Back to Journals » Journal of Pain Research » Volume 11
A predictive algorithm to identify genes that discriminate individuals with fibromyalgia syndrome diagnosis from healthy controls
Authors Lukkahatai N, Walitt B, Deandrés-Galiana EJ ![]() , Fernández-Martínez JL
, Fernández-Martínez JL ![]() , Saligan LN
, Saligan LN ![]()
Received 28 March 2018
Accepted for publication 16 July 2018
Published 21 November 2018 Volume 2018:11 Pages 2981—2990
DOI https://doi.org/10.2147/JPR.S169499
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Video abstract presented by Nada Lukkahatai.
Views: 268
Nada Lukkahatai,1 Brian Walitt,2 Enrique J Deandrés-Galiana,3 Juan Luis Fernández-Martínez,3 Leorey N Saligan2
1Acute and Chronic Care, Johns Hopkins School of Nursing, Baltimore, MD, USA; 2Symptoms Biology Unit, National Institute of Nursing Research (NINR), Bethesda, MD, USA; 3Group of Inverse Problems, Optimization and Machine Learning, Applied Mathematics, University of Oviedo, Oviedo, Spain
Objectives: Fibromyalgia syndrome (FMS) is a chronic and often debilitating condition that is characterized by persistent fatigue, pain, bowel abnormalities, and sleep disturbances. Currently, there are no definitive prognostic or diagnostic biomarkers for FMS. This study attempted to utilize a novel predictive algorithm to identify a group of genes whose differential expression discriminated individuals with FMS diagnosis from healthy controls.
Methods: Secondary analysis of gene expression data from 28 women with FMS and 19 age- and race-matched healthy women. Expression of discriminatory genes were identified using fold-change differential and Fisher’s ratio (FR). Discriminatory accuracy of the differential expression of these genes was determined using leave-one-out-cross-validation. Functional networks of the discriminating genes were described from the Ingenuity’s Knowledge Base.
Results: The small-scale signature contained 57 genes whose expressions were highly discriminatory of the FMS diagnosis. The combination of these high discriminatory genes with FR higher than 1.45 provided a leave-one-out-cross-validation accuracy for the FMS diagnosis of 85.11%. The discriminatory genes were associated with 3 canonical pathways: hepatic stellate cell activation, oxidative phosphorylation, and airway pathology related to COPD.
Conclusion: The discriminating genes, especially the 2 with the highest accuracy, are associated with mitochondrial function or oxidative phosphorylation and glutamate signaling. Further validation of the clinical utility of this finding is warranted.
Keywords: chronic pain, machine learning, medically unexplained symptoms, microarray
Introduction
Fibromyalgia syndrome (FMS) is a chronic and often debilitating condition that is characterized by persistent fatigue and widespread pain.1 Females are more at risk than men in developing FMS.2 Diagnosis relies on patients’ report of symptoms and the criteria that define the condition have been repeatedly changed over the years.3 Currently, there is no consensus on any definitive prognostic or diagnostic biomarker for FMS. It is generally accepted that the etiology of the condition involves a combination of genetic and environmental factors.
The subjective nature of the diagnostic criteria for FMS often makes an accurate and timely diagnosis of the condition a difficult task to achieve. In the absence of any measurable physiological change, clinicians must base their diagnosis on their patient’s self-reported symptoms, which often leads to misdiagnosis. There have been numerous studies that have attempted to locate a biological marker that can predict the risk of development or receipt of a definitive diagnosis of FMS.4–7 Presently, there is still no accepted marker that can reliably replace or complement patient symptom reports.
A previously published novel analytical algorithm identified functional groups of genes based on their differential expression prior to radiation therapy that identified individuals at risk of developing fatigue during radiation therapy.8 These groups of genes were then ranked according to their expression levels and predictive power to identify patients at risk for developing clinically significant fatigue related to radiation therapy for cancer. This analytical algorithm not only allows us to identify individuals at risk for specific conditions, but it may give insight into functional pathways that underpin these conditions.
The systemic nature of the FMS suggests that a single gene is most likely not the sole determining factor for this condition; it is more likely that there is a synergistically working group of genes contributing to the symptoms of FMS. This study utilized the aforementioned predictive analytical algorithm to identify a group of genes, based on their differential expression, that can discriminate individuals with the FMS diagnosis using 1990 or 2010 criteria from healthy controls. The study identified the functional pathways of the genes that have high discriminatory ability that may have a role in FMS.
Materials and methods
Study design, ethical standards, and clinical population
This is a reanalysis of previously published differentially expressed genes observed in women with FMS.5 That cohort was part of an institutional review board (IRB)-approved Medstar Health Research Institute protocol and included women who met the following criteria: 1) aged 18 years or older and 2) having an FMS diagnosis (either the 1990 or the 2010 American College of Rheumatology criteria) after a physical assessment by a rheumatologist. All participants were referred to the study for evaluation of FMS from community physicians, and their primary complaints were pain. These participants were also assessed for FMS-associated symptoms such as anxiety, irritable bladder, irritable bowel, pelvic pain, vulvodynia, headache, chest pain, and paresthesia. Many of the participants also met Fukuda criteria for ME/CFS.
Healthy women who were used as age- and race-matched controls for FMS participants were enrolled from a National Institutes of Health (NIH), IRB-approved protocol (NCT00888563). These healthy volunteers were excluded if they carried any pathologic clinical diagnosis, had pain and fatigue, had worked late evening and/or night shifts within the past month, had a severe psychiatric condition, were pregnant or lactating, reported consuming >300 mg of caffeine-containing beverages or >1 lb of chocolate a day, reported consuming >2 servings of alcohol-containing beverages every day, or had a detectable blood-alcohol content.
Written informed consents were obtained from all participants before administering study measures. All study measures were completed in 1 outpatient clinic visit.
