Back to Journals » Patient Preference and Adherence » Volume 20
A Qualitive Investigation of Geographic Disparities in Genetic Testing and Care Access for Amyotrophic Lateral Sclerosis: Insights From Patient Journey Mapping
Authors Sethi N, Levy OA, Richardson A, Harari OA, Sellati R
Received 2 October 2025
Accepted for publication 16 December 2025
Published 15 January 2026 Volume 2026:20 566747
DOI https://doi.org/10.2147/PPA.S566747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Johnny Chen
Nadia Sethi,1 Oren A Levy,2 Angela Richardson,3 Olivier A Harari,2 Rosemarie Sellati4
1Northeast ALS Consortium Patient Education, University of Florida, College of Pharmacy, Clinical Pharmacogenomics and Precision Medicine, Gainesville, FL, USA; 2Global Development, Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA; 3Department of Patient Engagement, IQVIA, Boston, MA, USA; 4Patient Advocacy, Regeneron Pharmaceuticals, Inc., Sleepy Hollow, NY, USA
Correspondence: Rosemarie Sellati, Patient Advocacy, Regeneron Pharmaceuticals, Inc., 1 Rockwood Road, Sleepy Hollow, NY, 10591, USA, Tel +1 914-348-5059, Email [email protected]
Purpose: Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disease that is associated with a high patient burden, reduced lifespan, and reduced quality of life. People living with ALS (PLwALS) often experience delays in diagnosis of ~1 year, and while current treatment options can slow disease progression, improve quality of life, and offer modest benefits in functional decline, they do not reverse neuronal damage. Defining and understanding the experiences of PLwALS can help identify gaps and barriers to optimal care.
Patients and Methods: This non-interventional study was intended to obtain insights on the patient experience from the perspective of PLwALS. We sought to develop an ALS patient journey map from initial presentation through to end-of-life care for 4 regions (North America, Asia-Pacific, Latin America, and Europe/Middle East/Africa). The map was based on results from a global audit of data sources (including publications, patient narratives, society guidelines, academic reports, industry white papers [n=104]), and one-to-one, 60-minute, semi-structured interviews with PLwALS (n=12) or those caring for PLwALS (n=2). The initial patient journey map was subsequently reviewed during 2 advisory sessions with patient advocates (7 PLwALS and 2 caregivers) in September and November 2023.
Results: We identified several barriers and challenges that most impact PLwALS, caregivers, and clinicians, with many similarities but also some differences across the regions.
Conclusion: These insights will enable targeted improvements in education, personalized care, resource allocation, care coordination, policy development, and funding, ultimately improving patient outcomes.
Keywords: amyotrophic lateral sclerosis, clinical care pathways, genetic testing, patient engagement, patient experience map, quality of life
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disease caused by deterioration of motor neurons in the spinal cord and brain that leads to high patient burden, reduced lifespan, and a negative impact on quality of life for people living with ALS (PLwALS) and their caregivers.1,2 In 2021, the estimated global prevalence of motor neuron disease, of which ALS is the most common type, was 273,000.3
ALS symptoms depend on the region affected.4 Typically, the initial symptom is progressive, unilateral weakness in an arm or leg (limb-onset ALS), resulting in difficulty performing daily tasks such as dressing and feeding.2 As the disease progresses, PLwALS may experience the inability to walk, speak, swallow, and breathe, and ultimately an early death.2,4 Some PLwALS present with bulbar-onset ALS and experience symptoms including dysarthria (slurring of speech), facial weakness, difficulty chewing, and dysphagia (difficulty swallowing) early in their disease, progressing to limb features later in the disease course.2 The current predicted survival for PLwALS is 2–5 years, although some can survive more than a decade from the initial diagnosis; those with bulbar‑onset ALS tend to have shorter survival times.2,4
Diagnosis of ALS requires a series of clinical examinations and neurological tests.5 However, due to the rarity and heterogeneity of the disease, alongside the lack of diagnostic markers and delays in referrals to neuromuscular specialists,5–7 the path to a formal diagnosis can be convoluted, with PLwALS frequently experiencing misdiagnoses and delays in diagnosis of approximately 1 year.6
Approximately 10% of ALS cases are familial.2,4 A breakthrough in the understanding of ALS came in 1993, when a mutation in the superoxide dismutase 1 (SOD1) gene was first implicated in its pathogenesis.8 SOD1 mutations are found in approximately 1–3% of sporadic ALS and 12–20% of familial ALS cases.2,4 Currently, variants in more than 120 different genes linked with ALS have been identified.2,9
Current treatment options do not reverse motor neuron damage or cure ALS, and offer only modest benefits in delaying functional decline.4,5 Until relatively recently, only 2 therapies (riluzole and edaravone) had been fully approved by the US Food and Drug Administration (FDA) for slowing ALS progression; several other therapies may be recommended to help relieve and manage ALS symptoms.10 In 2023, the FDA gave accelerated approval to tofersen, an antisense oligonucleotide specifically targeting SOD1, for the treatment of ALS caused by SOD1 mutations.10,11 Subsequently, tofersen has been granted approval in several other countries.12 Given the presence of known pathogenic variants, including in seemingly sporadic ALS,13 together with the approval of new targeted treatment options (eg, tofersen), genetic testing is now recommended for all PLwALS, regardless of family history.9,14 However, access to genetic testing and the methodologies used are inconsistent and vary geographically.9,14
While research continues to investigate new therapies, there is increasing recognition of the importance and value of understanding the patient experience, which has been defined as “the sum of all interactions…that influence patient perceptions across the continuum of care”.15 Defining and understanding the experiences PLwALS face can help identify gaps and barriers to optimal care. Input from PLwALS, caregivers, families, and healthcare professionals (HCPs) directly impacted by the disease is critical. Patient journey maps are increasingly being utilized to empower researchers in identifying the barriers and gaps in care that most impact patients and their caregivers.16 Patient journey mapping is an established approach in health-services research and patient-centered design, as it enables systematic identification of barriers, unmet needs, and decision points across the disease continuum. Its use is grounded in qualitative methodological principles outlined in the Consolidated Criteria for Reporting Qualitative Research (COREQ), which provides standards for rigor, reflexivity, and transparency in multi-source qualitative synthesis.17 The insights gained from developing these maps can be used to improve and enhance patient‑centric care by identifying gaps in communication and resources, delays in diagnosis, and challenges in receiving personalized and coordinated care.18,19
A complete patient journey map for PLwALS from initial symptom presentation to the end of life has yet to be elucidated. While prior qualitative and mixed-methods studies have examined aspects of the ALS care experience, including diagnostic delay, variability in access to multidisciplinary care, and caregiver burden, most have been conducted within single countries or clinical centers and do not address global or regional variability.7,20–25 These studies identified numerous difficulties, including delays in referrals to specialist services and the unsatisfactory delivery of diagnosis, which can negatively impact mental health and quality of life. While these studies have provided useful information on the diagnostic challenges PLwALS experience,7,20 there are differences not only in how PLwALS present and progress, but also how they are managed and treated around the world.
Building on these insights, our study sought to examine what regional differences exist between North America (NORAM), Asia-Pacific (APAC), Latin America (LATAM), and Europe/Middle East/Africa (EMEA) in the lived experience and care pathway for people diagnosed with ALS and their caregivers, and how system-level factors shape access to timely diagnosis, multidisciplinary support, and emerging genetic-based therapies. This global patient journey mapping exercise is grounded in a comparative, descriptive framework intended to synthesize evidence across regions and highlight actionable disparities that can inform advocacy, clinical practice, and policy.
Methods
Objectives
The primary objective of this study was to develop and refine ALS patient journey maps for NORAM, APAC, LATAM, and EMEA that accurately represent the perspectives of those living with or caring for someone with ALS. We sought to identify the barriers to optimal care, opportunities to improve the experience at various stages of the patient journey, and the barriers and challenges in the patient journey that are most bothersome to PLwALS. Finally, we sought to develop a better understanding of regional differences with respect to barriers to optimal care, access to genetic testing, and considerations for clinical trial participation.
