Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
A Pharmacogenetics-Based Approach to Managing Gastroesophageal Reflux Disease: Current Perspectives and Future Steps
Authors Eken E ![]() , Estores DS, Cicali EJ
, Estores DS, Cicali EJ ![]() , Wiisanen KK
, Wiisanen KK ![]() , Johnson JA
, Johnson JA ![]()
Received 2 March 2023
Accepted for publication 18 May 2023
Published 23 June 2023 Volume 2023:16 Pages 645—664
DOI https://doi.org/10.2147/PGPM.S371994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Eda Eken,1,2 David S Estores,3 Emily J Cicali,1,2 Kristin K Wiisanen,1,2 Julie A Johnson1,2
1Department of Pharmacotherapy and Translational Research, College of Pharmacy, University of Florida, Gainesville, FL, USA; 2Center for Pharmacogenomics and Precision Medicine, University of Florida, Gainesville, FL, USA; 3Division of Gastroenterology, Hepatology, and Nutrition, College of Medicine, University of Florida, Gainesville, FL, USA
Correspondence: Julie A Johnson, University of Florida, Department of Pharmacotherapy and Translational Research, Box 100486, Gainesville, FL, 32610-0486, USA, Tel +1-352-273-6310, Email [email protected]
Abstract: Proton pump inhibitors (PPIs) are commonly used medications to treat acid-related conditions, including gastro-esophageal reflux disease (GERD). Gastroenterology guidelines mention the importance of CYP2C19 in PPI metabolism and the influence of CYP2C19 genetic variations on variable responses to PPIs, but do not currently recommend the genotyping of CYP2C19 prior to prescribing PPIs. There are strong data to support the influence of CYP2C19 genetic variations on the pharmacokinetics of PPIs and clinical outcomes. Existing pharmacogenetic guideline recommendations for dose increases focus on H. pylori and erosive esophagitis indications, but PPIs are also the main therapy for treating GERD. Recent data suggest GERD patients being treated with a PPI may also benefit from genotype-guided dosing. We summarize the literature supporting this contention and highlight future directions for improved management of patients with GERD through precision medicine approaches.
Keywords: CYP2C19, precision medicine, proton pump inhibitors, omeprazole, pantoprazole, lansoprazole
Introduction
The burden of gastroesophageal reflux disease (GERD) imposed on the US health care system is quite significant. More than 4.3 million outpatient office visits have a primary diagnosis of GERD and esophagitis.1 Based on data from patients with health insurance (2016–2018) the annualized cost for patients with a primary diagnosis of GERD is $12,232 with $4277 attributed to drug cost.2
Proton pump inhibitors (PPIs) are the mainstay agents for treating GERD. Currently, there are 6 Food and Drug Administration (FDA)-approved PPIs available clinically in the US; these include first-generation benzimidazole derivative PPIs (ie, omeprazole, lansoprazole, pantoprazole) and second-generation benzimidazole derivative PPIs (ie, esomeprazole, dexlansoprazole, rabeprazole). Omeprazole and pantoprazole are number 8 and 20, respectively, in the top 200 most commonly prescribed medications in the US in 2020.3
Despite the best efforts of experts on GERD, its diagnosis is not straightforward. The American College of Gastroenterology (ACG) convened an expert panel in 2022 that defined GERD as the condition in which the reflux of gastric contents into the esophagus results in symptoms and/or complications. GERD is objectively defined by the presence of characteristic mucosal injury seen at endoscopy and/or abnormal esophageal acid exposure demonstrated on a reflux monitoring study.4 This definition is further complicated by the difficulty of discerning symptom(s) or condition(s) related to GERD. Katzka et al summarized the complexities of the different GERD manifestations (ranging from reflux esophagitis to cough); described the danger of oversimplification of symptom generation; and the danger of overprescribing PPIs for various GERD manifestations without adequate justification for dose escalation and continued use.5
Neither typical symptoms attributed to GERD (heartburn or regurgitation) nor a trial of PPI for symptoms (ie, omeprazole 20 mg before meals daily for 4 to 8 weeks) are sensitive or specific enough to establish a diagnosis of GERD.6 The American Gastroenterological Association (AGA) panel of experts recommend a more personalized approach, starting with a therapeutic trial of standard doses of a PPI (no particular PPI is recommended), lasting 4 to 8 weeks, after which nonresponse or incomplete response is followed by an upper endoscopy done following discontinuation of PPIs. Endoscopy findings that indicate a GERD diagnosis serve as the main basis for further treatment considerations.6
Assessment of severity among patients with reflux esophagitis has been standardized and validated using the Los Angeles (LA) classification.7 This classification system relies on the presence, extent, and degree of involvement of the esophageal lumen circumference graded from least to most severe (ie, grade A to grade D, respectively). According to recent expert consensus statements,8,9 LA grade A esophagitis does not provide conclusive evidence for GERD because it is frequently encountered in asymptomatic healthy individuals. LA grade B esophagitis can be diagnostic of GERD in the presence of typical GERD symptoms and PPI response, whereas high-grade esophagitis (LA grades C or D), is considered confirmatory evidence for GERD. For patients with mild forms of GERD (eg, no reflux esophagitis worse than LA grade B), it may be possible to decrease or even eliminate PPI therapy, whereas patients with severe reflux esophagitis (LA grade C or D) may require continuous long-term PPI therapy or invasive anti-reflux surgery, in addition to optimization of lifestyle factors.4,6
In 2022, language referring to the CYP2C19 genotype was added to the ACG guidelines for GERD treatment stating that “genetic differences in CYP2C19 metabolism affect PPI response; however, genetic testing in this regard has no established role in practice”.4 Contrary to this statement, there are numerous published clinical trials supporting genotype-guided PPI therapy with implementation reported in various institutions.10–14 The same ACG guidelines continue to mention “If one is considering a PPI switch, changing to a PPI that does not rely on CYP2C19 for primary metabolism (ie, rabeprazole) might be considered”.4 We propose that the data herein support the use of CYP2C19 genotype data to guide prescribing decisions with PPIs and that this will be increasingly utilized by physicians who treat GERD in the future. The data we summarize support the latter statement from ACG but suggest that esomeprazole might also be considered as an alternative PPI, as the data suggest minimal differences with esomeprazole based on CYP2C19 genotype.