Measures
Polysymptomatic distress syndrome in FMS was measured by the Widespread Pain Index (WPI) and Symptoms Severity Scale (SSS).9 WPI measures the number of bodily areas (total =19) where participants experienced pain over the past week. SSS is a 4-item, 0–3 rating scale (total =12) used to measure severity of unrefreshed waking, cognitive problems, fatigue, and other somatic symptoms (eg, irritable bowel syndrome, numbness/tingling, dizziness, constipation, nausea). Higher scores for both instruments indicate widespread painful bodily locations and more severe symptoms, respectively.9 The WPI and SSS have been validated in previous studies and are currently used as part of the 2010 FMS diagnostic criteria.10
The number of tender points reported (conducted by applying <4 kg on 18 bodily areas) and participant’s pain threshold (average kilogram tolerated on the 18 bodily areas) were measured using a portable dolorimeter, a reliable tool to measure tenderness in FMS.11 These measures are consistent with the 1990 FMS diagnostic criteria.12
Pain intensity (4 items) and pain interference (7 items) were measured using the Brief Pain Inventory-Short Form (BPI-SF), which has a numeric rating scale of 0 or no pain/interference to 10 or pain as bad as you can imagine/complete interference.13 The internal consistency of the BPI-SF as measured by Cronbach’s α for pain intensity is 0.88 and 0.87 for pain interference.14
Discriminatory algorithm development and pathway analysis
A previously published predictive algorithm identified genes that discriminated fatigue from nonfatigue subjects, based on the differential expression of raw microarray transcripts from whole-blood RNAs collected from subjects before and during radiation therapy for nonmetastatic prostate cancer.8 Using the same predictive algorithm, this study used fold-change differential (FC) and Fisher’s ratio (FR) of raw microarray gene expression data from PBMC RNAs of FMS subjects and age- and gender-matched healthy controls to generate a reduced base of most discriminatory genes (learning phase). With 1% tail of FC (over- and underexpressed genes), we selected a final set of 897 highly discriminatory probes with FC in the intervals of –3.88, –0.29 and 0.35, 5.52, and a FR >0.9.
The genomic probes were then ranked by decreasing discriminatory power as measured by their FR. With 2 bioinformaticians (EDAG, JLFM), who were blinded to the diagnosis (FMS vs healthy control) of the sample, the discriminatory accuracy of the genes was established via leave-one-out-cross-validation (LOOCV) to estimate how accurately the model (classifier) correctly identified individuals with FMS from healthy controls. LOOCV is a well-established method in which a single sample from the original dataset serves as the validation data (sample test) and the remaining samples as training data. The class assignment is based on a nearest-neighbor classifier (k-NN) in the reduced set of genetic probes, that is, the class with the minimum distance in the reduced base to the sample test is assigned to the sample test. The average LOOCV discriminatory accuracy is calculated by iterating over all the samples. This algorithm serves to find the small-scale signature with the highest discriminatory accuracy. To analyze the stability of the minimum-scale signatures, a set of 10,000 random 75/25 hold-out simulations were performed. This methodology serves to analyze the stability of the accuracy of this small-scale signature at diagnosis. Ingenuity Pathway analysis (Ingenuity® Systems, www.ingenuity.com, Redwood City, CA, USA) identified the functional networks of the discriminatory genes. Hold-out sampling based in bootstrapping was applied to identify the functional pathways related to the differentially expressed genes.
Results
Demographic and clinical characteristics of sample
The original study5 enrolled 28 women with FMS (42.9±6.9 years old) and 19 age-matched healthy controls (39.9±7.4 years old). The mean body mass index (BMI) of women with FMS (25.2±5.0) was slightly lower than the mean BMI of the healthy volunteers (26.8±6.1). Women with FMS reported higher pain intensity (4.4±1.9) and pain interference (4.7±2.8) than healthy controls (pain intensity =1.1±1.3; pain interference =0.0±0). Based on American College of Rheumatology diagnostic criteria, the women with FMS reported widespread tenderness with an average tender-point score of 14.6±3.5 of 18 bodily sites. They also reported moderate-to-high fatigue (2.2±0.9 of 3), unrefreshed waking (2.2±0.8 of 3), and a moderate number of somatic symptoms (2.0±0.7 of 3) (Table 1). The medications used by the study participants are also described in Table 2.
