Back to Journals » International Journal of General Medicine » Volume 10
A prospective study of the association between sickle cell disease and hepatobiliary effects in Bahrain
Authors Abdulla MA ![]() , Almoosa FJ, Almoosa RJ, Al Qamish J
, Almoosa FJ, Almoosa RJ, Al Qamish J ![]()
Received 17 April 2017
Accepted for publication 21 June 2017
Published 4 August 2017 Volume 2017:10 Pages 221—226
DOI https://doi.org/10.2147/IJGM.S139833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Maheeba A M Abdulla,1 Fajer Juma Almoosa,1 Rana Juma Almoosa,1 Jehad Al Qamish2
1Department of Internal Medicine, Salmaniya Medical Complex, 2Department of Internal Medicine, Ibn Al Nafees Hospital, Manama, Bahrain
Background: Sickle cell disease is a genetic disorder that leads to abnormally high levels of hemoglobin sickling in erythrocytes. Patients suffer debilitating and severe complications that affect multiple organs, though mainly the liver, gallbladder, spleen, bones, and kidneys. It has a significant impact on morbidity and mortality rates and is associated with substantial health care costs.
Methods: For this study, the researchers prospectively reviewed the charts of 154 sickle cell disease patients who had been treated in Salmaniya Medical Complex hospital in Bahrain between September 2013 and December 2014.
Results: The results obtained showed that 95% of patients had sickle cell hemoglobin type. Of the sample patients, 80% exhibited bone pain, with all patients exhibiting bone crisis. Biochemical tests revealed reduced hemoglobin concentration, elevated bilirubin, compromised liver function, and lower white blood cell counts. The incidence of hepatitis B and C was very low, at 0% and 9%, respectively. There were a large number of hospital admissions, with 11 days as an average length of stay. The most common ultrasound findings in this study were hepatomegaly, hepatosplenomegaly, cholelithiasis, gallbladder sludge, and splenomegaly.
Conclusion: Sickle cell disease had profound negative effects on multiple organs, with a particularly large number of complications related to the hepatobiliary system.
Keywords: sickle cell disease, hemoglobin types, bone pain, liver function, hepatobiliary complications
Introduction
Sickle cell disease (SCD) belongs to a group of genetic disorders affecting red blood cells. It is an autosomal recessive disorder caused by a single amino acid substitution – valine for glutamic acid – in the sixth position of the β-globin chain, which gives rise to certain hemoglobin (Hb) abnormalities.1–5 It is most prevalent in the tropical and subtropical regions, and 300,000 new cases are being diagnosed annually.4,6,7 It has significant effects on morbidity and mortality rates and is associated with substantial health care costs.3,8,9
The sickled red blood cells are a major contributing factor to the development of vaso-occlusion, leading to endothelial damage, anemia, formation of pigment gallstones, cholelithiasis, and multiple organ complications.3 Numerous acute as well as chronic complications occur in SCD, which can manifest as early as the first few months of life.8 Among the acute complications, vaso-occlusive crisis, associated with severe pain, is one of the most common and most difficult to manage.8 Other acute complications include acute chest syndrome, acute stroke, priapism, various hepatobiliary complications, splenic sequestration, and acute renal failure.8
The spleen, gallbladder, and liver are frequently affected in SCD.1 Splenic infarction and atrophy are very common and may lead to further complications such as bacterial infections and functional asplenia.1 The wide range of hepatic dysfunctions that occur in SCD patients are not only a result of the sickling process but also a result of multiple blood transfusions that these patients undergo in their lifetimes.9,10 These transfusions may lead to infections such as hepatitis B and/or C, other chronic or acute infections, iron overload, occurrence of pigment stones, cholecystitis, and obstruction of the biliary tract.1,10,11 Chronic hemolysis leads to elevated bilirubin levels (mainly unconjugated), elevated plasma aspartate transaminase (AST) and plasma alanine transaminase (ALT), and altered liver function test results.1,10 Among the other frequently diagnosed complications, liver infarction has been found in as many as 34% of patients, acute sickle hepatic crisis in 10% of patients presenting with pain crisis, gallstones in 58% of patients, and cholelithiasis in 26%–58% of SCD homozygous patients.1,10 Intrahepatic cholestasis is a severe and possibly fatal complication of hepatic crisis and, importantly, liver cirrhosis has been reported to occur in ~18% of patients and leads to death in ~11% of those cases.1 SCD sufferers also very frequently experience musculoskeletal symptoms, such as bone pain, which is a result of a vascular necrosis of the bones (osteonecrosis).12,13 The bone necrosis is believed to be caused by repeated vaso-occlusion, reduced blood flow to the bone tissue, and a resulting death of the tissue.12,13