CYP2C19 Polymorphisms, Phenotypes, and Population Frequencies
To facilitate clinical implementation of pharmacogenetic information and utilization of pharmacogenetic data across institutions, the Clinical Pharmacogenetics Implementation Consortium (CPIC) established standard terminology for allele functional status and phenotypes for pharmacogenes, including CYP2C19.15 CPIC also develops structured, evidence-based, clinical practice guidelines for drugs affected by pharmacogenetics (ie, gene-drug pairs). In 2020, CPIC published guidelines on use of CYP2C19 with PPIs, with the recommendations based on the available body of literature, severity of clinical consequences, availability of alternative therapies and whether a prescribing change (drug choice or dose) is warranted.16
Allele Functionality
The most common no-function CYP2C19 polymorphism is the CYP2C19*2 variant allele.17 The polymorphism rs4244285 (c.681G>A) defines the *2 allele and leads to a single base-pair change, resulting in a synonymous guanine to adenosine (G>A) polymorphism in exon 5 that creates a splice site, altering the mRNA reading frame and rendering the protein non-functional.18 Other, less frequent, no-function alleles include CYP2C19 *3,*4, *5, *6, *7, and *8.17 Of these alleles, CYP2C19 *3 and *8 are more commonly seen. CYP2C19 *3 results from the polymorphism rs4986893 (c.636G>A) at position 636 on exon 4 and creates a premature stop codon.
In 2006, Sim et al reported a novel variant allele, CYP2C19*17.19 The defining polymorphism is rs12248560, which is a double variation (c.-806C>T and −3402C>T) in the promotor region leading to increased gene transcription and, consequently, increased CYP2C19 protein and increased metabolism via CYP2C19.18
Genotype frequency data of 2.29 million 23andMe participants, assessing ethnic distribution of the three most common CYP2C19 alleles (*2, *3, and *17), revealed that 58.3% of overall participants expressed of at least one increased-function or no-function CYP2C19 allele.20 The overall frequencies of *2, *3, and *17 were 15.2%, 0.3%, and 20.4%, respectively, but varied by ethnicity. The *2 allele was most prevalent in East Asian (28.4%) and Native Hawaiian and Other Pacific Islander (27.6%) populations, approximately half of which was seen in other populations (ranging from 11.9% (Middle Eastern) to 17.5% (African American)). The *3 allele was most common in the Native Hawaiian and Other Pacific Islander (6.5%) and East Asian (6%) populations, whereas the *17 variant was least commonly observed (5.5% and 3.7%, respectively) in these populations. The frequency of the *17 allele varied amongst all other populations, ranging from approximately 16% (Hispanic/Latino) to 22% (African American).20