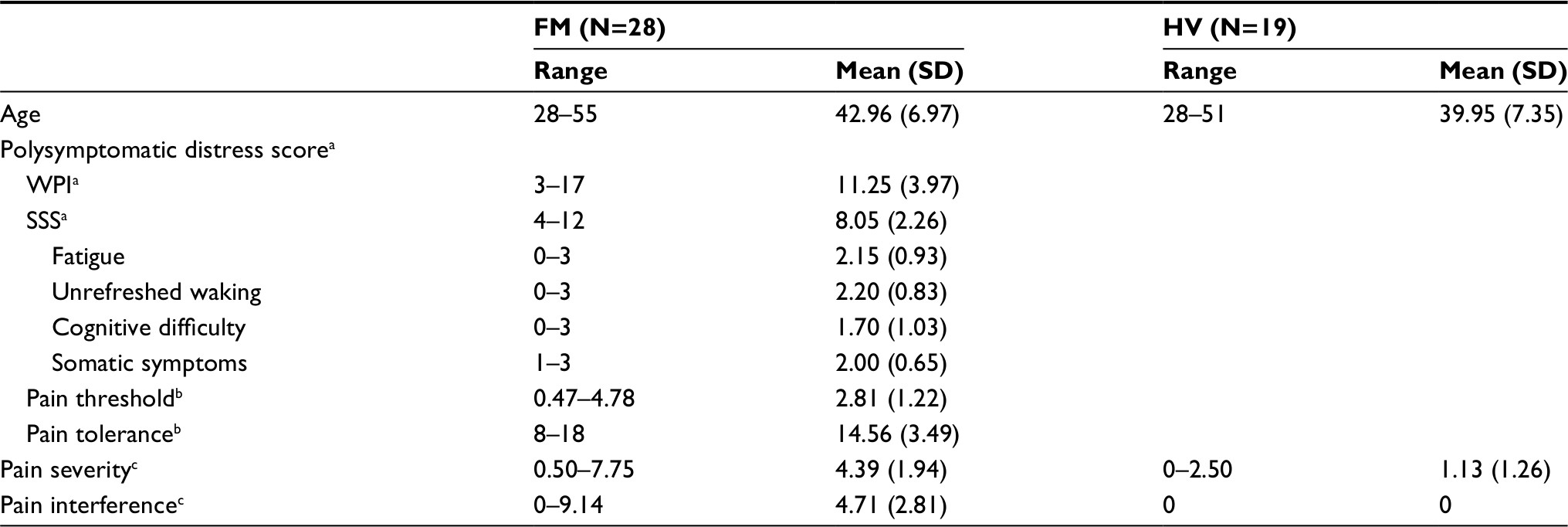 | Table 1 Demographic characteristics of sample Notes: aMeasured by the polysymptomatic distress survey – the 2010 fibromyalgia diagnostic instrument. bMeasured by the dolorimeter. cMeasured by the BPI. Abbreviations: BPI, Brief Pain Inventory; FM, fibromyalgia; HV, healthy volunteer; SSS, Symptom Severity Scale; WPI, Widespread Pain Index. |
 | Table 2 Medication used by study participants Abbreviations: cr, controlled release; OTC, over-the-counter. |
The shortest list with the highest discriminatory accuracy was composed of 57 genes (minimum-size signature). Table 3 shows a list of these discriminatory genes ranked in decreasing FR, their associated FR and mean LOOCV accuracy. MRPL4 and SLC38A were the 2 genes whose differential expressions gave the highest accuracy in discriminating FMS subjects from healthy controls at 85.11%. The probe index that guided the selection of these 57 discriminatory genes is described in Figure 1. The stability analysis of the small-scale signature is shown in Figure 2. It can be concluded that the minimum-scale signature is quite stable since the median discriminatory accuracy is very close to the LOOCV accuracy.