Development of the Patient Journey Map Framework
In this study, we employed a patient journey mapping methodology to synthesize insights across published literature, global desktop research, and semi-structured interviews. An initial global ALS patient journey map was created based on results from a global audit and patient advocate interviews, grounded in qualitative methodological principles outlined in the COREQ. In addition, our analytic process aligned with framework-based methods commonly used in applied health research, in which diverse data streams are organized into predefined and emergent categories to support comparison across regions and health-system contexts.26,27
Global Audit
Secondary research was conducted across 2022 and 2023 to identify data on the PLwALS experience. Data sources included publications, PLwALS narratives, society guidelines, academic reports, industry white papers, and interviews with PLwALS or those caring for PLwALS (Supplemental Table S1 and Table 1).
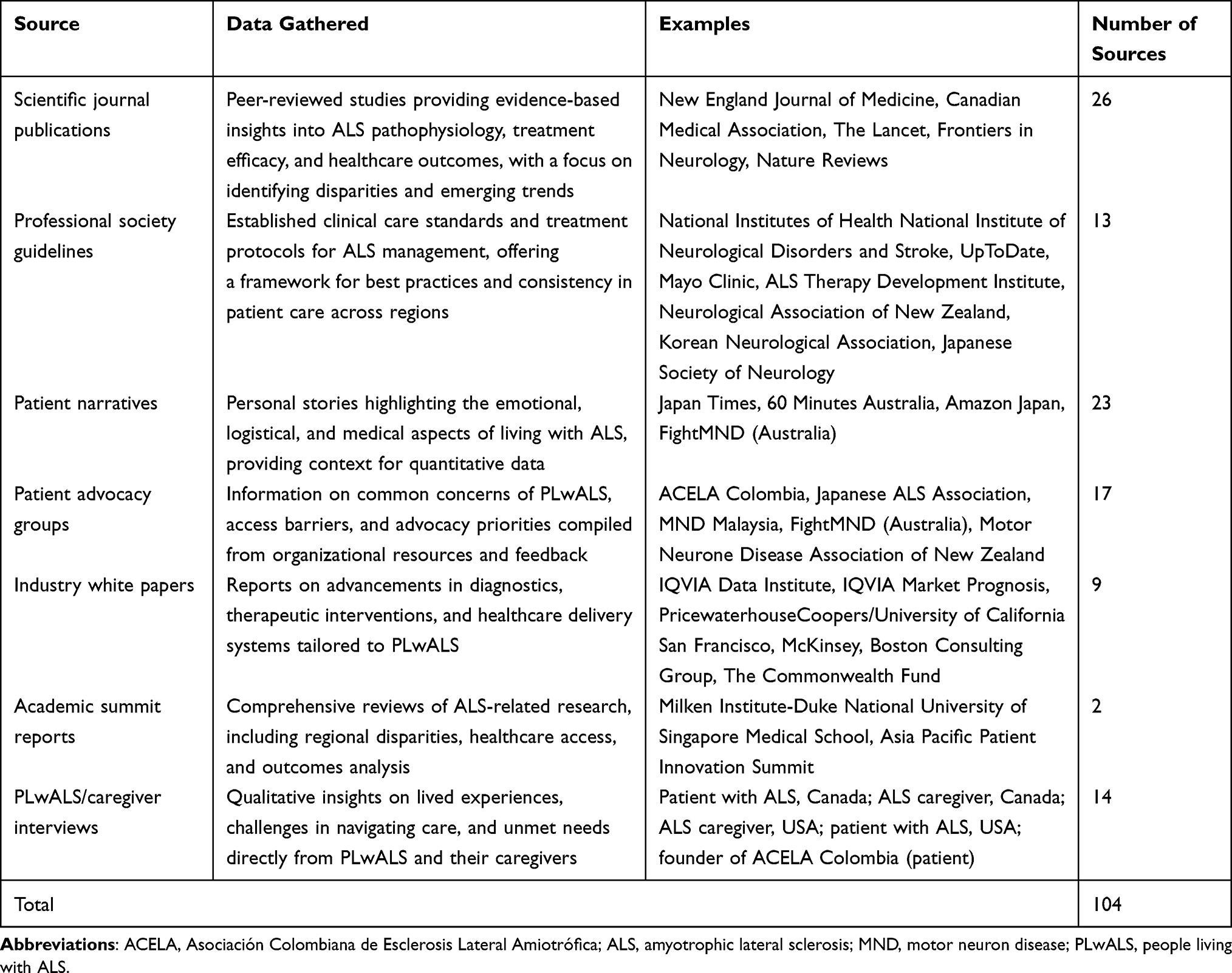 |
Table 1 Global Sources Used to Define the ALS Patient Experience |
Patient and professional organizations with an interest in ALS or neuromuscular diseases were identified using search engines (Google and Bing). Research was conducted across the 4 geographical regions, with search engine locations set to the market being analyzed and using the native language of that market (Figure 1). Search terms used included, [“amyotrophic lateral sclerosis” or “ALS”] and [“patient support”, “patient education”, “education”, “support”, “advocacy group,” or “patient group”]. Organizations identified in the search were assessed and included if they were a registered non-profit providing education and/or support services to people impacted by ALS. Data were also included from organizations identified as partners of groups identified in the initial search. Websites and platforms hosted by for-profit organizations were excluded.
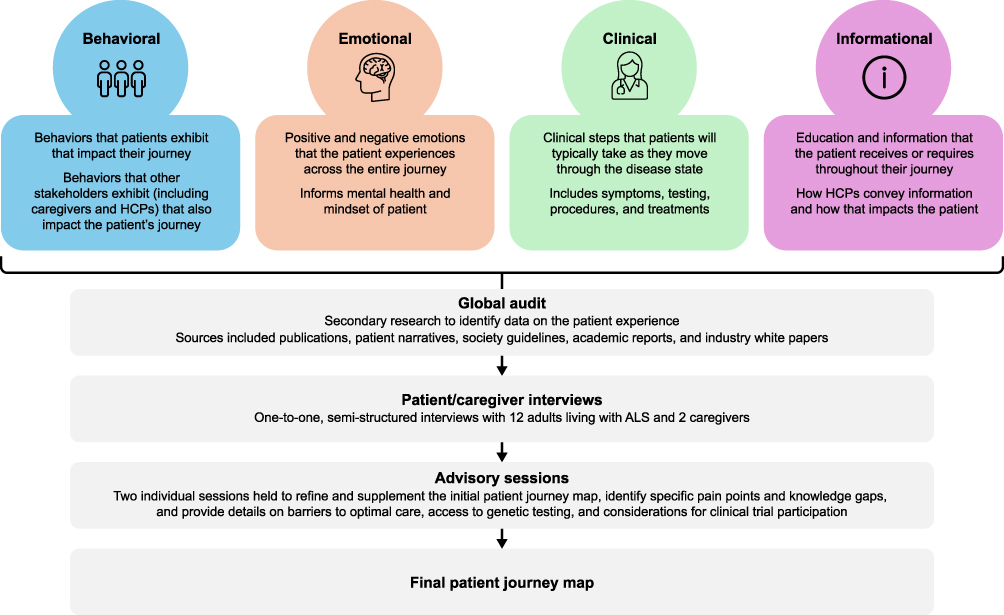 |
Figure 1 Development of the patient journey framework. Abbreviations: ALS, amyotrophic lateral sclerosis; HCP, healthcare professional. |
Data sources that were deemed to provide an authentic and accurate perspective of the patient experience were further evaluated for their reliability and validity. Relevant content from these sources was used to establish the initial ALS patient journey framework.
Patient Advocate Interviews
Interviewee Samples and Recruitment
Interviewees were identified through the leading patient organizations for the region, and all had experience engaging with the healthcare industry. Recruitment materials were disseminated by advocacy organizations, and interested individuals contacted the research team directly to volunteer. All interviewees were aged ≥18 years and either had a confirmed ALS diagnosis (including individuals with known SOD1 or other genetic variants where available) or had cared/were caring for someone with a confirmed ALS diagnosis. A purposive sampling approach was used to capture diverse perspectives across gender, genetic status, and disease type. The final sample comprised a total of 12 PLwALS and 2 caregivers, including2 female caregivers, 2 female participants with SOD1-ALS, 2 male participants with SOD1-ALS, 4 female participants with sporadic ALS, and 4 male participants with sporadic ALS. Four PLwALS and 1 caregiver were from the ALS Association (USA), 3 PLwALS and 1 caregiver were from ALS Canada (Canada), 1 PLwALS was from the European Organisation for Professionals and People with ALS (EUpALS; Europe), 1 PLwLAS was from the International Alliance of ALS/MND Associations (EUMEA, LATAM, and APAC), and 3 PLwALS were jointly affiliated with the EUpALS and the International Alliance of ALS/MND Associations. Among the interviewees, only 1 PLwALS was involved in reviewing the questionnaire as well as participating in the research; the caregiver who assisted with refinement of the questionnaire did not participate in the study. All other individuals from the patient organizations participated only in the research and were not involved in the questionnaire development.