CYP2C19 Phenotypes
The predicted CYP2C19 phenotypes based on CYP2C19 genotype are shown in Table 1. Two normal function CYP2C19 *1 alleles classify patients as normal metabolizers (NMs).16 Patients with one normal function and one non-functional allele (eg CYP2C19 *1/*2 or *1/*3) or one non-functional and one gain of function allele (eg CYP2C19 *2/*17) are considered intermediate metabolizers (IMs). Patients with a diplotype containing two non-functional alleles (eg CYP2C19 *2/*2) are considered poor metabolizers (PMs). Those with one normal function and one gain of function (CYP2C19 *1/*17) are classified as rapid metabolizers (RMs) and two gain of function alleles (CYP2C19 *17/*17) are ultrarapid metabolizers (UMs).
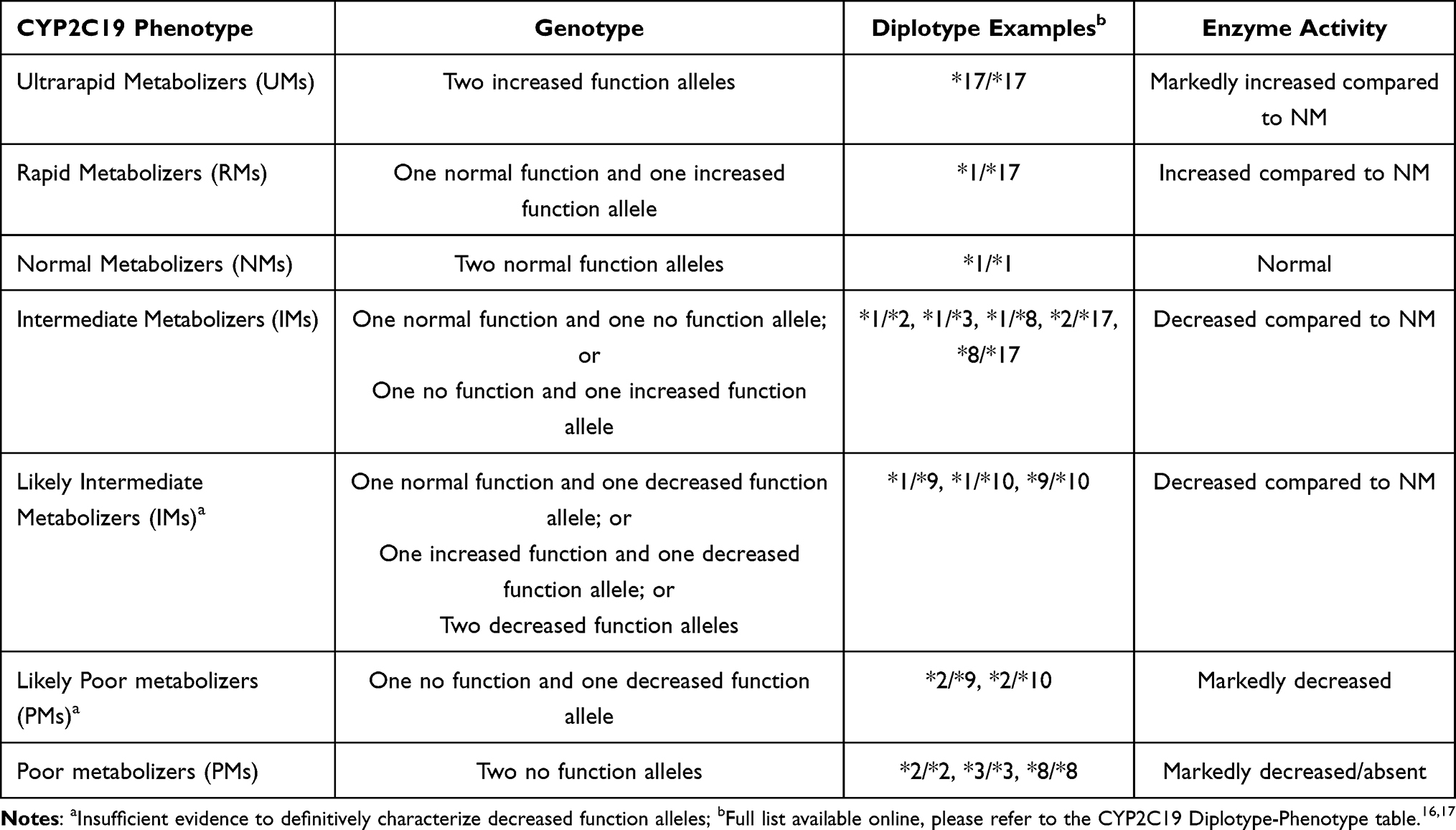 |
Table 1 CPIC Phenotype and Genotype Definitions with Associated Enzyme Activity |
Ethnic distribution of the various phenotypes are summarized in Figure 1, based on data extracted from CPIC’s phenotype frequency table.21 The highest prevalence of CYP2C19 PM and IM phenotypes are observed in those of East Asian ancestry making up about 13% and 46%, respectively. In contrast, RM and UM phenotypes are rarely observed in those of East Asian ancestry but are more common in those of other continental ancestries. As a result, the majority of humans carry a CYP2C19 variant allele that leads them to have a non-NM phenotype. Specifically, 48–67% of African Americans, East Asians, and European and Latino ancestries have a non-NM phenotype, whereas among those of Native American ancestry, the NM phenotype predominates, at about 68%.17,21
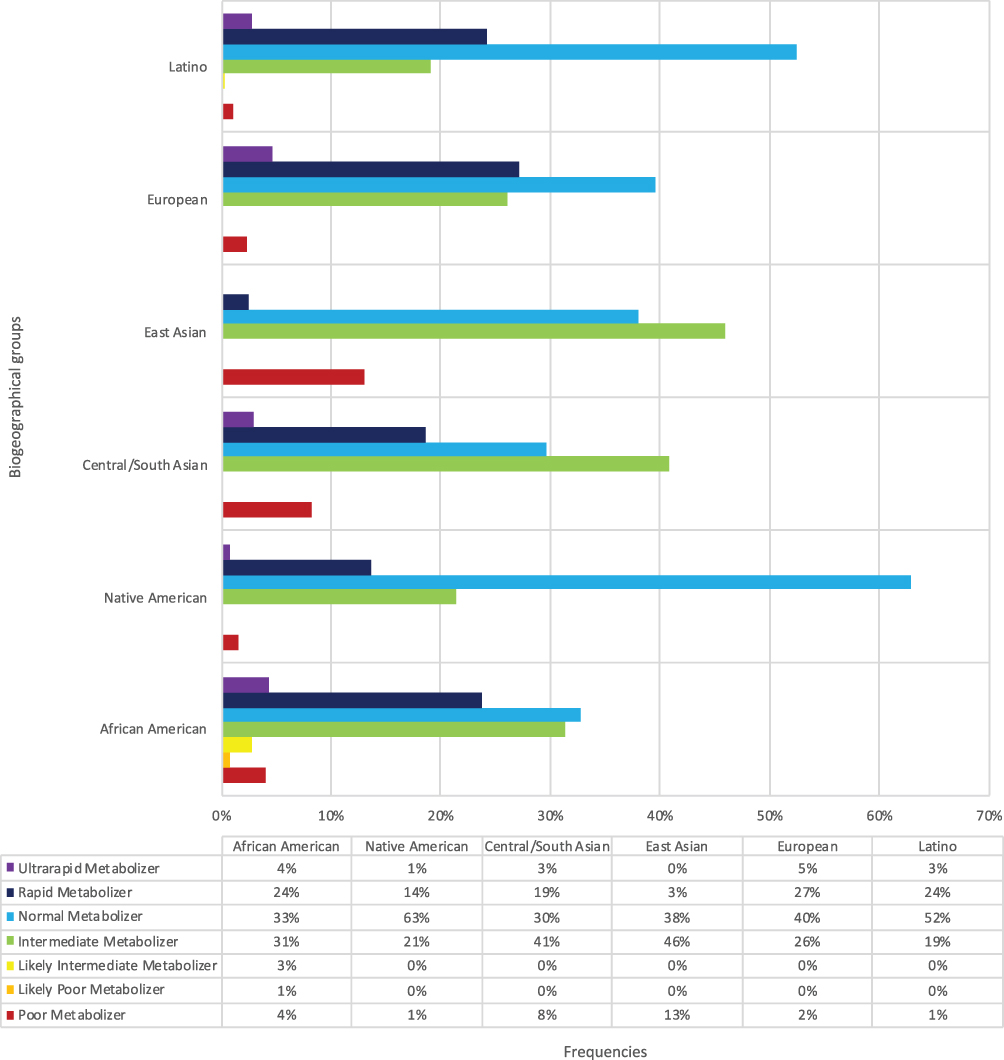 |
Figure 1 Frequencies of CYP2C19 phenotypes in biogeographical groups. |