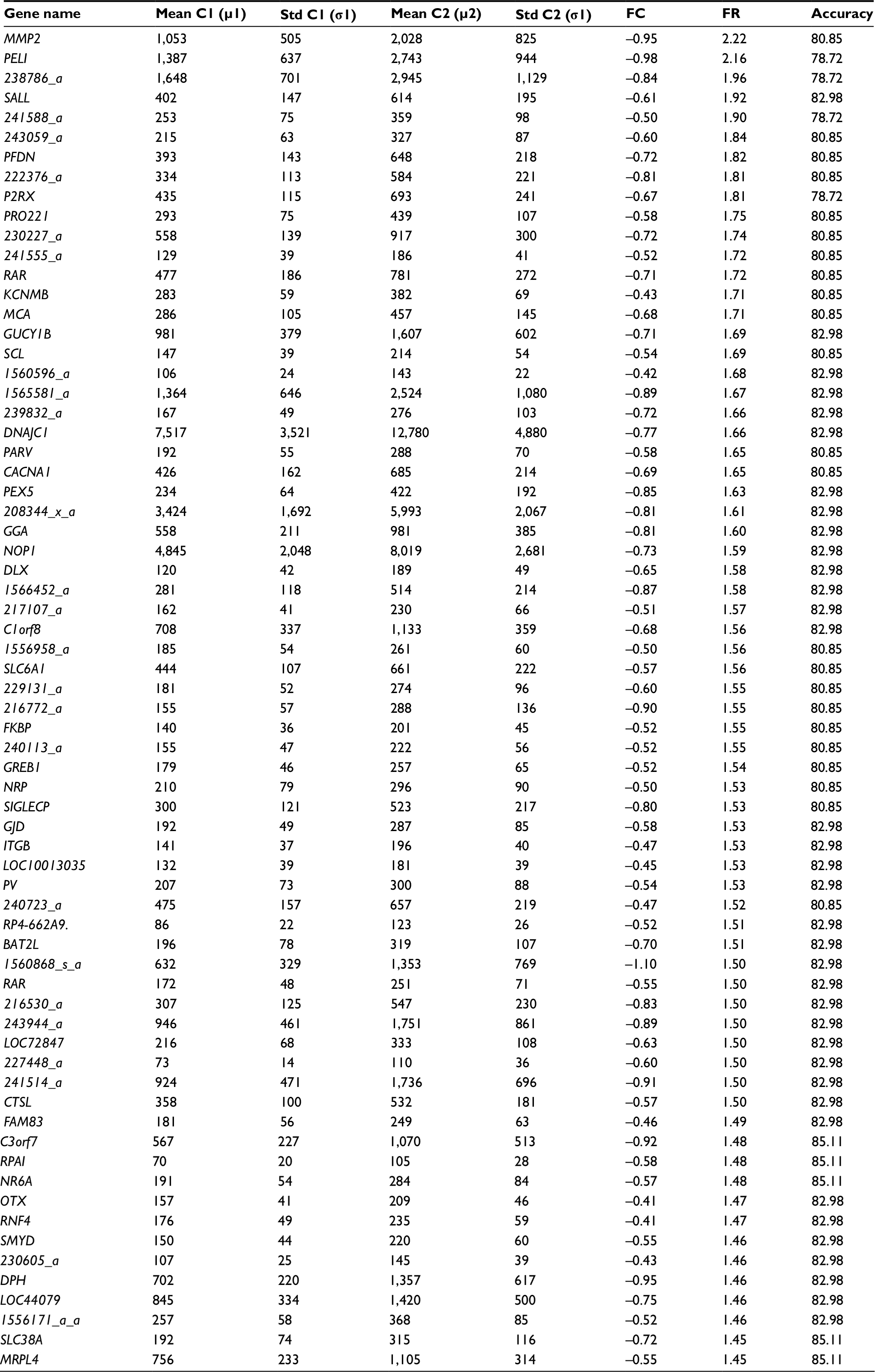 | Table 3 List of discriminatory genes Notes: Minimum small-scale list of genes/probes providing the highest LOOCV discriminatory accuracy at 85.11%. C1 stands for women with FMS and C2 for healthy controls. The mean (μ1, μ2) and the SD (σ1, σ2) of the expressions of these genes in both groups (FMS and controls) are shown, as well as the LOOCV accuracy (Acc [%]) of the genes when genes were added to the equation one at a time. Overexpression implies that the average expression of a particular gene from the FMS cohort is higher than the expression of that gene in the healthy cohort (underexpression means the opposite direction of expression). It can be observed that all the genes in this minimum-size signature are underexpressed. Abbreviations: FC, fold-change differential; FMS, fibromyalgia syndrome; FR, Fisher’s ratio; LOOCV, leave-one-out-cross-validation. |
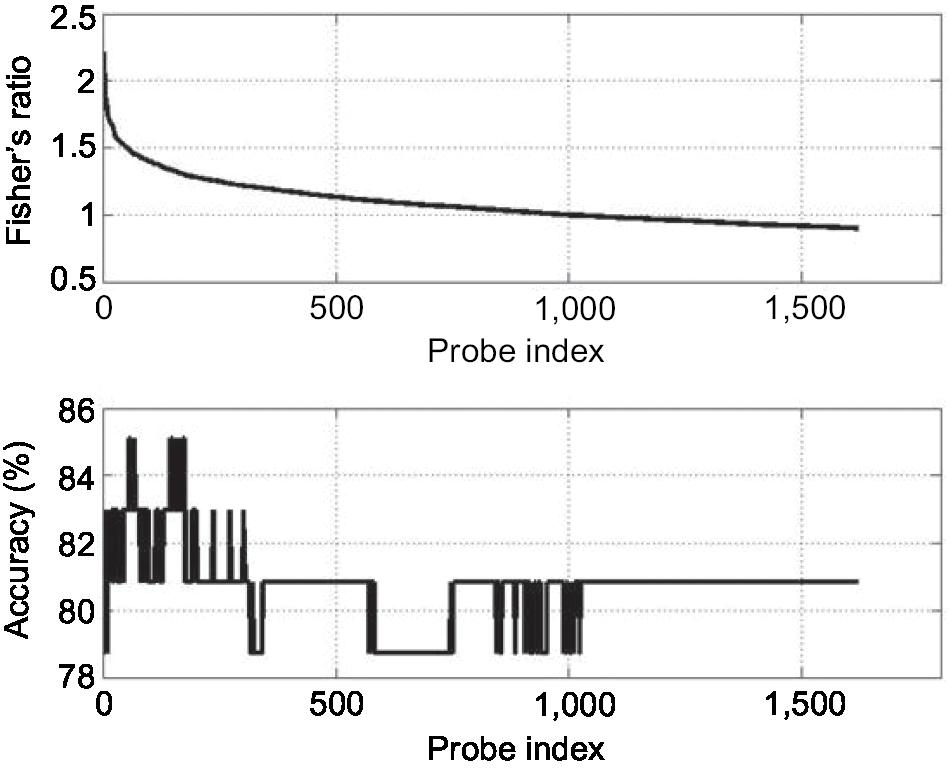 | Figure 1 Fisher’s ratio curve and accuracy of the list of discriminatory genes. Notes: The minimum-size list of genes with the highest LOOCV discriminatory accuracy (85.1%) contains the first 57 genes shown in Table 3. Abbreviation: LOOCV, leave-one-out-cross-validation. |
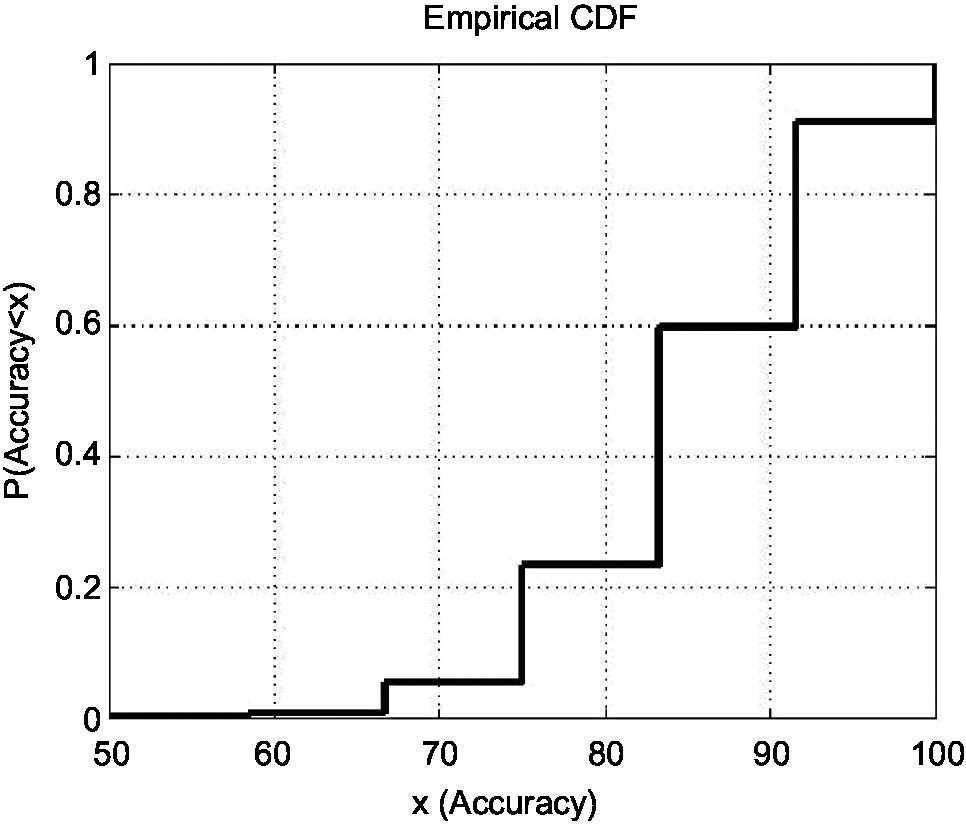 | Figure 2 Stability analysis of the small-scale signature (57 most discriminatory genes). Notes: This figure shows the CDF of the discriminatory accuracy obtained after 10,000 random 75/25 hold-out simulations. It can be observed that the median accuracy (percentile 50) is around 83%, with the lower