Interview Structure
One-to-one, 60-minute interviews were conducted virtually across 2022 and 2023 using secure video conferencing platforms with the 14 adult participants in either English or Spanish, depending on the interviewee’s native language. The semi-structured interviews followed a predefined discussion guide (see Supplementary Materials) covering clinical care experiences, emotional and psychological impacts, informational needs, and navigation challenges across the ALS care pathway, including questions about the path to diagnosis, treatment, provider interactions, and lifestyle impact. The aim of these interviews was to gather first-hand perspectives on the lived experience and unmet needs of PLwALS, and to contribute, along with the data from the global audit, to the initial patient journey map. The discussion guide was reviewed and refined by a cross-functional team of subject matter experts, including individuals with PhDs in public health and social work, individuals with MDs in neurology, nonprofit outcome researchers, scientists investigating neurological diseases, a PLwALS, and a caregiver for a PLwALS. This ensured that the content of the questionnaire reflected both clinical and lived experience perspectives.
Interviews were conducted by 2 trained qualitative researchers employed by the external research agency supporting the project (IQVIA). Both interviewers had professional experience conducting patient and caregiver interviews, and held relevant disciplinary backgrounds, including research science, social work, and public health education. Their combined training and experience supported the consistent application of the discussion guide and contributed to minimizing interviewer bias. Verbatim transcripts were generated for analysis.
Analysis
An inductive–deductive hybrid analytical strategy was used to allow themes to emerge organically from interviewee narratives while also aligning with predefined domains relevant to ALS, including clinical, emotional, informational, and logistical dimensions of the care experience. Interview summaries and transcripts were reviewed manually by the research team, who identified recurring patterns, concepts, and experiential touchpoints. Theme development was supported through triangulation and cross-review among team members to confirm consistency, resolve interpretive differences, and enhance analytical credibility. The thematic findings were synthesized into the patient journey map, with insights organized into visual categories that reflected lived experiences across the 4 regions.
Refinement of the Patient Journey Map with Patient Experts
The initial ALS patient journey map was reviewed during 2 advisory sessions with patient experts (7 PLwALS and 2 caregivers) held virtually using secure videoconferencing platforms on September 21 and November 6, 2023. During these meetings the patient experts: (1) refined and supplemented the map with additional details; (2) identified specific pain points and knowledge gaps that were most bothersome to PLwALS, caregivers, and their families; and (3) provided detailed perspectives on barriers to optimal care, access to genetic testing, and considerations for clinical trial participation.
Thematic Coverage
Given the focused aims of this project, thematic convergence was observed across interviews and advisory board discussions, with no substantially new domains emerging in later sessions. The sample allowed for the identification of consistent patterns across clinical, emotional, informational, and logistical domains. The analysis reached a point of practical saturation, after which additional interviews were unlikely to yield new high-level thematic categories relevant to the study objectives.
Ethics
The study was conducted in accordance with international guidelines, including the Declaration of Helsinki. This study was non-interventional and intended to obtain insights on the patient experience from the perspective of PLwALS. Clinical research ethics committee or independent review board approval was not required. Patient experts were compensated for their time per fair market value for patient advocates in their respective countries. All patients provided informed consent, including the publication of anonymized responses and quotes.
Results
Development of the Initial Patient Journey Map Framework
The global audit identified data from 104 different sources (Table 1 and Supplemental Table S1) and provided published evidence on barriers, access challenges, diagnostic patterns, and treatment experiences, which established the initial structure of the framework. The type of content covered by the sources varied substantially from video interviews from a patient perspective on progression and end-of-life care, to guidelines, journal articles, and white papers providing clinical perspectives on testing, diagnosis, and treatment. Patient advocacy groups (PAGs) across the globe (including MND New Zealand, the Korean Neurological Association, and the Japanese Society of Neurology) provided numerous resources to support PLwALS, caregivers, and clinicians across the patient journey.
Refinement of the Patient Journey Map With Patient Experts
The initial ALS patient journey map (Supplemental Figure S1) was reviewed and refined by the patient experts, and focused on 4 themes: clinical; emotional; behavioral; and informational (Figure 1). Overall, the patient experts found the map provided a good representation of the patient experience. However, they were able to provide insights that helped to refine the NORAM‑focused journey and highlight areas of unmet need (Figure 2). Key insights at each of the main stages of the patient journey are summarized below; key quotes from PLwALS and their caregivers are summarized in Table 2.
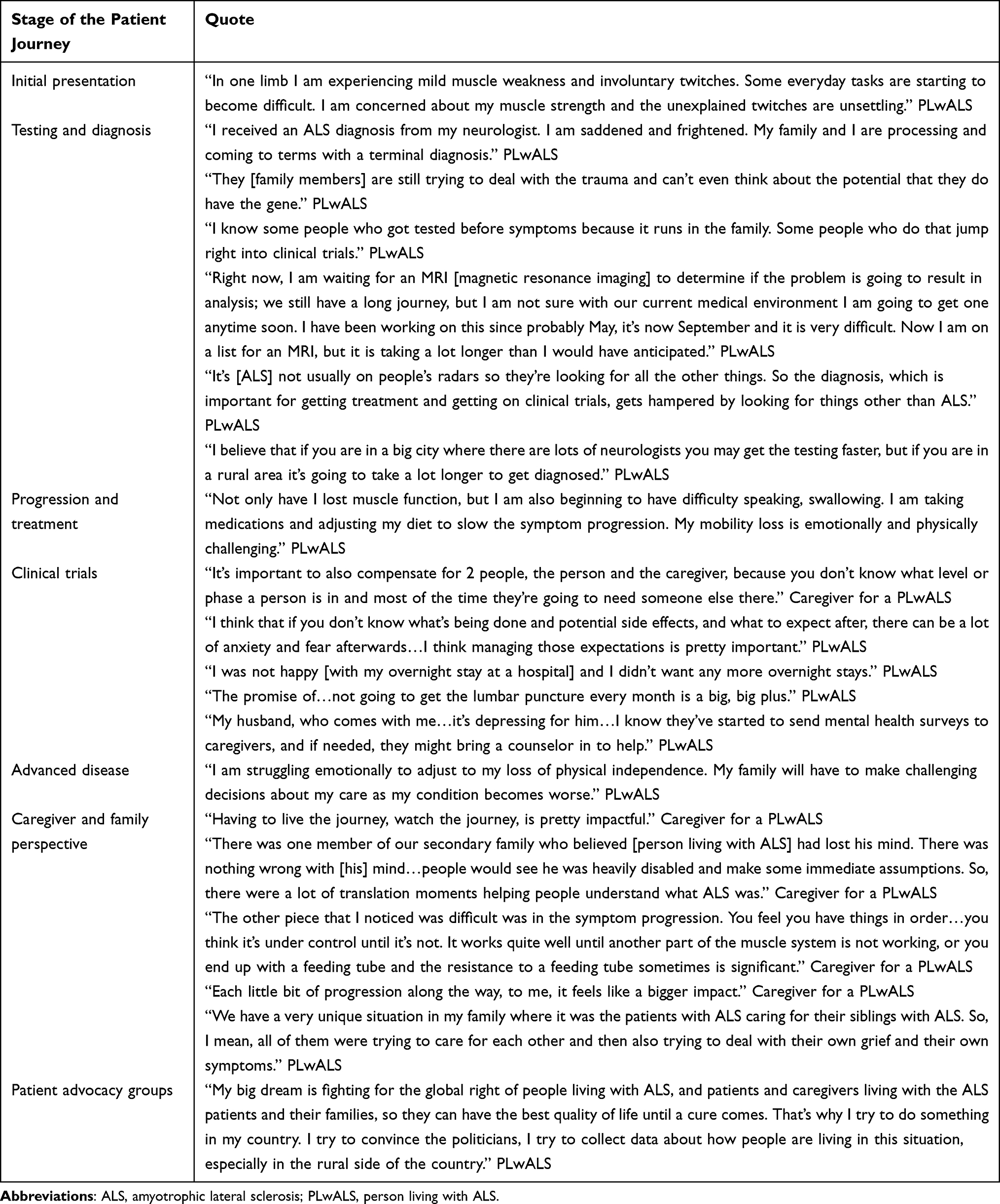 |
Table 2 Key Quotes From PLwALS and Caregivers on Their Experience of Living with ALS |
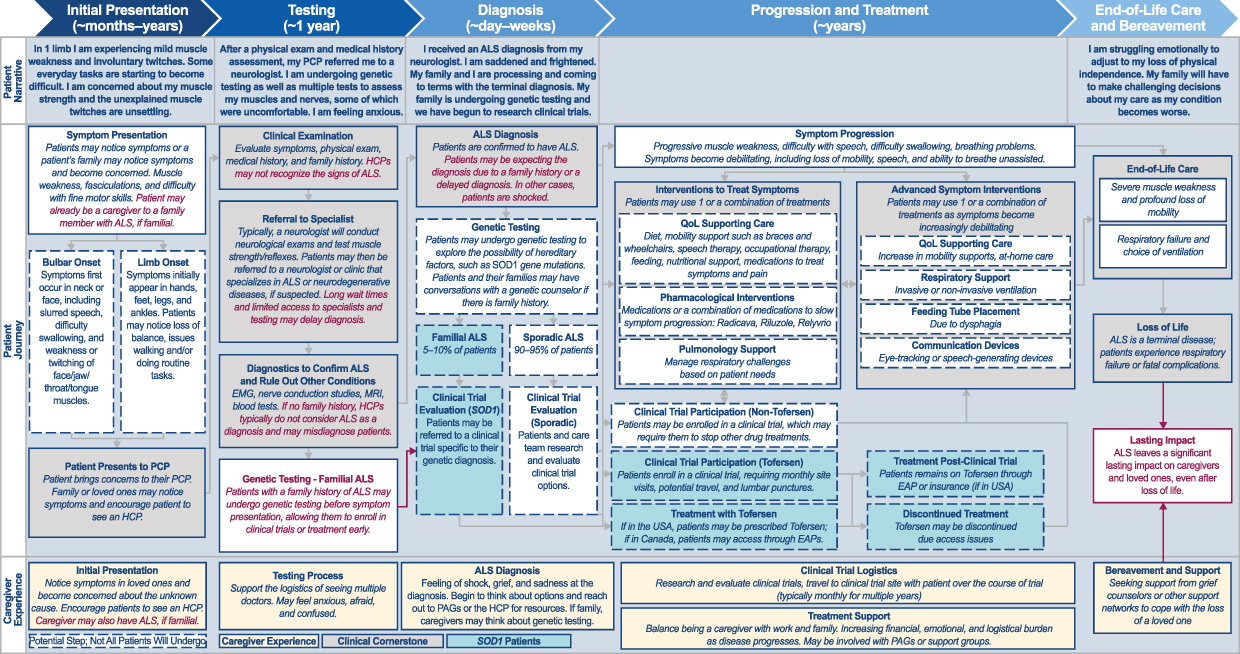 |
Figure 2 Updated ALS patient journey refined by patient advocates (NORAM-focused). Abbreviations: ALS, amyotrophic lateral sclerosis; EAP, employee assistance program; EMG, electromyography; HCP, healthcare professional; MRI, magnetic resonance imaging; NORAM, North America; PAG, patient advocacy group; PCP, primary care physician; QoL, quality of life; SOD, superoxide dismutase. Notes: Red text indicates changes/differences versus the initial patient journey map (Supplemental Figure S1). |