It is important to note the nomenclature associated with CYP2C19 phenotype classification when analyzing literature published prior to CPICs standardization of phenotypes in 2017.15 Numerous studies have classified NMs as extensive metabolizers (EMs),15 often because the CYP2C19*17 allele was not tested or studies were conducted prior to discovery of this variant allele in 2006.19 Additionally, studies often grouped together RMs and UMs and referred to these phenotypes as UMs. However, in 2017, CPICs final consensus of phenotype terms codified an explicit distinction between the two phenotypes based on pharmacokinetic (PK) and pharmacodynamic (PD) outcomes.15,16 Therefore, the literature summarized herein and in Table 2 and Table 3, uses CPICs updated phenotype terminology as opposed to the original reported phenotypes. In addition, early studies documenting the relationship between CYP2C19 polymorphisms and PK/PD were conducted mainly in those of Asian ancestry, in which the CYP2C19 *17 haplotype is the least prevalent among these individuals.17,21
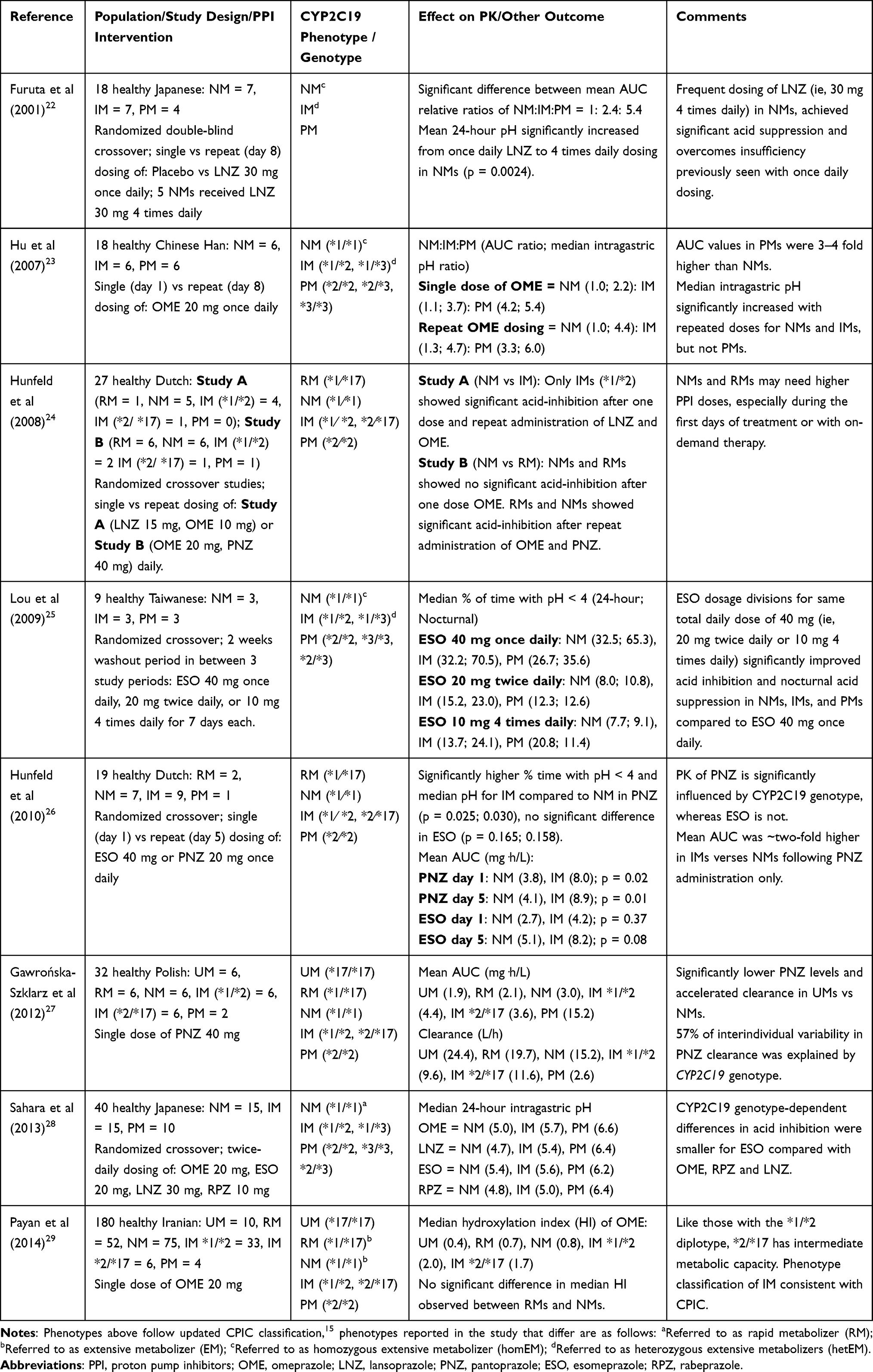 |
Table 2 Summary of Selected CYP2C19-PPI Pharmacokinetic Studies |
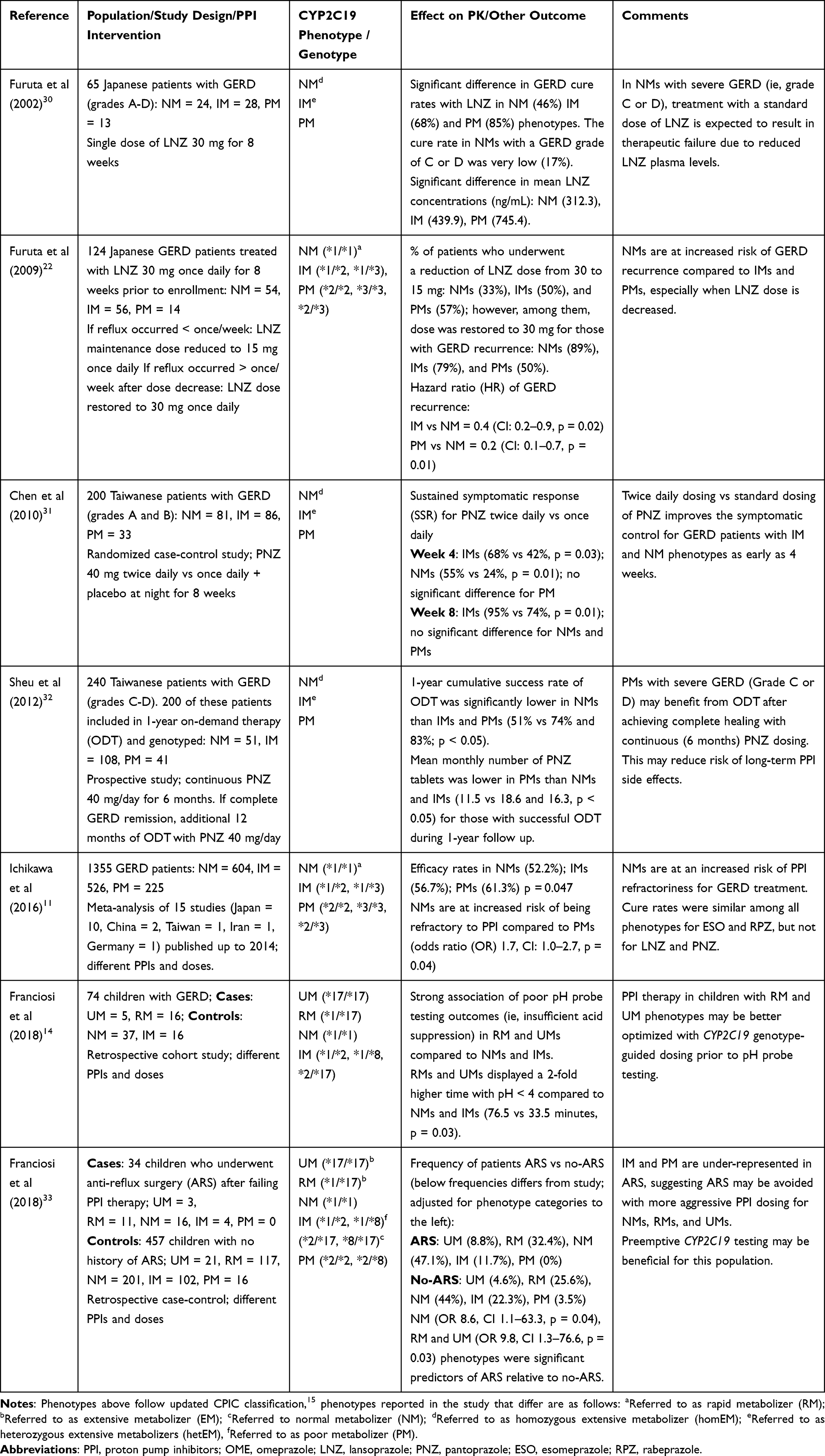 |
Table 3 Summary of Selected CYP2C19-PPI Outcome-Related Studies |
Influence of CYP2C19 Genetic Variation on PK and Clinical Response of PPIs
PPIs are prodrugs that inhibit the final step of gastric acid secretion. PPIs are weak bases and require an extremely acidic medium, like that of the gastric parietal cells, in order to be activated. Once activated by protonation, they bind to cysteine residues of the H+/K+─ATPase proton pumps, irreversibly inhibiting these pumps via covalent bonding, and rendering them nonfunctional. Synthesis of new functional proton pumps takes about 54 hours, explaining the extended duration of action despite the short half-life (approximately 90 minutes) of PPIs.16,34 All PPIs are enzymatically inactivated in the liver primarily by the cytochrome P450 2C19 (CYP2C19) enzyme, and to a lesser extent by CYP3A4. However, rabeprazole is unique in that it is primarily nonenzymatically cleared.
The metabolic parameters seen among the clinically available PPIs differ due to various influences of CYP2C19 polymorphisms. Greatest to least, dependence on CYP2C19 for their metabolism are: omeprazole > lansoprazole > dexlansoprazole > pantoprazole > esomeprazole > rabeprazole. Similarly, CYP2C19 genetic variability affects PPI metabolism to varying degrees in a clinical setting. In vitro and in vivo data suggest that CYP2C19 is responsible for approximately 90% of omeprazole clearance, and, to a lesser extent, approximately 70% of esomeprazole (ie S-isomer of omeprazole) clearance. Reports have documented a smaller magnitude of association between CYP2C19 genotype influence on PK parameters for both esomeprazole25,28 and rabeprazole.35–38