The laboratorical tests most frequently showed elevated (unconjugated) bilirubin levels, which correlated with lactate dehydrogenase levels, suggesting that hemolysis, and not liver disease, was the cause of hyperbilirubinemia.1 Investigations regarding the concentrations of different Hb types have tended to indicate a lack of typical adult Hb type A (HbA), >90% concentration of Hb type S (HbS), and <10% concentration of fetal Hb (HbF) type F.8,14 The HbF type is frequently elevated in the sickle cell sufferers compared to the general population and has been shown to have protective properties, with patients exhibiting milder disease symptoms.14 The incidence of glucose-6-phosphate dehydrogenase (G6PD) deficiency in sickle cell patients is not different to that reported in the general population but, if present, has more profound negative effects in sickle cell patients than in the normal population.15,16 Other laboratorical tests in sickle cell patients have shown elevated platelet and white blood cell (WBC) count, and increased platelet hyperactivity.5,17,18
Due to the many comorbidities and complications of the disease, SCD patients undergo surgery more often and at an earlier age than members of the general population, which also puts them at an increased risk of further life-threatening or chronic complications.3 The most frequent procedures performed on these patients are cholecystectomies, splenectomies, and total hip replacement.3,19,20
The specific aim of this study was to investigate the frequency and pattern of hepatobiliary abnormalities in SCD patients from Bahrain.
Methods
A prospective analysis of sickle cell anemia patients admitted to Salmaniya Medical Complex, Bahrain, between September 2013 and December 2014 was conducted in order to study the incidence of hepatobiliary abnormalities. Demographic information, including age, gender, nationality, weight, height, body mass index, vaccination history, smoking and alcohol status, age at the time of diagnosis, duration of disease, presence of other comorbidities, and family history of SCD, was collected. During the course of patients’ admission, details including number of admissions, outcomes of hospitalization, and symptoms – with specific focus on bone and abdominal pain – were also recorded.
The diagnosis of SCD was established with a positive sickling test result, where the trait was identified by Hb electrophoresis. Hb capillary electrophoresis was conducted using a mini capillary instrument (Minicap; Sebia Inc., Norcross, GA, USA). Briefly, the sickling test was carried out by adding a drop of EDTA blood to five drops of freshly prepared reducing agent (2:3 sodium dithionate and disodium hydrogen phosphate). The slide was examined microscopically under a cover slip. Immediate sickling of erythrocytes occurs in positive cases.
Liver function tests included assessments of bilirubin, albumin, ALT, AST, and alkaline phosphatase (ALP). Prothrombin time was calculated, and results were expressed as an international normalized ratio (INR). Full blood count tests (Hb, WBC, and platelets) were performed using an automated blood counter, and patients were also tested for G6PD deficiency, hepatitis B surface antigen (HbsAg), and hepatitis C antibodies (anti-HCV). Confirmation of hepatitis B or C virus infections was done by polymerase chain reaction for all the patients with positive preliminary test results.
All patients were subjected to abdominal ultrasonographic examination in order to collect data on the status of the gall bladder, bile ducts, liver, and spleen. Further, the patients were subjected to endoscopic retrograde cholangiopancreatography (ERCP) with or without sphincterotomy to obtain data on bile duct stones, biliary leak, or biliary strictures (benign or malignant); colecystectomy (open or laparoscopic) to obtain data on gall stones; and splenectomy for other reasons, such as acute splenic sequestration crisis, hypersplenism, massive splenic infarction, and splenic abscess.