and upper quartiles being 75% and 92%. The minimum and maximum discriminatory accuracies achieved were 50% and 100%. Therefore, it can be concluded that the minimum-scale signature is quite stable to the partial lack of data since the median discriminatory accuracy is very close to the LOOCV accuracy. This numerical experiment serves to analyze the discriminatory power of this small-scale signature at diagnosis. Abbreviations: CDF, cumulative distribution function; LOOCV, leave-one-out-cross-validation. |
Associated functional pathways
The top 3 canonical pathways that are associated with the discriminating genes are related to hepatic fibrosis/hepatic stellate cell activation, oxidative phosphorylation, and airway pathology in COPD (Figure 3). The Akt pathway, also known as protein kinase B pathway, was the main pathway in the first network that connected the discriminatory genes (Figure 4A). The NF-kB signaling pathway was the main pathway that connected all the discriminatory genes in the second network (Figure 4B). The third network was dominated by proinflammatory cytokines such as interferon α, interferon β, and IL-12 (Figure 4C).
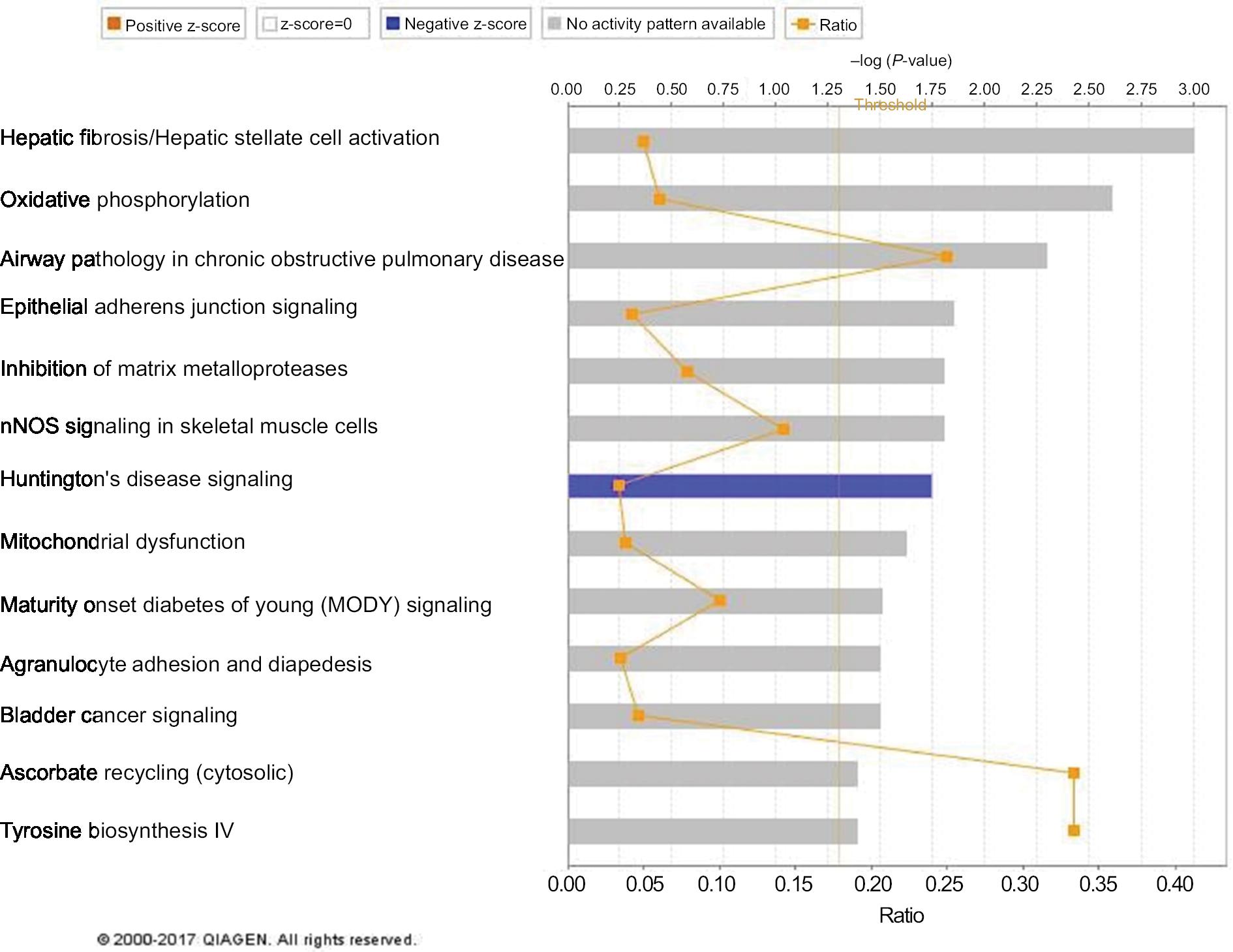 | Figure 3 Canonical pathways of the discriminatory genes. Notes: The 57 discriminatory genes are associated with the above canonical pathways. The pathways are ranked according to the P-value. |