Differences in PK parameters and clinical efficacy of various PPIs among the range of CYP2C19 genotypes have been well documented; namely, the area under the concentration versus time curve (AUC) and clearance (Table 2). For example, in 2012, Gawrońska-Szklarz et al showed that after a single oral dose of pantoprazole 40 mg, IMs had a significant increase in AUC compared to NMs (4.38 ± 1.00 mg⋅h/L vs 3 ± 1.02 mg⋅h/L; p < 0.05).27 Similarly, Hunfeld et al found that the mean AUC was about two-fold higher in IMs than NMs following a single dose of pantoprazole 40 mg (7.95 mg⋅h/L vs 3.82 mg⋅h/L; p = 0.023), and following repeat administration of pantoprazole 40 mg daily (8.90 mg⋅h/L vs 4.12 mg⋅h/L; p = 0.010).26 In contrast, and consistent with the lower influence of CYP2C19 on its metabolism, there were only about 60% increases in AUC following single dose of esomeprazole 40 mg (4.24 mg⋅h/L vs 2.68 mg⋅h/L; p = 0.369) and 5 days of therapy (8.22 mg⋅h/L vs 5.10 mg⋅h/L; p = 0.079). This study illustrates the difference in magnitude of the influence of the IM phenotype on pantoprazole versus esomeprazole, though it should be noted that the lack of statistical differences with esomeprazole likely are the result of a Type 2 statistical error, as there were only 7 participants in each study group.
The aforementioned study by Gawrońska-Szklarz et al documented that 57% of interindividual variability in pantoprazole clearance was explained by CYP2C19 genotype status.27 Specifically, the study demonstrated significantly lower plasma concentrations and accelerated elimination (clearance) in UMs vs NMs 3 hours after single dose oral administration of pantoprazole 40 mg. The lowest population oral clearance was associated with PM (3.68 L/h) and the highest oral clearance was observed in UM phenotype (31.13 L/h); while the IM phenotype had an observed oral clearance of 8 L/h.27 The study by Hunfeld et al, concluded that on day 1 and day 6, all studied PPIs demonstrated the same pattern between genotype and AUC, with highest AUC in PMs, and lowest in RMs (ie, in the absence of UMs).24 Consistent with the mathematical and biological relationship between clearance and AUC, clearance showed the same genotypic trend as the AUC, with lowest clearance in PMs and highest in RMs.