Data are presented as mean ± standard deviation for continuous variables and as percentages for categorical variables. As this was a prospective analysis, informed consent was obtained from all patients. The institutional ethics board of Salmaniya Medical Complex approved the study protocol.
Results
The average age of patients in the cohort was 31 years, with the youngest patient being 18 years old and the oldest 57 years. Females represented 64% (N=99) of the total sample. The average length of hospital stay varied from 1 to 119 days, and the number of admissions was similarly spread out, from as little as 2 to 259. The majority (95%) of patients presented with sickle cell (SS) Hb, 80% presented with bone pain, and all (100%) were diagnosed with bone crisis (Table 1).
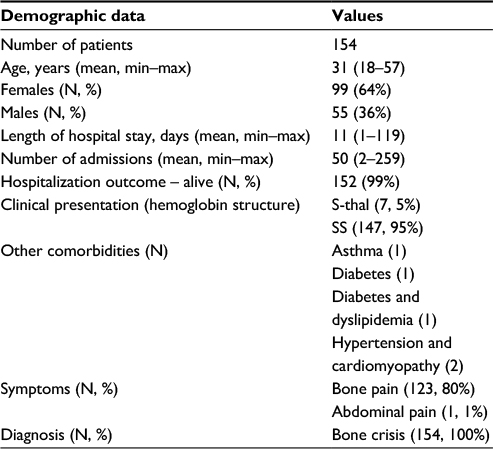 | Table 1 Diagnostic tests and procedures performed Abbreviations: S-thal, hemoglobin S/thalassemia; SS, hemoglobin S – sickle cell. |
Liver function and other laboratorical tests
The hematological tests revealed that 88% of patients had Hb levels below the normal range (<12.0 g/dL), with a mean level of 9.3 in the cohort (range 5.7–13 g/dL). The mean level of HbS was 73 g/dL. The patients in the cohort had raised HbF, with an average level of 16 g/dL (range 1.1–75 g/dL), and HbA with an average of 4 g/dL (range 0–56 g/dL). HbS was identified in 79% of patients (range 22%–99%), HbF in 17% (range 1%–51%), and HbA in 4% (range 0%–61%).The analysis of bilirubin levels showed that only 9% (N=14) of the cohort had bilirubin levels within the normal range, with an average bilirubin level of 33 µmol/L in the cohort, which is above the normal range (5–21 µmol/L). The albumin levels in the cohort were also altered, with 20% (N=31) of the cohort having albumin levels below the normal range (<35 g/L). Other hematological test results also showed that 50% (N=77) of the patients had elevated WBC (>9.6×109/L), 29% (N=45) had below normal platelet count, and 15% (N=23) had above the normal platelet count range of 150–400 (×109/L). The majority of patients (86%) did not have G6PD deficiency. However, 73% (N=112) had elevated ALT (≥33 U/L), 19% (N=30) elevated ALP (>136 U/L), and 60% (N=93) elevated GGT (>55 U/L). All the 154 patients in the cohort were negative for hepatitis B, but 14 patients (9%) were positive for hepatitis C (Table 2).