Initial Presentation
When PLwALS first notice symptoms, they generally present to their primary care physician (PCP); however, some may present to emergency care if their symptoms are sudden and severe. During this stage, PLwALS describe feeling scared and confused, and will usually seek further information online or from HCPs.
Testing and Diagnosis
Our research found that the path to an ALS diagnosis is often extended due to lack of disease awareness, availability of specialized medical experts, and access to healthcare. PLwALS are generally referred to a neurologist by their PCP, at which point they will undergo a series of tests which can include neuroimaging, electrophysiological assessment, and bloodwork. The overall process can be lengthy, and during this period PLwALS describe feelings of dread, uncertainty, and anxiety. Once a diagnosis has been confirmed, PLwALS often report feeling scared about their future as well as guilty about potentially passing this genetic condition to their children.
Genetic Testing
PLwALS may experience a range of familial, emotional, clinical, and financial impacts associated with genetic testing. For example, a family history or personal experience of ALS may deter individuals from testing.
Individuals described feeling shame and dread upon receiving confirmation that their ALS is associated with a genetic mutation, and guilt about the hereditary nature of the disease. Others may use the results of genetic testing to inform family planning decisions, carefully taking into consideration their values, morals, and the potential risks before deciding whether to have children or not. Furthermore, clinicians and PLwALS commented that, until recently, they questioned the value of genetic testing due to the lack of available and effective treatment options. However, genetic testing in a family member of a person living with genetic ALS can now allow for clinical monitoring before symptom onset, which could allow PLwALS to enroll in clinical trials or begin treatment earlier,28 potentially extending life expectancy and improving quality of life.
Despite recommendations that genetic testing should be offered to all PLwALS,9,14 our research found that it is not covered by insurance policies in all countries and therefore PLwALS and their families may incur significant out-of-pocket costs associated with testing. A confirmed genetic mutation may also affect the availability and cost of future health and life insurance coverage. Given these challenges, PLwALS and their families need to take into consideration many factors before deciding whether genetic testing, if available, is the right option for them.
Progression and Treatment
As symptoms progress, PLwALS may start treatment to slow progression and/or manage symptoms of the disease. During this stage, PLwALS can experience worry, concern about the unknown, and even grief.
As the disease progresses and symptoms increase/worsen, PLwALS may need to stop work and make modifications to their homes, eg, ramps for wheelchairs, both of which can have a substantial financial impact.
Clinical Trials
Our research observed that PLwALS and their families consider clinical trial outcomes and the potential impact on quality of life when deciding whether to pursue clinical trial enrollment. While PLwALS are hopeful that the investigational treatments will slow progression, they can also feel anxious about a lack of efficacy, unknown side effects, and stopping any treatments they are currently receiving. Furthermore, invasive clinical trial procedures (eg, lumbar punctures) can discourage PLwALS from participating in clinical trials.
Our research also noted that caregivers and family members play a critical role in supporting PLwALS during clinical trials. Having a family member attend clinical trial visits can improve the experience, and caregivers are often needed to support PLwALS in completing all clinical study procedures.
Improving Clinical Trial Participation and Retention
PLwALS are more motivated to enroll in clinical trials if they believe they will experience alleviation or potential reversal of symptoms; however, they fear being placed on a placebo arm or undergoing a washout period, which can increase their reluctance to participate.
Our research also found that evidence of improvement in quality of life may be more or just as compelling and meaningful to PLwALS and caregivers than solely clinical endpoints. Patient experts suggested that, to drive awareness and motivation to participate, clinical trials must minimize participant burden by limiting invasive procedures and making them logistically accessible (eg, minimal overnight stays).29
The patient experts further suggested that at-home assessment tools (eg, wearable devices with suitable privacy safeguards) and appropriate resources at clinical trial sites can help minimize barriers to clinical trial enrollment. Items such as blankets, food, and charging cords would make things more convenient for PLwALS who must stay overnight. It was emphasized that support items should meet the individual needs of PLwALS and cater to the particular needs at the specific point the PLwALS were at in their disease journey (eg, providing a water bottle with a straw to PLwALS with upper limb weakness).
Our findings also highlighted the need to increase education, mental health support, and clear communication regarding what to expect during and after the trial to better prepare PLwALS and their caregivers for the clinical trial journey both medically and logistically. Furthermore, some caregivers are also PLwALS who will witness the disease progression in their family and go through the process themselves. They too need to better understand the clinical trial to help avoid unwanted anxiety and fear.
The patient experts further suggested that social support programs can help drive a sense of camaraderie among PLwALS and caregivers, thereby improving clinical trial recruitment and retention.