Higher PPI exposure (ie AUC, plasma concentration levels) observed in IM and PM phenotypes is also associated with a significant increase in acid suppression, compared to NMs.23,24,26,36,39,40 At standard doses, the effect of PPIs on acid suppression (ie, as measured by median intragastric pH) is highest among PMs, followed by IMs, NMs, RMs, and lastly, UMs, associated with the least acid-inhibitory effect.35,37,41–43 The majority of studies conducted on the impact of CYP2C19 metabolism on the PK of PPIs, were published in the early 2000s, prior to the identification of the CYP2C19*17 variant allele. While there are fewer trials of the influence of CYP2C19*17 on PPI exposure, existing data reveal compelling evidence of CYP2C19*17 leading to increased gene expression and enzyme activity. Enhanced PPI clearance observed in RMs and UMs lead to lower PPI levels, therefore, these phenotypes are at an increased risk for PPI treatment failure due to insufficient acid suppression.24,26,27
There has also been interest in understanding how the combination of a gain-of-function and no-function allele would impact metabolism. Specifically, would the no-function allele dominate in the phenotype or would the two essentially offset one another, leading to relatively normal metabolism and an NM phenotype. Payan et al evaluated this question, testing the combination of no function and gain of function alleles (CYP2C19*2/*17) on CYP2C19 activity via hydroxylation index of omeprazole (the ratio of omeprazole to its major metabolite, hydroxyomeprazole, in serum 3 hours post-dose).29 The median hydroxylation index of omeprazole was 1.74 in CYP2C19*2/*17 and 1.98 in CYP2C19*1/*2 carriers (P = 0.33) indicating no significant difference between the two diplotypes. This suggests the loss of function *2 allele has much greater impact on the phenotype than the gain of function *17 allele. Therefore, it was concluded that like those with the *1/*2 diplotype, *2/*17 has intermediate metabolic capacity and a phenotypic classification of the *2/*17 diplotype as IMs, which is consistent with the CPIC classification. Of note, in this same study, there was no significant difference in median hydroxylation index observed between RMs and NMs, (0.71 and 0.78, respectively; P > 0.05) and UMs had a median hydroxylation index of 0.36, demonstrating very high metabolic capacity.
Additional Polymorphisms Associated with PK and Clinical Response of PPI’s
A retrospective analysis, conducted by Kee et al, identified novel variants that might influence PPI metabolism (and response). Specifically, they found a significant association of omeprazole treatment failure in refractory GERD with CYP2C19 UM phenotype inferred specifically by CYP2C:TG/TG (p = 0.03) (TG haplotypes [rs11188059 and rs2860840]), but not with CYP2C19 *17/*17.44,45 Interestingly, CYP2C19 *17 was absent in all CYP2C:TG homozygotes, suggesting that these might be additional genetic variants that influence PPI metabolism. This study suggests further exploration of the presence of undiscovered genetic variants within the CYP2C locus and other regions that may induce clinically relevant CYP2C19 UM phenotypes. Larger studies validating these findings are necessary to implement PPI dose adjustments for other genetic variations in the CYP2C locus.
In a study of genetic variability in the protein target of PPIs, Zhang et al concluded that gastric H+/K+─ATPase (rs2733743) polymorphisms (eg, a single nucleotide polymorphism [SNP] of the gastric hydrogen-potassium adenosine triphosphatase [H+/K+-ATPase] α-subunit) may have a greater effect than CYP2C19 polymorphisms on gastric acid suppression efficiency and may be related to the inter-individual differences in gastric acid inhibition achieved by PPIs.46–48 However confirmation studies are necessary to implement these findings clinically.
P-glycoprotein (P-gp) is responsible for PPI absorption in the small intestine and is highly polymorphic.11 Genetic polymorphisms in the P-gp enzyme coded by the ATP-binding cassette, sub-family B (MDR/TAP), member 1 gene (ABCB1; previously multidrug resistance transporter gene 1 [MDR1]) affects the pharmacokinetics and pharmacodynamics PPIs. The influence of the ABCB1 3435C>T (rs1045642) polymorphism on the PK/PD of lansoprazole, evaluated by Kodaira et al, found that lansoprazole plasma levels were higher in individuals with the ABCB1 3435 TT genotype than in those with the CT or CC genotypes.49 However, the study concluded that the influence of ABCB1 polymorphisms on the PK/PD of PPIs was smaller than that of CYP2C19 polymorphisms and could be disregarded in the clinical setting.
Collectively these studies highlight that work should continue to discover and validate other genetic variants that might have clinically relevant influence on PPI pharmacokinetics and/or efficacy.
Clinical Outcomes for GERD Treatment
Patients with decreased CYP2C19 functionality (ie, IMs and PMs) are said to be “therapeutically advantaged” compared to NMs, RMs, UMs at traditional doses. Specifically, increased PPI exposure in IMs and PMs has been linked to improved acid suppression (ie, higher intragastric pH and longer time with pH > 4.0) and treatment success; however, prolonged acid suppression potentiates a greater risk for PPI-associated adverse effects in the setting of chronic therapy (> 12 weeks).
Concerns regarding the long-term safety of PPIs have been raised as emerging literature has associated several adverse effects with long-term PPI use.50,51 Specifically, the FDA-approved PPI package inserts52–56 include the following adverse effects in the “Warning and Precautions” section: increased risk for osteoporosis-related fractures (eg, in hip, wrist, or spine) with long term use,57 increased risk of Clostridium difficile-associated diarrhea,58 hypomagnesemia with prolonged treatment,59 cyanocobalamin (vitamin B-12) deficiency with daily use longer than 3 years. In addition, long term PPI-use has been reported to be associated with increased risk of dementia, gastroenteritis,60,61 acute62–65 and chronic66–68 kidney disease, pneumonia (eg, highest risk at 1 month regardless of dose),69 mortality,70 and more recently, COVID-19-associated mortality.71
A longitudinal (median 5.7 years) observation cohort study conducted by Xie et al observed the risk of death among US veterans prescribed PPIs.70 Compared with those exposed to PPIs for ≤30 days, the study found a significant increase in mortality associated with duration of exposure >90 days; 91–180 days (HR 1.17 [1.13 to 1.20]), 181–360 days (HR 1.31 [1.27 to 1.34]) and 361–720 days (HR 1.51 [1.47 to 1.56]).70 These findings provide support to CPIC guideline recommendations (further discussed in the therapeutic guidance section below), of considering a dose reduction in patients with IM and PM phenotypes (and thus higher PPI exposure) who receive chronic PPI treatment for longer than 12 weeks.16
In contrast, PPI treatment failure and GERD recurrence rates are commonly seen in patients who have insufficient acid inhibition due to CYP2C19 normal and increased function phenotypes (Table 3).11,14,22,30,32 Current CPIC guidelines have clear recommendations for increasing the dose of omeprazole, lansoprazole, pantoprazole and dexlansoprazole (though a slightly weaker recommendation for the latter than the first three) for patients who are normal, rapid, and ultrarapid metabolizers of the CYP2C19 enzyme. However, this dose increase is recommended for specific indications (ie, Helicobacter pylori infection and erosive esophagitis).16 CPIC guidelines have yet to add recommendations on GERD-specific PPI dose adjustments.