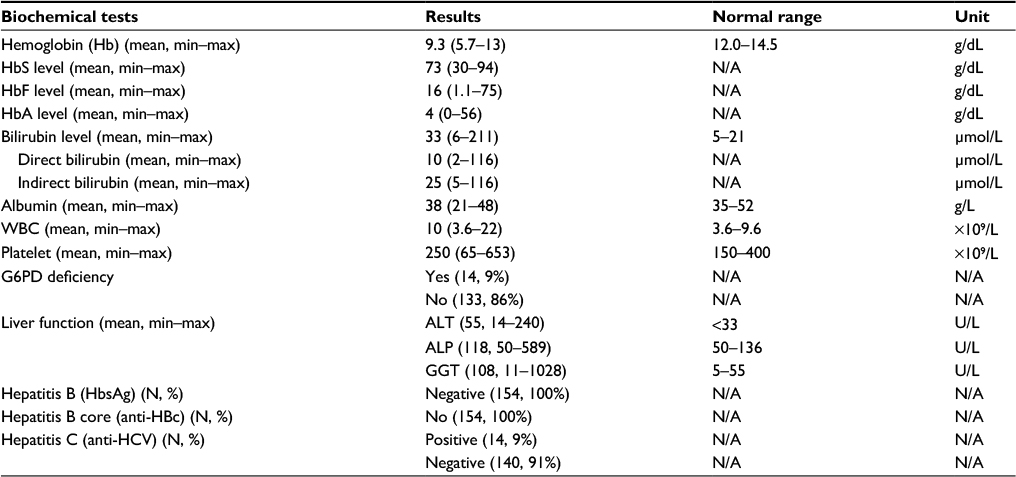 | Table 2 Hematological and biochemical profiles of the study cohort Abbreviations: WBC, white blood cell; G6PD, glucose-6-phosphate dehydrogenase; ALT, alanine aminotransferase; ALP, alanine phosphatase; GGT, γ-glutamyltransferase; HbS, hemoglobin type S; HbA, adult hemoglobin type A; HbF, fetal hemoglobin type F; HbsAg, hepatitis B surface antigen; N/A, not applicable. |
Ultrasound scanning (US) was performed in 42% (N=65) of cases. Normal US results were obtained for 22% (N=14) of patients. Hepatomegaly was the most common finding of the US, with 15 cases being diagnosed (23%) and hepatosplenomegaly was the second most common condition observed (20%), followed by cholelithiasis (15%), gallbladder sludge (15%), and splenomegaly (12%) (Table 3, Figure 1).
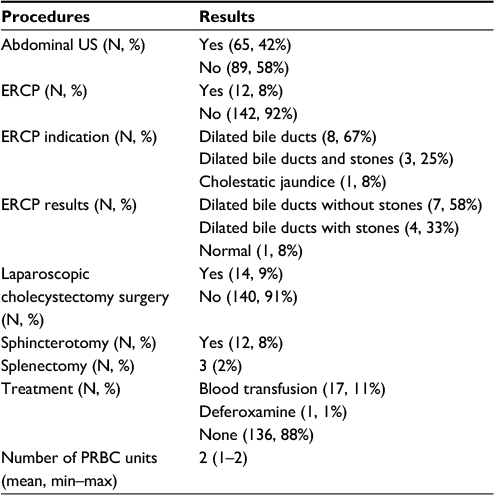 | Table 3 Diagnostic tests and procedures performed Abbreviations: US, ultrasound scanning; ERCP, endoscopic retrograde cholangiopancreatography; PRBC, packed red blood cells. |
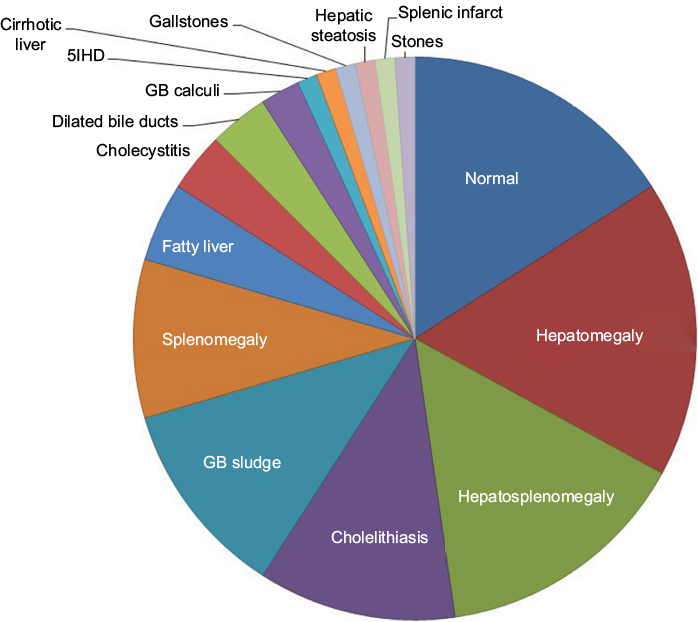 | Figure 1 Abdomen ultrasound scanning results: a pie chart showing the types and frequency of pathological findings. Abbreviations: GB, gallbladder; infart, infarction; 5IHD, type 5 intrahepatic bile duct. |
ERCP was performed in 12 cases (8%), and all of the patients who had ERCP exam underwent a sphincterotomy (N=12). The most common indication for a patient to be referred to ERCP was dilated bile ducts (67%), and the results of the procedure showed that seven of the patients had dilated bile ducts without stones (58%) (Table 3).