Our research found that the rare and heterogeneous nature of ALS has necessitated innovation in clinical trial design. For example, traditional randomized control trials are associated with high operating costs and relatively large patient enrollment, but innovative trial designs, such as multi-arm/multi-stage platform trials, as well as increased digitalization, can accelerate the clinical development process while utilizing fewer participants. Inclusion criteria should also be carefully considered and based on specific prognosticators (eg, genetic mutation status) or disease staging to allow PLwALS to receive tailored investigational treatment. Furthermore, while clinician assessments are the current primary and secondary outcomes, they require travel to the study site, which can be difficult for some PLwALS. Therefore, patient-reported outcomes supplemented by digital records may reduce patient burden and optimize the trial experience, while maintaining the rigor of data collection. Finally, HCPs must be educated and aware of which clinical trials are available, and who to contact for enrollment.
Advanced Disease
As the disease advances, severe muscle weakness, profound loss of mobility, and the need for mechanical ventilation can lead to feelings of grief for PLwALS and their families.
Perspective of the Caregiver and Family
Like PLwALS, caregivers and family members can experience emotional, financial, and interpersonal impacts during the ALS journey, and may go through a series of similar emotions including shock, grief, worry, anxiety, fear, confusion, and sadness.
In general, family members and caregivers provide vital day-to-day support for PLwALS. Not only do caregivers help with activities of daily living, managing medications and medical equipment, and mobility issues, but they frequently advocate for the PLwALS at medical appointments and socially. Consequently, they need to balance being a caregiver with work and other family commitments, which can add to the financial and emotional burden of ALS.
While family members can be extremely supportive, some may not understand the disease, which can result in tense relationships and additional stress. The physical presentations of the disease may result in PLwALS being stigmatized, and caregivers may find themselves having to explain the disease to family members, friends, and even strangers.
The daily impact of ALS on the caregivers and family becomes even more significant as the disease progresses. As symptoms affecting speech, breathing, and mobility become more impactful, often in an unpredictable pattern, caregivers must work with PLwALS to navigate their loss of independence, frequently increasing their own burden. Caregivers can subsequently experience grief, including anticipatory grief (eg, grieving upcoming losses of function and quality of life), burnout, and fear, alongside the increased logistical and financial burden.
In familial ALS, it is common for PLwALS to care for siblings, parents, and children while also managing their own condition. These PLwALS experience substantial emotional and physical burden, and often feel guilt if their own children have the condition.
Comparison of the ALS Patient Journey Across Geographical Regions
Several key aspects of the patient journey were identified and investigated further to elucidate similarities and disparities across the geographical regions, including access to and sentiments on genetic testing and clinical trial participation. These are summarized in Figure 2 for NORAM, and in Figure 3 for APAC, LATAM, and EMEA.
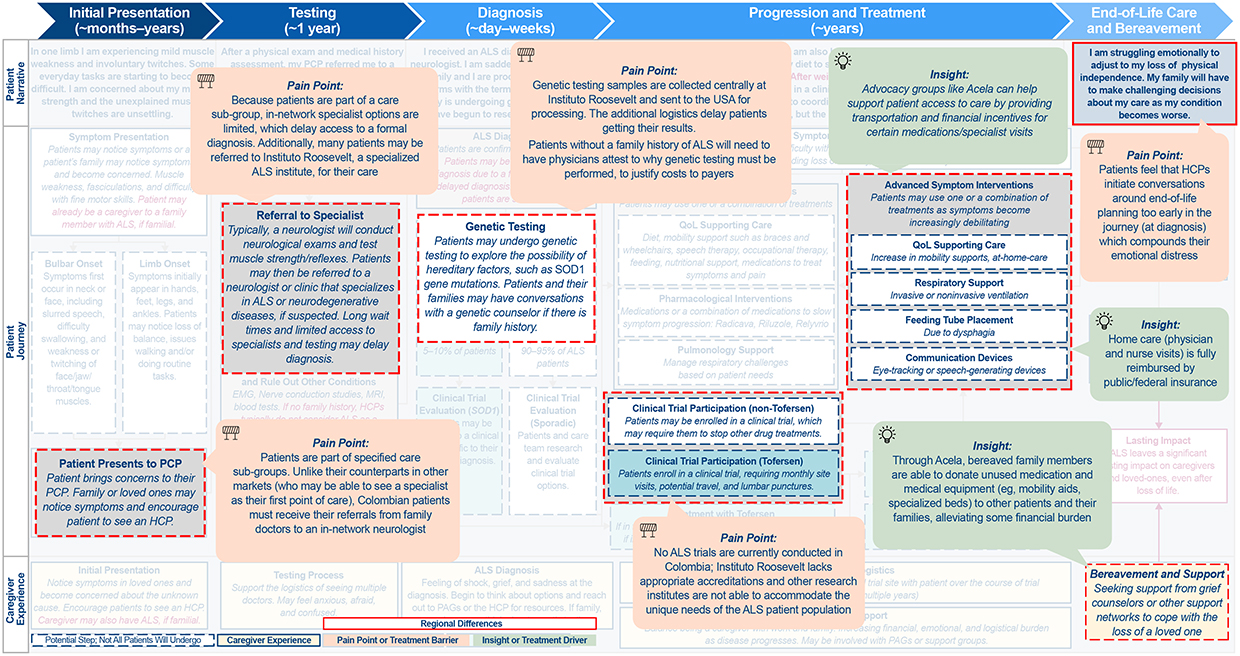
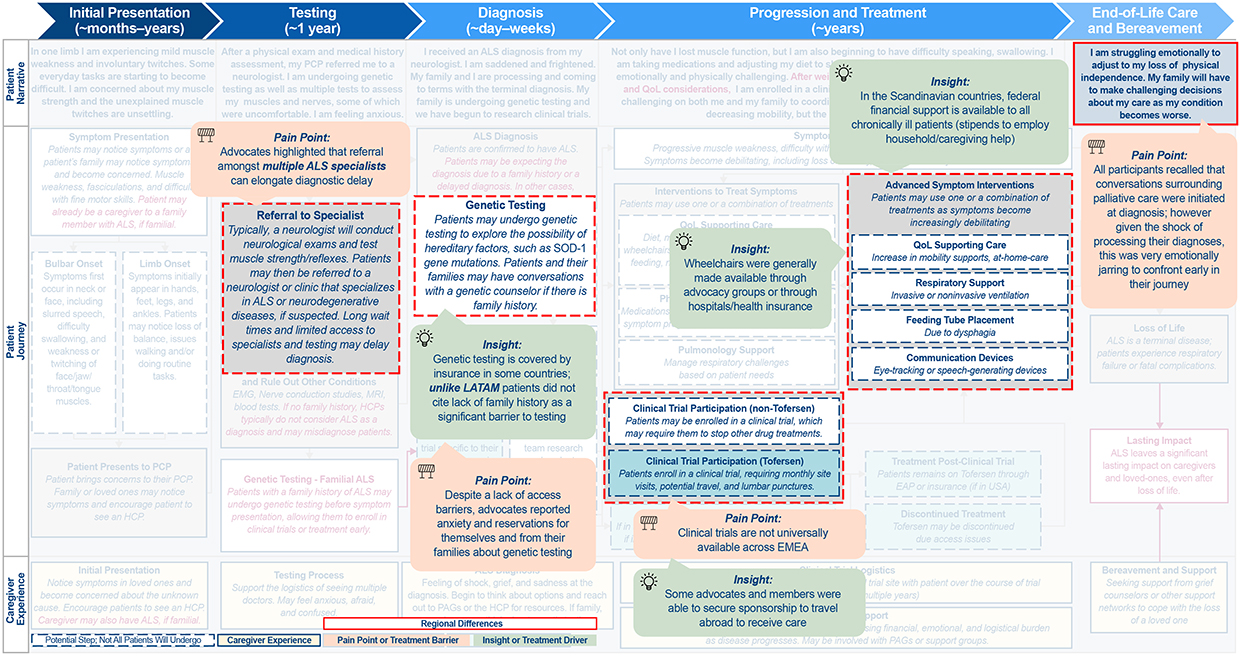
Figure 3 Continued. Figure 3 Continued. Figure 3 Differences in the ALS patient journey in (A) APAC, (B) LATAM, and (C) EMEA. Abbreviations: ALS, amyotrophic lateral sclerosis; APAC, Asia-Pacific; EMEA, Europe/Middle East/Africa; GP, general practitioner; HCP, healthcare professional; LATAM, Latin America; MND, motor neuron disease; NZ, New Zealand; PAG, patient advocacy group; PCP, primary care physician; POC, point of care; QoL, quality of life; SOD, superoxide dismutase. Notes: Highlighted boxes indicate key differences between regions.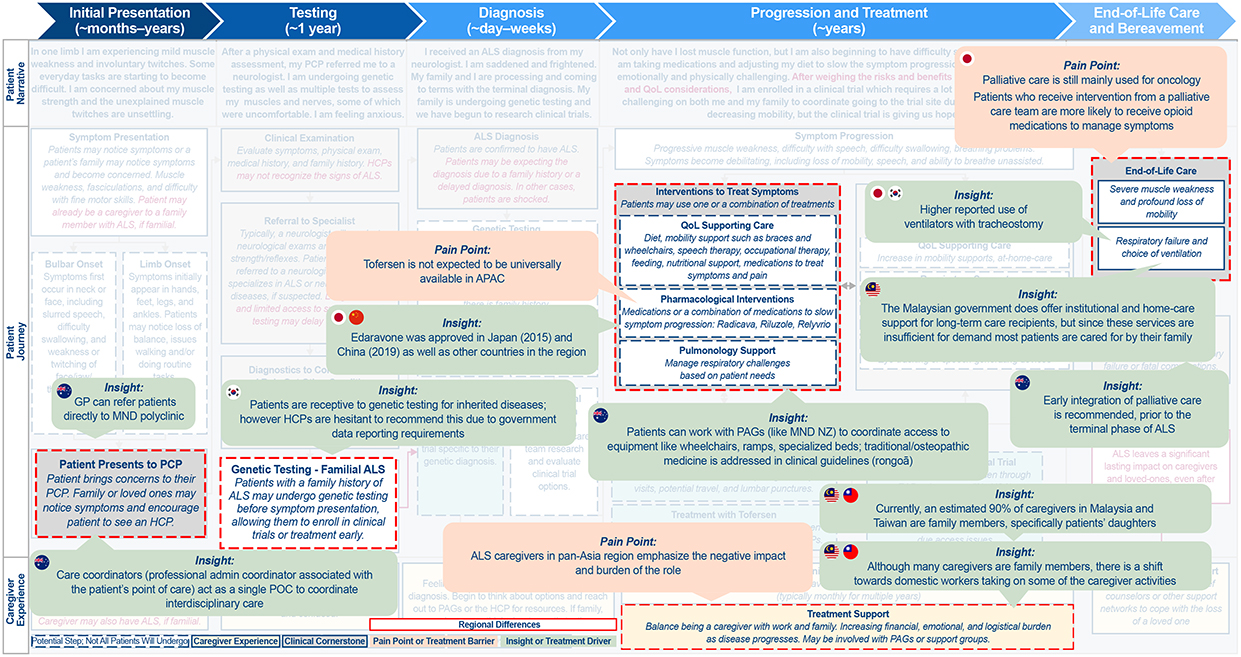
As already noted, PLwALS, caregivers, and family members typically express different emotions across the course of the patient journey; in general, these were found to be consistent across the different regions. Other similarities included barriers to diagnosis and lack of HCP awareness and expertise. Consistently, PLwALS will see multiple HCPs before receiving a diagnosis, and there can be a limited number of specialists, often with long wait times for each. Tools such as thinkALS™,30 which has been developed by the ALS Association to aid clinicians in the diagnosis of ALS, can help speed up diagnosis and reduce wait times.
The journey to an ALS diagnosis is further compounded by its rarity and a lack of awareness of the disease, which was noted across all regions. Many clinicians may not recognize the symptoms of ALS, or mistake symptoms for other disorders. It is common for PLwALS to have at least 1 misdiagnosis.