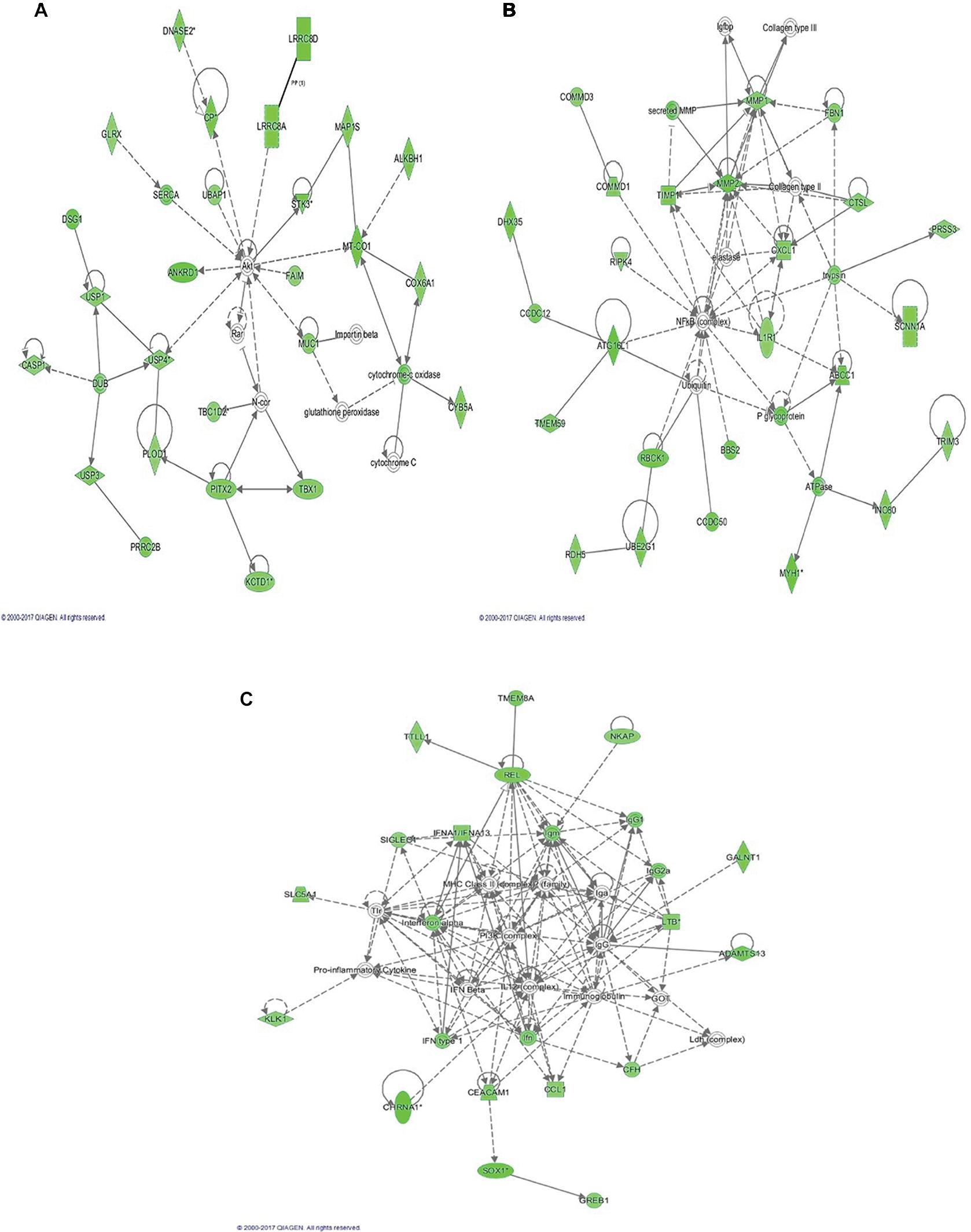 | Figure 4 Functional networks of the discriminatory genes. Notes: (A) Network dominated by AKt pathway4; (B) network dominated by NF-kB signaling; (C) network dominated by the proinflammatory cytokines. |
Discussion
This is the first study that attempted to utilize a predictive algorithm using FC, FR, and LOOCV to identify a list of genes that can discriminate individuals with FMS diagnosis from healthy controls. The study used a novel algorithm to advance our initial search for a potential prognostic or diagnostic biomarker for FMS, with the goals to advance our understanding of the biologic underpinnings of FMS and to strategically plan for future investigations.
The study participants with FMS had similar pain and fatigue scores as previously reported in studies involving FMS women.15,16 However, the FMS cohort enrolled in this study had lower BMI compared to the healthy controls, which conflicts with a previous report.17 This observation suggests that the FMS participants in this study may represent a different subpopulation of FMS patients or may be related to the geographic catchment of the patients. Metro Washington DC, where the study participants came from, tends to have a lower BMI than other regions of the United States.18 Future studies should explore the influence of BMI in identifying predictive markers for FMS.