Laparoscopic cholecystectomy surgery was performed in fourteen (9%) cases and splenectomy in three cases (2%).The majority of the patients in the cohort did not receive any treatment (88%), and those who did (N=17) received blood transfusion with an average of two packed red blood cells units or deferoxamine (N=1) (Table 3).
Discussion
This study prospectively reviewed 154 charts of patients with SCD. The data showed that 95% of the patients in the cohort had homozygous sickle cell (SS) Hb type, and all the 154 patients were diagnosed with bone crisis, which presented as bone pain in 80% of cases. Musculoskeletal complications are among the most common complications in sickle cell patients.12,13 These are severe and have potentially devastating effects on patients’ health and quality of life; yet there is no standardized treatment currently available.13 Of the various treatment management options, total arthroplasty is the most effective.12
The vast majority (88%) of patients had Hb levels below the normal range. The concentration of HbA was below the normal range but slightly higher (at 4%) than that reported earlier for sickle cell patients (~0%).8 Similarly, the concentration of the protective HbF in this cohort was higher than that reported in SCD patients of earlier studies (17% in this cohort versus <10% in earlier reports).14 The patients had increased bilirubin and albumin levels and WBC count, as had been established in the earlier studies.1,5,17,18 The liver function tests also showed typically abnormal results, with ALT and GGT outside the normal ranges. On a positive note, the incidence of hepatitis B and C in this cohort was very low, at 0% and 9%, respectively. Previous studies have reported prevalence rates of ~1.8% and 12.5%, respectively, for hepatitis B and C infections among SCD patients.21 The reasons for this discrepancy might include better and more efficacious screening of the transfused blood, increased vaccination rates, and the lower number of transfusions received by the patients in this cohort.
SCD patients tend to undergo numerous hospital visits, surgeries, and other clinical procedures compared to the normal population.3 A similar trend was identified in this cohort. The surgery types and diagnostic procedures most frequently performed in this cohort were ultrasound of the abdomen, ERCP, cholecystectomy, sphincterotomy, and splenectomy. Contrary to the previously published data reporting a large number of blood transfusions among SCD patients, only 17 transfusions were performed in this cohort.10 The reasons for the difference include a relatively low sickle cell Hb concentration and a lack of splenic sequestration, aplasia, or vaso-occlusive events.22
The findings of the abdomen ultrasound procedure were similar to those reported in earlier studies, with only 14 of the 65 performed ultrasounds being normal.23 Hepatomegaly, splenomegaly, and cholelithiasis were the most common ultrasound findings. These findings are also similar to those reported in earlier studies.23
Conclusion
This cohort differed from those in previously published reports with regard to the lower number of blood transfusions, lower prevalence of hepatitis B and C infections, and enhanced concentrations of HbF and HbA. As expected, the SCD had profound negative effects on multiple organs, with a particularly large number of complications related to the hepatobiliary system, reflected in the number of abdomen ultrasound scan findings, poor liver function test results, and high bilirubin levels.
Disclosure
The authors report no conflicts of interest in this work. Further, none of the authors of this paper are related to each other in any way that could create a conflict of interest.