Due to the relatively recent discovery of the CYP2C19*17 allele, studies on effects of CYP2C19 rapid and ultrarapid metabolism on PPI systemic exposure and efficacy are limited, though in the historical literature, these three phenotypes would have been captured under the extensive metabolizer (EM) phenotype. However, in the literature where the CYP2C19*17 allele is tested, there is growing support for the association of increased PPI clearance and an increased risk of therapeutic failure associated with the CYP2C19*17 allele. For example, a retrospective cohort study of 74 children with symptomatic GERD found a strong association of poor pH probe testing outcomes (ie, insufficient acid suppression) in RM and UM phenotypes (cases) compared to individuals carrying normal and/or no function alleles (controls). Specifically, Franciosi et al found that compared to the control group, RMs and UMs displayed a 2-fold higher percentage (P = 0.04) and duration of time (P = 0.03) in which pH was below 4.14
GERD recurrence is particularly of concern for RMs and UMs, but more recently, NMs were also found to be at increased risk of therapeutic failure (Table 3). Compelling evidence from a meta-analysis of 19 studies revealed NMs (ie, classified as RMs in the meta-analysis, however refers to CYP2C19 *1/*1 diplotype) were at a significantly higher risk of being refractory to PPI therapy than PMs (OR: 1.661, 95%; CI: 1.023–2.659; P = 0.040), irrespective of other confounding variables.11 A previous study showed that cure rates for GERD are also significantly influenced by CYP2C19 genotype and grade of GERD before treatment.30 GERD was classified as cured when endoscopy findings revealed no mucosal break at the end of the 8-week treatment period. The cure rates by genotype reported in the study were 45.8% for NMs; 67.9% for IMs; and 84.6% for PMs. The NM metabolizer phenotype was also shown to have lower plasma concentrations of PPI, which correlated with reduced acid suppression, and the cure rates in NMs with GERD grade of C or D were extremely low (16.7%). The study demonstrated that CYP2C19 genotype status is one of the predictable determinants for outcomes with PPI-based GERD therapy along with assessing the grade status of GERD prior to therapy. If CYP2C19 status is determined before treatment with PPI’s, an optimal dose can be prescribed, in particular in patients with NM, RM, and UM phenotypes and severe GERD, individuals for whom prescription of the typical dose of PPI’s usually result in therapeutic failure. Determining the therapeutic strategy a priori by making use of genotyping results would be expected to increase GERD cure rates with the initial treatment.
Several studies have also examined the impact of CYP2C19 activity on refractory GERD symptoms using standard doses versus multiple daily dose PPI regimens.11 Although limited, existing data suggest multiple (ie, double, or even quadruple) daily dosing of PPIs decreases CYP2C19 genotype-dependent differences in acid inhibition.25,28,31,72 Chen et al examined the difference in sustained symptomatic response (SSR) rates between CYP2C19 NM, IM, and PM phenotypes on pantoprazole 40 mg twice daily (80 mg total daily dose) verses standard dosing (40 mg once daily).31 In the RCT, 200 patients were classified (via panendoscopy) as being either LA grade A or B in GERD severity. For IMs, a significantly higher rate of SSR was observed in the twice-daily pantoprazole group vs standard-dosed group on week 4 (67.5% vs 41.9%, p = 0.027) and on week 8 (94.9% vs 73.7%, p = 0.013). Similarly, NMs had a significantly higher rate of SSR in the twice-daily group vs the standard-dosed group on week 4 (55% vs 23.7%, p = 0.006), however no significant difference was noted on week 8 (82.1% vs 68.4%, p = 0.194). Of note, the SSR in PMs on weeks 2, 4, and 8 were not significantly different between the two treatment groups (p > 0.05).31 These data suggest that in CYP2C19 IMs, double-dosed pantoprazole is associated with improved SSR rates as compared to standard dosing after 8 weeks. Moreover, in CYP2C19 IMs and NMs, accelerated control of GERD (grade A or B) symptoms was observed as early as four weeks after initiating therapy.
Additionally, Sahara et al compared the acid-inhibitory effects of four PPIs dosed twice daily (omeprazole 20 mg, lansoprazole 30 mg, rabeprazole 10 mg, and esomeprazole 20 mg) in CYP2C19 NM, IM and PM phenotypes.28 In H. pylori-negative CYP2C19 NMs, median 24-h intragastric pH with esomeprazole 20 mg twice daily was significantly higher than that with omeprazole, rabeprazole or lansoprazole; concluding that esomeprazole dosed twice daily is preferred, over the other PPIs tested, in CYP2C19 NMs who are at risk of being unresponsive to PPI therapy at standard once daily doses.28 Of note, acid inhibition attained by all four PPIs in IMs and PMs was sufficient for GERD treatment. Similarly, Lou et al found that esomeprazole dosage divisions for the same total daily dose of 40 mg (ie, 20 mg twice daily or 10 mg four times daily) resulted in significant clinical improvement in acid inhibition (median intragastric pH >4 and median percent of time the nocturnal pH was <4) in CYP2C19 NM, IM, and PM phenotypes compared to esomeprazole 40 mg once daily.25 Specifically, esomeprazole, controlled nocturnal gastric acid secretion more effectively when it was divided into 2 (20 mg twice daily) or 4 doses (10 mg four times daily).25 Therefore, in long-term maintenance therapy for GERD, divided daily doses of esomeprazole are recommended and may be more effective in controlling nocturnal acidity for CYP2C19 NMs who are refractory to standard PPI doses. This recommendation may be further extrapolated to RM and UM phenotypes who are at a higher risk of treatment failure compared to NMs. Additionally, Furuta et al observed that gastric acid inhibition in CYP2C19 NMs treated with lansoprazole 30 mg once daily was therapeutically insufficient for GERD treatment.30 Another study conducted by Furuta et al revealed that frequent dosing of lansoprazole (ie, 30 mg four times daily) in NMs, achieved significant acid suppression and overcomes the insufficiency previously seen with once daily dosing.72
While multiple daily dosing of PPIs demonstrated improved acid suppression and response rates in patients with refractoriness to PPIs, it is important to consider in the clinical setting the challenges with patient adherence to a multiple daily dosing regimen and the associated consequences of poor adherence.