The downregulation of MRPL4 and SLC38A highly discriminated individuals with FMS from healthy controls using this novel predictive algorithm. MRPL4 or the mitochondrial ribosomal protein L4 gene encodes mitochondrial ribosomal proteins that help in protein synthesis within the mitochondrion.18 A prior report showed reduced mitochondrial chain activities and bioenergetic levels with increased levels of oxidative stress in the skin of FMS patients.19,20 A previous report observed similar differential expressions of MRP and SLC genes in patients with chronic fatigue conditions (chronic fatigue syndrome, cancer, and HIV patients) compared to matched controls.21 Our finding suggests that downregulation of MRPL4 may diminish mitochondrial chain activities by reducing the synthesis of ribosomal proteins within the mitochondria, contributing to the symptoms (eg, pain, fatigue, and depression) experienced by individuals with FMS similar to what was found in a previous review.22
SLC38A or sodium-coupled neutral amino acid transporter encodes several proteins that are involved with uptake of nutrients, production of energy, chemical metabolism, detoxification, and neurotransmitter cycling.22 For example, one of the proteins it encodes is SLC38A1, an important transporter of glutamine.23,24 Glutamine is a precursor of an excitatory synaptic neurotransmitter, glutamate.24 Glutamate has been reported to play an important role in fatigue and depression,25,26 prominent symptoms reported by FMS patients. A recent review describes an association of increasing cerebral glutamate levels with dysregulation of pain processing in the central nervous system of FMS patients.27 The downregulation of SLC38A may reduce the concentration of sodium-coupled neutral amino acid transporters reducing the transport of glutamine in synapses contributing to the fatigue, pain, and depressive symptoms reported by individuals with FMS.
The Akt pathway was one of the major pathways that was associated with the discriminatory genes. The Akt pathway regulates several events such as apoptosis, protein synthesis, cell proliferation, and energy metabolism that have been associated with symptoms reported by FMS patients.28–31 The direct upstream kinase responsible for activating the Akt pathway is the PDK.32 During the winter season, animal hibernators have a markedly suppressed mitochondrial activity through the reduction of oxidative phosphorylation and inhibition of PDK.33,34 During the active state in the summer, there is increased expression of PDK in skeletal muscles and white adipose tissues of animals,35,36 suggesting the role of the Akt pathway through PDK in fuel utilization and metabolic suppression. Our findings showed downregulation of these discriminatory genes that are associated with the Akt pathway suggesting attenuation of metabolic rate may have a role in FMS.
The other 2 functional pathways, NFκB signaling pathway and the proinflammatory cytokines, signal the potential role of inflammation in FMS. A recent finding revealed the evidence of systemic inflammation and neuroinflammation in the plasma and cerebrospinal fluid of FMS patients.37 Our findings revealed downregulation of the FMS discriminatory genes that are associated with inflammation, such as interferon α (Figure 4C). Our previous study showed that interferon signaling and interferon regulatory activation factor pathways distinguished FMS patients with varying pain levels.38 This finding suggests that the discriminatory genes identified in this study may further classify the different subsets of FMS patients.
The aim of this paper was to conduct an exploratory attempt to show that the previously published novel predictive algorithm can potentially generate a list of genes that can discriminate, based on their differential expression, FMS patients from healthy individuals. We have forgone a multiple-comparisons correction, because the differential expression of many of the genes are influenced by each other or by common upstream signaling pathways. Thus, we would expect to see many rise and fall in tandem, and their measurements should not be treated as independent variables. In fact, this interdependence may be what leads to clustering of the symptoms experienced by FMS patients.
Follow-up studies are warranted to address several limitations of the study. There is a very high likelihood that the list of genes generated in this study maybe artifacts of over training on a single cohort of a small number of patients. For the findings to have clinical and diagnostic utilities, the predictive genes must be validated in a larger, independent cohort of patients with FMS. Further, future studies should account the overlap of FMS and ME/CFS diagnoses in the symptom presentation of study participants and for the presence of other medical comorbidities. In addition, the selected differentially expressed genes were obtained from raw microarray data, basing on our previous finding that using raw microarray data can better generate genetic signatures that are associated with functional pathways that are a priori known for specific medical conditions.39 Future studies should consider using preprocessing techniques, such as Robust Microarray Average, to determine if similar discriminatory genes can be identified.
Conclusion
Our results do not have any clinical implications as presented and may not hold up to more extensive field testing; however, this study will be used to inform future biomarker studies. The study findings can initiate the investigation of functional pathways that may play a role in FMS and the development of a potential prognostic or diagnostic tool for FMS.
Significance and innovation
- This is the first study to employ a novel predictive mathematical algorithm to analyze gene expression data.
- The result revealed potential significant roles of Akt pathway, NFκB signaling pathway, and the proinflammatory cytokines in discriminating individuals with FMS from healthy controls.
Acknowledgments
The authors express gratitude to Dr Joan Austin for editing this paper. This study is part of a collaborative activity of National Institute of Nursing Research (NINR) and the Medstar Research Institute as approved by the Office of Human Subjects Research (OHSR) of the National Institutes of Health (OHSR IRB-Exempt #4966) and is funded in part by a MedStar grant (NCT00888563) and the Division of Intramural Research of the NINR.
Disclosure
The authors report no conflicts of interest in this work.