PLwALS’ location can also impact access to specialists, testing, and healthcare resources, with rural areas typically having fewer specialists and testing capabilities, further increasing wait time and limiting access for PLwALS who live in these areas.
NORAM
The primary challenges in ALS care in NORAM center around the complex healthcare system, and include navigating appointments with specialists and treatment facilities, as well as access and reimbursement for treatment and care. While genetic testing is available in NORAM, PLwALS and family members have mixed sentiments about being tested. Since the availability of genetic-specific treatments (commercially and within clinical trials), some PLwALS with a family history of ALS may now actively seek out genetic testing, even if they are not experiencing symptoms. Some familial PLwALS may also encourage their family members to get genetic testing pre-symptomatically as there is now a potential benefit to early diagnosis. However, for others, a history of family trauma related to the disease may deter individuals from genetic testing. In these instances, individuals may associate a genetic diagnosis with feelings of shame, guilt, and dread. It should also be recognized that genetic testing may affect current and future health and life insurance policies, which can also deter PLwALS from testing.
Through our research we observed that HCPs in NORAM typically focus on management options that preserve quality of life. A specific example of this is the move to mechanical ventilation. The patient experts described how communicating is of great importance to PLwALS, so, where possible, HCPs delay invasive ventilation to prevent a decrease in quality of life.31 Organizations (ie, manufacturers and healthcare systems) in NORAM are also increasingly conscious of the patient voice and have moved towards a patient-centric model of care, marketing, and communication, often including PLwALS in the drug development process and emphasizing the importance and value of shared decision-making. PAGs in NORAM play a vital role beyond traditional support, actively driving clinical research, shaping policy, and ensuring patient-centric drug development. The Northeast ALS Consortium is a leading example, with a mission to “accelerate the development of new treatments through innovative research and working collaboratively with people living with ALS and all stakeholders in the ALS research community”. ALS Canada reinforces this commitment through initiatives like the Canadian ALS Learning Institute, which educates and empowers individuals affected by ALS to engage in policy and research efforts that drive patient-centered innovation.32 Similarly, the ALS Association plays a critical role in shaping policies and improving access to care as demonstrated by their state public policy priorities, which focus on ensuring affordability of essential services, equipment, and treatments.33 Together, these organizations exemplify the broader role of PAGs in NORAM bridging research, industry collaboration, and policy to improve outcomes for PLwALS.
APAC
APAC was found to be distinct from NORAM with respect to clinical practice, patient advocacy structure, and cultural attitudes (Figure 3A). APAC covers a diverse region, and so PLwALS have varying access to disease-modifying therapy (eg, tofersen), as well as differing approaches to palliative care, including initiation and withdrawal of life support. For example, while in NORAM palliative care is focused around preserving quality of life, in APAC the focus is more around prioritizing ease of care and comfort.
While attitudes toward patient-centric care are shifting, HCPs in APAC are still the primary contact due in part to regional variation in socioeconomic status, health literacy, and culture. Similar to NORAM, PAGs in APAC predominantly hold a traditional role and tend to focus on navigating pre-existing social services to improve access to care and awareness. The International Alliance of ALS/MND Associations plays a critical role in this landscape, bringing together numerous country-specific organizations that work at a regional level to educate and empower PLwALS. Their Patient and Caregiver Advisory Council serves as a platform for education and engagement, helping to inform the community while also advancing drug development in collaboration with industry partners.34
In contrast to NORAM, in APAC there remains a strong culture of collectivism, filial piety, and use of traditional medicines, and familial caregiving is common practice. Consequently, caregivers may find themselves trapped in a “caring dilemma,” where they are motivated by culture to sustain their caregiving practices but simultaneously resent the unavoidability of their situation.35 Collectively, the practices and beliefs in APAC can make it challenging to directly engage PLwALS in their own care, therefore innovation with regard to clinical trial design and execution is necessary.
