medicines.org.uk/emc/medicine/10039. Accessed January 27, 2016. | ||
Salmon M, Luttmann MA, Foley JJ, et al. Pharmacological characterization of GSK573719 (umeclidinium): a novel, long-acting, inhaled antagonist of the muscarinic cholinergic receptors for treatment of pulmonary diseases. J Pharmacol Exp Ther. 2013;345(2):260–270. | ||
Partridge MR, Karlsson N, Small IR. Patient insight into the impact of chronic obstructive pulmonary disease in the morning: an internet survey. Curr Med Res Opin. 2009;25(8):2043–2048. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.