References
Wolfe F. New American College of Rheumatology criteria for fibromyalgia: a twenty-year journey. Arthritis Care Res (Hoboken). 2010;62(5):583–584. | ||
Aparicio VA, Ortega FB, Carbonell-Baeza A, et al. Are there gender differences in quality of life and symptomatology between fibromyalgia patients? Am J Mens Health. 2012;6(4):314–319. | ||
Wolfe F, Häuser W. Fibromyalgia diagnosis and diagnostic criteria. Ann Med. 2011;43(7):495–502. | ||
Jones KD, Gelbart T, Whisenant TC, et al. Genome-wide expression profiling in the peripheral blood of patients with fibromyalgia. Clin Exp Rheumatol. 2016;34(2 Suppl 96):S89–S98. | ||
Lukkahatai N, Walitt B, Espina A, Wang D, Saligan LN. Comparing Genomic Profiles of Women With and Without Fibromyalgia. Biol Res Nurs. 2015;17(4):373–383. | ||
Docampo E, Escaramís G, Gratacòs M, et al. Genome-wide analysis of single nucleotide polymorphisms and copy number variants in fibromyalgia suggest a role for the central nervous system. Pain. 2014;155(6):1102–1109. | ||
Buskila D, Sarzi-Puttini P, Ablin JN. The genetics of fibromyalgia syndrome. Pharmacogenomics. 2007;8(1):67–74. | ||
Saligan LN, Fernández-Martínez JL, deandrés-Galiana EJ, Sonis S. Supervised classification by filter methods and recursive feature elimination predicts risk of radiotherapy-related fatigue in patients with prostate cancer. Cancer Inform. 2014;13:141–152. | ||
Wolfe F, Clauw DJ, Fitzcharles MA, et al. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010;62(5):600–610. | ||
Wolfe F. How to use the new American College of Rheumatology fibromyalgia diagnostic criteria. Arthritis Care Res (Hoboken). 2011;63(7):1073–1074. | ||
Tunks E, McCain GA, Hart LE, et al. The reliability of examination for tenderness in patients with myofascial pain, chronic fibromyalgia and controls. J Rheumatol. 1995;22(5):944–952. | ||
Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160–172. | ||
Cleeland CS, Ladinsky JL, Serlin RC, Nugyen CT. Multidimensional measurement of cancer pain: comparisons of US and Vietnamese patients. J Pain Symptom Manage. 1988;3(1):23–27. | ||
Kapstad H, Rokne B, Stavem K. Psychometric properties of the Brief Pain Inventory among patients with osteoarthritis undergoing total hip replacement surgery. Health Qual Life Outcomes. 2010;8:148. | ||
Lukkahatai N, Walitt B, Espina A, Gelio A, Saligan LN. Understanding the Association of Fatigue With Other Symptoms of Fibromyalgia: Development of a Cluster Model. Arthritis Care Res (Hoboken). 2016;68(1):99–107. | ||
Vincent A, Benzo RP, Whipple MO, McAllister SJ, Erwin PJ, Saligan LN. Beyond pain in fibromyalgia: insights into the symptom of fatigue. Arthritis Res Ther. 2013;15(6):221. | ||
Okifuji A, Bradshaw DH, Olson C. Evaluating obesity in fibromyalgia: neuroendocrine biomarkers, symptoms, and functions. Clin Rheumatol. 2009;28(4):475–478. | ||
Wei X, Zhang Y, Fu Z, Zhang L. The association between polymorphisms in the MRPL4 and TNF-α genes and susceptibility to allergic rhinitis. PLoS One. 2013;8(3):e57981. | ||
Sánchez-Domínguez B, Bullón P, Román-Malo L, et al. Oxidative stress, mitochondrial dysfunction and, inflammation common events in skin of patients with Fibromyalgia. Mitochondrion. 2015;21:69–75. | ||
Castro-Marrero J, Cordero MD, Sáez-Francas N, et al. Could mitochondrial dysfunction be a differentiating marker between chronic fatigue syndrome and fibromyalgia? Antioxid Redox Signal. 2013;19(15):1855–1860. | ||
Filler K, Lyon D, Bennett J, et al. Association of Mitochondrial Dysfunction and Fatigue: A Review of the Literature. BBA Clin. 2014;1:12–23. | ||
Schweikhard ES, Ziegler CM. Amino acid secondary transporters: toward a common transport mechanism. Curr Top Membr. 2012;70:1–28. | ||
Schiöth HB, Roshanbin S, Hägglund MG, Fredriksson R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol Aspects Med. 2013;34(2–3):571–585. | ||
Jenstad M, Quazi AZ, Zilberter M, et al. System A transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cereb Cortex. 2009;19(5):1092–1106. | ||
Saligan LN, Luckenbaugh DA, Slonena EE, Machado-Vieira R, Zarate CA Jr. An assessment of the anti-fatigue effects of ketamine from a double-blind, placebo-controlled, crossover study in bipolar disorder. J Affect Disord. 2016;194:115–119. | ||
Lener MS, Niciu MJ, Ballard ED, et al. Glutamate and Gamma-Aminobutyric Acid Systems in the Pathophysiology of Major Depression and Antidepressant Response to Ketamine. Biol Psychiatry. 2017;81(10):886–897. | ||
Pyke TL, Osmotherly PG, Baines S. Measuring Glutamate Levels in the Brains of Fibromyalgia Patients and a Potential Role for Glutamate in the Pathophysiology of Fibromyalgia Symptoms: A Systematic Review. Clin J Pain. 2017;33(10):944–954. | ||
Strozyk E, Kulms D. The role of AKT/mTOR pathway in stress response to UV-irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int J Mol Sci. 2013;14(8):15260–15285. | ||
Kim HS, Skurk C, Maatz H, et al. Akt/FOXO3a signaling modulates the endothelial stress response through regulation of heat shock protein 70 expression. FASEB J. 2005;19(8):1042–1044. | ||
Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. | ||
Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13(3):234–244. | ||
Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–269. | ||
Stieler JT, Bullmann T, Kohl F, et al. The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS One. 2011;6(1):e14530. | ||
Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358(Pt 1):69–77. | ||
Buck MJ, Squire TL, Andrews MT. Coordinate expression of the PDK4 gene: a means of regulating fuel selection in a hibernating mammal. Physiol Genomics. 2002;8(1):5–13. | ||
Glueck SB, Heldmaier G. Settling down for a long winter’s nap: focus on “coordinate expression of the PDK4 gene: a means of regulating fuel selection in a hibernating mammal.” Physiol Genomics. 2002;8(1):3–4. | ||
Bäckryd E, Tanum L, Lind AL, Larsson A, Gordh T. Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J Pain Res. 2017;10:515–525. | ||
Lukkahatai N, Majors B, Reddy S, Walitt B, Saligan LN. Gene expression profiles of fatigued fibromyalgia patients with different categories of pain and catastrophizing: a preliminary report. Nurs Outlook. 2013;61(4):216–224.e2. | ||
Deandrés-Galiana, EJ, Fernández-Martínez JL, Saligan LN, Sonis ST. Impact of microarray preprocessing techniques in unraveling biological pathways. J Comput Biol. 2016;23(12):957–968. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.