LATAM
LATAM was found to be most distinct from NORAM with respect to healthcare infrastructure and policy (Figure 3B). Although PLwALS in LATAM have a defined referral pathway, they face administrative hurdles and access to genetic testing that can result in delays accessing appropriate care. Furthermore, this care is often provided in urban areas, which can be difficult for PLwALS in rural and suburban areas to access. Even in urban areas, clinical trials are not universally available due to lack of accreditation at local ALS specialized treatment centers. In addition, historic allegations of political corruption and abuse of public funding further contribute to an ongoing healthcare crisis in this region.36 As in APAC, there is a strong belief that PLwALS should be cared for at home by families rather than by HCPs. PAGs in LATAM tend to provide support for PLwALS and their families by providing educational, administrative, and financial resources, as well as supporting social and emotional needs. In Colombia, La Asociación Colombiana de Esclerosis Lateral Amiotrófica provides guidance on navigating healthcare systems, offering legal assistance, psychosocial counseling, and interdisciplinary care programs, and advocating for improved access to treatment and assistive technologies.37
EMEA
EMEA was found to be distinct from NORAM with respect to access to genetic testing and the complexity of individual countries’ specific requirements for clinical research and drug approval/coverage (Figure 3C). Availability of genetic testing differs by country and, as in NORAM, PLwALS have varying receptivity to testing.
EMEA-based PAGs and advocates focus on raising disease awareness, supporting and navigating research and development, and influencing public policy. The EUpALS exemplifies this approach by uniting 28 ALS associations across 22 European countries to defend the rights of all European PLwALS, with an emphasis on advocating for European harmonization of access to ALS clinical trials and future medicines.38
Similar to LATAM, clinical trials are not universally available in all EMEA countries due to lack of accreditation at local ALS specialized treatment centers. While PLwALS may have the opportunity to travel abroad for treatment or to participate in clinical trials, this is costly and logistically challenging, so not routinely done.
Discussion
The research summarized here provides a unique perspective of the experience of ALS from initial presentation through to end-of-life care, with direct input from those affected by the disease (PLwALS, caregivers, advocates, clinicians, and disease experts). Throughout the patient journey, PLwALS, caregivers, and families experience emotional, financial, and interpersonal impacts, irrespective of geographical location. Our research also highlighted that current unmet needs are focused on care, treatment, and genetic testing. Specifically, there is a lack of disease awareness, a need for more timely diagnoses, and an opportunity for shared decision-making tools across all regions studied.
Previous research of the ALS patient journey has been limited to the path to diagnosis. A study using retrospective chart reviews demonstrated that many barriers exist that delay referral to specialist services, and suggested that comprehensive data recording and collection using multiple data sources can be used to reconstruct the timelines of the ALS patient journey and thus expedite referral to specialists.20 The lived experiences of PLwALS and their family/caregivers during the diagnosis stage of the patient journey have also been reported.7,39 Separate studies identified numerous difficulties, including failure to recognize the significance of some symptoms and unsatisfactory delivery of diagnosis,39 as well as the psychological distress and impact on quality of life experienced by both PLwALS and their caregivers.7,39 Collectively, these studies highlighted the need for a more streamlined and empathetic diagnostic process to provide better support for those affected by ALS, an aspect we also identified during our own research. We found that the journey to receiving an ALS diagnosis is fraught with challenges and can be long and complex due to barriers to accessing healthcare. These barriers to diagnosis can be significant, often spanning many years, and were consistently found across the 4 different geographical regions investigated.
However, we also observed substantial differences in ALS experiences between geographical regions that impact care. In NORAM, reliance on insurance coverage, variability in access to multidisciplinary care, and cost-related barriers shaped many of the reported challenges and unmet needs, whereas in EMEA, perceptions were more often influenced by differences in referral pathways, availability of specialized centers, and regional resource constraints. These contextual factors were incorporated into the patient journey framework to acknowledge how system-level differences shape patient and caregiver needs. One of the main differences was genetic testing. Genetic testing is recommended for PLwALS and their family members with appropriate counselling and support;14,40 while it is generally accessible in NORAM, it is limited in other regions due to issues with infrastructure, coverage, and access to facilities. Irrespective of access, attitudes to genetic testing vary. While some PLwALS opt for testing so that they can access clinical trials and treatments earlier or make informed family planning decisions, others refrain from testing due to worries about the impact on relationships, emotional status, and future health and life insurance coverage. The primary difference in APAC compared with NORAM was that in many APAC countries HCPs play a more central decision‑making role in PLwALS’ care, with limited input from PLwALS. In LATAM, we found that the primary difference versus NORAM was the lack of healthcare infrastructure to provide innovative treatment and care. In both APAC and LATAM, there remains a culture of filial piety, with family members expected to care for PLwALS at home, adding to the familial/caregiver burden. While EMEA shares many similarities with NORAM, the region has complex country‑specific requirements for clinical research and drug approval/coverage, ultimately impacting the healthcare PLwALS receive.
Our research is line with prior research and guidance documents that report that early diagnosis, access to genetic testing, and access to multidisciplinary and palliative care are integral to successful ALS healthcare models, and that delays or restrictions to any of these may have a detrimental effect on patient morbidity and mortality.24,25,41,42 Collectively, these findings further highlight the problems of care fragmentation and health system inequalities in ALS.
A strength of our research is that the initial patient journey map, which was developed through findings from a global audit of multiple sources, was refined and adapted by patient experts. We believe this supported a better understanding of the disease and the authentic ALS patient journey, and highlighted the distinct challenges and unmet needs across the different geographic regions studied. Our work advances the field by providing a comparative, cross-regional perspective that has been largely absent from prior single-center or single-country studies to date, thereby offering new insights into system-level drivers of inequity that can inform advocacy, clinical strategy, and policy development.
Our study included PLwALS and their caregivers from across the 4 different regions; however, only a relatively small number were interviewed in each region. While the interviews and advisory sessions provided a wealth of information on the patient journey, due to the heterogeneity of ALS and the differences between regions and even within regions, it is possible that some useful information may have been missed that could have made our understanding of the ALS patient journey even more robust.
Conclusion
Through the development of an ALS patient journey map, we were able to identify barriers and challenges that most impact PLwALS, caregivers, and clinicians. While only a relatively small number of those affected by ALS were interviewed, these personal insights will help enable the implementation of targeted education and improvements in personalized care, appropriate resource allocation, care coordination, policy development, and funding, which can ultimately improve quality of life and outcomes for PLwALS. There is also an opportunity to increase general knowledge and awareness of ALS and to collaborate with patient and professional communities to help improve the ALS lived experience.
Abbreviations
ALS, amyotrophic lateral sclerosis; APAC, Asia-Pacific; EMEA, Europe/Middle East/Africa; EUpALS, European Organisation for Professionals and People with ALS; FDA, United States Food & Drug Administration; HCP, healthcare professional; LATAM, Latin America; NORAM, North America; PAG, patient advocacy group; PCP, primary care physician; PLwALS, people living with amyotrophic lateral sclerosis; SOD1, superoxide dismutase 1.
Data Sharing Statement
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, and statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the product and indication has been approved by major health authorities (eg Food and Drug Administration, European Medicines Agency, Pharmaceuticals and Medical Devices Agency, etc.), if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Requests should be submitted to Available from: https://vivli.org/.
Acknowledgments
The authors would like to thank the ALS Association, ALS Canada, EUpALS, ALS-Therapy Development Institute, Northeast ALS Consortium, and International Alliance of ALS/MND Associations. Medical writing assistance, under the direction of the authors, was provided by Prime Group of Companies (Knutsford, UK), funded by Regeneron Pharmaceuticals, Inc., according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M22-1460). The authors had unrestricted access to study data, were responsible for all content and editorial decisions, and received no honoraria related to the development of this publication.
Author Contributions
All authors made a significant contribution to the work reported, whether that was in the conception, study design, execution, acquisition of data, or analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. To ensure methodological rigor and minimize potential industry-related bias, an external subject-matter expert with no affiliation to the sponsor was included as a co-author; their independent expertise contributed additional validity and credibility to the analysis and interpretation presented.
Funding
This study was funded by Regeneron Pharmaceuticals, Inc. The sponsors were involved in the study design and the collection, analysis and interpretation of data, as well as in data checking information provided in the manuscript.
Disclosure
NS is a shareholder in AbbVie, and a past employee of the ALS Therapy Development Institute; they have also received personal fees from Trace Neuroscience and personal fees from Cytokinetics, outside the submitted work.
OL, OH, and RS are employees of and shareholders in Regeneron Pharmaceuticals, Inc.
AR is an employee of IQVIA. The authors report no other conflicts of interest in this work.
This work in this article has not been previously presented or published.