References
Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol. 2010;8(6):483–489; quiz e470. | ||
Farrell K, Dent L, Nguyen ML, Buchowski M, Bhatt A, Aguinaga Mdel P. The relationship of oxygen transport and cardiac index for the prevention of sickle cell crises. J Natl Med Assoc. 2010;102(11):1000–1007. | ||
Adam S, Jonassaint J, Kruger H, et al. Surgical and obstetric outcomes in adults with sickle cell disease. Am J Med. 2008;121(10):916–921. | ||
Abraham A, Jacobsohn DA, Bollard CM. Cellular therapy for sickle cell disease. Cytotherapy. 2016;18(11):1360–1369. | ||
Okpala I. The intriguing contribution of white blood cells to sickle cell disease – a red cell disorder. Blood Rev. 2004;18(1):65–73. | ||
Asnani MR, Quimby KR, Bennett NR, Francis DK. Interventions for patients and caregivers to improve knowledge of sickle cell disease and recognition of its related complications. Cochrane Database Syst Rev. 2016;10:CD011175. | ||
Bello-Manga H, DeBaun MR, Kassim AA. Epidemiology and treatment of relative anemia in children with sickle cell disease in sub-Saharan Africa. Expert Rev Hematol. 2016;9(11):1031–1042. | ||
Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. | ||
Simon E, Long B, Koyfman A. Emergency medicine management of sickle cell disease complications: an evidence-based update. J Emerg Med. 2016;51(4):370–381. | ||
Banerjee S, Owen C, Chopra S. Sickle cell hepatopathy. Hepatology. 2001;33(5):1021–1028. | ||
Berry PA, Cross TJ, Thein SL, et al. Hepatic dysfunction in sickle cell disease: a new system of classification based on global assessment. Clin Gastroenterol Hepatol. 2007;5(12):1469–1476; quiz 1369. | ||
Naseer ZA, Bachabi M, Jones LC, Sterling RS, Khanuja HS. Osteonecrosis in sickle cell disease. South Med J. 2016;109(9):525–530. | ||
Marti-Carvajal AJ, Sola I, Agreda-Perez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane Database Syst Rev. 2014(7):CD004344. | ||
Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. | ||
Bienzle U, Sodeinde O, Effiong CE, Luzzatto L. Glucose 6-phosphate dehydrogenase deficiency and sickle cell anemia: frequency and features of the association in an African community. Blood. 1975;46(4):591–597. | ||
Benkerrou M, Alberti C, Couque N, et al. Impact of glucose-6-phosphate dehydrogenase deficiency on sickle cell anaemia expression in infancy and early childhood: a prospective study. Br J Haematol. 2013;163(5):646–654. | ||
Curtis SA, Danda N, Etzion Z, Cohen HW, Billett HH. Elevated steady state WBC and platelet counts are associated with frequent emergency room use in adults with sickle cell anemia. PLoS One. 2015;10(8):e0133116. | ||
Kenny MW, George AJ, Stuart J. Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism. J Clin Pathol. 1980;33(7):622–625. | ||
Al-Mulhim AS, Alshehri MH. Laparoscopic cholecystectomy in adult patients with sickle cell disease. Surg Laparosc Endosc Percutan Tech. 2012;22(5):454–458. | ||
Ghmaird A, Alnoaiji MM, Al-Blewi S, Zaki S, El-Lewi A, Ahmad N. Splenectomy in patients with sickle cell disease in Tabuk. Open Access Maced J Med Sci. 2016;4(1):107–111. | ||
Ghafourian-Boroujerdnia M, Assarehzadegan MA, Zandian K. Hepatitis B and C infections and different genotypes of HCV among sickle cell anemia patients in Ahvaz, South-Western Iran. Jundishapur J Microbiol. 2013;6(5):e6368. | ||
Marouf R. Blood transfusion in sickle cell disease. Hemoglobin. 2011;35(5–6):495–502. | ||
Balci A, Karazincir S, Sangun O, et al Prevalence of abdominal ultrasonographic abnormalities in patients with sickle cell disease. Diagn Interv Radiol. 2008;14(3):133–137. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.