References
1. Rosa Silva JP, Santiago Júnior JB, Dos Santos EL, de Carvalho FO, de França Costa IMP, de Mendonça DMF. Quality of life and functional independence in amyotrophic lateral sclerosis: a systematic review. Neurosci Biobehav Rev. 2020;111:1–19. doi:10.1016/j.neubiorev.2019.12.032
2. Hulisz D. Amyotrophic lateral sclerosis: disease state overview. Am J Manag Care. 2018;24(Suppl 15):S320–S326.
3. GBD 2021. Nervous System Disorders Collaborators. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024;23(4):344–381. doi:10.1016/s1474-4422(24)00038-3.
4. Nowicka N, Juranek J, Juranek JK, Wojtkiewicz J. Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int J Mol Sci. 2019;20(11):2616. doi:10.3390/ijms20112616
5. Xu X, Shen D, Gao Y, et al. A perspective on therapies for amyotrophic lateral sclerosis: can disease progression be curbed? Transl Neurodegener. 2021;10(1):29. doi:10.1186/s40035-021-00250-5
6. Paganoni S, Macklin EA, Lee A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5–6):453–456. doi:10.3109/21678421.2014.903974
7. O’Brien MR, Whitehead B, Jack BA, Mitchell JD. From symptom onset to a diagnosis of amyotrophic lateral sclerosis/motor neuron disease (ALS/MND): experiences of people with ALS/MND and family carers - a qualitative study. Amyotroph Lateral Scler. 2011;12(2):97–104. doi:10.3109/17482968.2010.546414
8. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364(6435):362. doi:10.1038/364362c0
9. Nijs M, Van Damme P. The genetics of amyotrophic lateral sclerosis. Curr Opin Neurol. 2024;37(5):560–569. doi:10.1097/WCO.0000000000001294
10. Ansari U, Alam M, Nadora D, et al. Assessing the efficacy of amyotrophic lateral sclerosis drugs in slowing disease progression: a literature review. AIMS Neurosci. 2024;11(2):166–177. doi:10.3934/Neuroscience.2024010
11. Biogen MA Inc. QALSODY (tofersen) prescribing information. Available from: www.biogencdn.com/us/pdfs/qalsody-prescribing-information.pdf.
12. European Medicines Agency. Qalsody (tofersen). Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/qalsody.
13. Brown CA, Lally C, Kupelian V, Flanders WD. Estimated prevalence and incidence of amyotrophic lateral sclerosis and SOD1 and C9orf72 genetic variants. Neuroepidemiology. 2021;55(5):342–353. doi:10.1159/000516752
14. Roggenbuck J, Eubank BHF, Wright J, et al. Evidence-based consensus guidelines for ALS genetic testing and counseling. Ann Clin Transl Neurol. 2023;10(11):2074–2091. doi:10.1002/acn3.51895
15. The Beryl Institute. Defining patient and human experience. Available from: https://theberylinstitute.org/defining-patient-experience/.
16. Bulto LN, Davies E, Kelly J, Hendriks JM. Patient journey mapping: emerging methods for understanding and improving patient experiences of health systems and services. Eur J Cardiovasc Nurs. 2024;23(4):429–433. doi:10.1093/eurjcn/zvae012
17. Tong A, Sainsbury P, Craig J. Consolidated criteria for reporting qualitative research (COREQ): a 32-item checklist for interviews and focus groups. Int J Qual Health Care. 2007;19(6):349–357. doi:10.1093/intqhc/mzm042
18. Bolz-Johnson M, Meek J, Hoogerbrugge N. “Patient journeys”: improving care by patient involvement. Eur J Hum Genet. 2020;28(2):141–143. doi:10.1038/s41431-019-0555-6
19. LaVela SL, Gallan AS. Evaluation and measurement of patient experience. Patient Exp J. 2014;1(1):28–36. doi:10.35680/2372-0247.1003
20. Galvin M, Madden C, Maguire S, et al. Patient journey to a specialist amyotrophic lateral sclerosis multidisciplinary clinic: an exploratory study. BMC Health Serv Res. 2015;15:571. doi:10.1186/s12913-015-1229-x
21. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7(11):639–649. doi:10.1038/nrneurol.2011.153
22. Rooney J, Byrne S, Heverin M, et al. A multidisciplinary clinic approach improves survival in ALS: a comparative study of ALS in Ireland and Northern Ireland. J Neurol Neurosurg Psychiatry. 2015;86(5):496–501. doi:10.1136/jnnp-2014-309601
23. McMackin R, Bede P, Ingre C, Malaspina A, Hardiman O. Biomarkers in amyotrophic lateral sclerosis: current status and future prospects. Nat Rev Neurol. 2023;19(12):754–768. doi:10.1038/s41582-023-00891-2
24. de Almeida FEO, Do Carmo Santana AK, de Carvalho FO. Multidisciplinary care in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Neurol Sci. 2021;42(3):911–923. doi:10.1007/s10072-020-05011-2
25. Paganoni S, Nicholson K, Leigh F, et al. Developing multidisciplinary clinics for neuromuscular care and research. Muscle Nerve. 2017;56(5):848–858. doi:10.1002/mus.25725
26. Gale NK, Heath G, Cameron E, Rashid S, Redwood S. Using the framework method for the analysis of qualitative data in multi-disciplinary health research. BMC Med Res Method. 2013;13(1):117. doi:10.1186/1471-2288-13-117
27. Greenhalgh T, Papoutsi C. Studying complexity in health services research: desperately seeking an overdue paradigm shift. BMC Med. 2018;16(1):95. doi:10.1186/s12916-018-1089-4
28. Benatar M, Heiman-Patterson TD, Cooper-Knock J, et al. Guidance for clinical management of pathogenic variant carriers at elevated genetic risk for ALS/FTD. J Neurol Neurosurg Psychiatry. 2025;96(3):e334339. doi:10.1136/jnnp-2024-334339
29. Sellati R, Rouse E, Levy O. Opportunities to reduce patient burden in clinical trials for people living with ALS. Presented at the 35th International Symposium on ALS/MND, December 5–7, 2024, San Diego, CA, USA. Abstract CLT-49. 2024:249–250.
30. ALS Association. thinkALS™ Tool. Available from: https://www.als.org/thinkals/thinkals-tool.
31. Ozeki-Hayashi R, Nakazawa E, Truog R, Akabayashi A. Initiation and withdrawal of invasive ventilation for patients with amyotrophic lateral sclerosis: a narrative literature review. J - Multidisciplinary Scientific J. 2022;5(3):402–409.
32. Zizzi C, Seabury J, Rosero S, et al. Patient reported impact of symptoms in amyotrophic lateral sclerosis (PRISM-ALS): a national, cross-sectional study. EClinicalMedicine. 2023;55:101768. doi:10.1016/j.eclinm.2022.101768
33. ALS Association. State Public Policy Priorities. Available from: https://www.als.org/our-priorities/state-public-policy-priorities.
34. International Alliance of ALS/MND Associations. PALS and CALS Advisory Council. Available from: https://www.als-mnd.org/about-us/committees-advisory-councils/pals-and-cals-advisory-council/.
35. Ng R, Indran N. Societal narratives on caregivers in Asia. Int J Environ Res Public Health. 2021;18(21):11241. doi:10.3390/ijerph182111241
36. Llumpo A, Montagu D, Brashers E, Foong S, Abuzaineh N, Feachem R. Lessons from Latin America: the early landscape of healthcare public-private partnerships. Healthcare Public-Private Partnership Series. 2025;2025:1
37. Acela. Progamas. Available from: https://www.acelaweb.org/programas/.
38. European Organisation for Professionals and People with ALS (EUPPALS). Mission statement. Available from: https://als.eu/node/11.
39. Galvin M, Corr B, Madden C, et al. Caregiving in ALS - a mixed methods approach to the study of burden. BMC Palliat Care. 2016;15(1):81. doi:10.1186/s12904-016-0153-0
40. Benatar M, Stanislaw C, Reyes E, et al. Presymptomatic ALS genetic counseling and testing. Neurology. 2016;86(24):2295–2302. doi:10.1212/WNL.0000000000002773
41. Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10(5–6):310–323. doi:10.3109/17482960802566824
42. Mercadante S, Al-Husinat L. Palliative care in amyotrophic lateral sclerosis. J Pain Sympt Manage. 2023;66(4):e485–e499. doi:10.1016/j.jpainsymman.2023.06.029
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.