Treatment Guidelines
General GERD Treatment Guidance
Most patients with GERD have nonerosive disease, with about 20% of patients with uncontrolled GERD progressing to erosive esophagitis. PPIs have demonstrated efficacy in healing erosive esophagitis and preventing the development and recurrence of GERD symptoms in long-term users. The updated AGA and ACG guidelines highlight the superiority of PPIs, compared to H2-receptor antagonists (H2RAs), in preventing the development and recurrence of GERD symptoms in long-term users and demonstrating efficacy in healing erosive esophagitis.4,73 For patients experiencing GERD symptoms, gastroenterology guidelines recommend an 8-week trial of once-daily PPI. If symptoms persist beyond 8 weeks, an esophagogastroduodenoscopy is recommended to confirm a diagnosis of GERD. Maintenance PPI therapy is only recommended for patients with GERD complications (ie, LA grade C or D erosive esophagitis and Barrett’s esophagus).6,51 For GERD patients requiring maintenance therapy, it is clinically important to use the lowest doses for the shortest duration to treat the underlying gastrointestinal disorders, especially in IMs and PMs where reduced drug clearance is predicted to increase PPI-associated adverse effects.
AGA recently provided an update on PPI de-escalation and de-prescribing. The expert panel concluded that PPI dose de-escalation for GERD can be effective for the majority of patients.51 This recommendation was based on a trial of 117 GERD patients using higher-than-standard PPI doses, where 80% of patients successfully stepped down to standard PPI doses without significant symptom recurrence or the need to increase the PPI dose again.74 Additionally, AGA recommends use of as-needed H2RAs and/or antacids for treatment of upper gastrointestinal (GI) symptoms after PPI withdrawal. This recommendation is based on the Inadomi et al finding that approximately 50% of all patient with uncomplicated GERD who discontinued PPIs, successfully remained off PPIs 6 months later; 75% of these patients were using H2Ras or antacids after PPI withdrawal.75 Severe persistent symptoms lasting greater than 2 months after PPI discontinuation may suggest the presence of a continuing indication for PPI therapy.51
Guidance for GERD Therapy Based on CYP2C19 Phenotype
Dutch Pharmacogenetics Working Group (DPWG)
The objective of the DPWG is to develop pharmacogenetics-based therapeutic recommendations based on a systematic literature review and to optimize drug use in patients whose genotypes are known.76 In 2018, DPWG updated their 2011 guidelines and developed specific dosing recommendations for CYP2C19 PM, IM, and UM phenotypes.76,77 No action is recommended for IM and PM phenotypes taking first generation PPIs (ie, omeprazole, lansoprazole, pantoprazole) as the DPWG mentions higher plasma concentration of PPIs results in an increase in the therapeutic effectiveness, without an increase in the side effects. For CYP2C19 UMs undergoing H. pylori eradication therapy with omeprazole, lansoprazole, or pantoprazole they recommend a 3-fold, 4-fold, and 5-fold dose increase, respectively. For other indications, they recommend to be alert to reduced effectiveness and, if necessary, increase PPI dose as recommended for H. pylori treatment. For all indications DPWG recommends advising UM phenotype patients to report persisting symptoms of dyspepsia.
Second-generation PPIs, esomeprazole and rabeprazole, are mentioned in the guidelines but do not have recommendations due to the lack of data available for UMs, whereas, dexlansoprazole is not mentioned in the guidelines.77 No action is recommended for IM and PM phenotypes taking rabeprazole as DPWGs systemic review of literature showed that the higher plasma concentration of rabeprazole does not result in an increase in side effects; although esomeprazole is also associated with higher plasma concentrations in these phenotypes, the guideline mentions there is insufficient evidence to support an increased risk of side effects.
Clinical Pharmacogenetics Implementation Consortium (CPIC)
CPIC is an international consortium of volunteers established to address barriers to clinical implementation of pharmacogenetic tests by creating evidence-based gene-drug clinical practice guidelines. The CPIC CYP2C19-PPI guidelines currently have the strongest prescribing recommendations for omeprazole, lansoprazole, pantoprazole (ie, CPIC level A, moderate or strong prescribing action recommended) and optional recommendations for dexlansoprazole (ie, CPIC level B). With inconsistent evidence linking CYP2C19 genotype with variability in esomeprazole and rabeprazole plasma concentrations and effectiveness, CPIC currently does not have prescribing action recommendations for these PPIs (ie, CPIC level C). Additionally, recommendations apply to both oral and IV PPI formulations.
In general, CPIC recommends initiating PPIs at standard doses for NMs and RMs; however, the guidelines note that a large body of evidence reports that standard doses are associated with eradication failure in H. pylori infection and lower erosive esophagitis healing rates in NMs and RMs as compared to IMs and PMs.16 Therefore, CPIC also recommends that a dose increase of 50–100% in CYP2C19 NM and RM phenotype individuals who are being treated for H. pylori infection or erosive esophagitis be considered in order to optimize clinical efficacy. Given that evidence has emerged since the publication of the guidelines suggesting a similar challenge for GERD patients who are NM or RM, we recommend clinicians also consider dose increases in GERD patients when initiating therapy. Further, given this emerging evidence, one might anticipate that GERD will be added in the CPIC PPI update as an indication for dose modification in RM and NM.
The low PPI exposure documented in patients who are CYP2C19 UMs compared to NMs, IMs, and PMs led to the CPIC recommendation to increase the starting daily dose by 100% in CYP2C19 UMs, regardless of indication. Additionally, the guidelines recommend divided daily dosing for NMs, RMs, and UMs, regardless of indication, to maximize the likelihood of therapeutic plasma concentrations and therapeutic efficacy. These patients should also be monitored for PPI efficacy.
In contrast to NM, RM, or UM phenotypes, who may need higher or more frequent PPI doses, or alternative treatment with a drug less dependent on CYP2C19 metabolism, those with IM and PM phenotypes may achieve sufficient acid inhibition with standard once-daily PPI doses. Once efficacy is achieved, CPIC recommends a 50% daily dose reduction for chronic PPI therapy (extending beyond 12 weeks) to reduce the risk of adverse effects associated with prolonged acid suppression.16,34 If a dose reduction is made, it is recommended to monitor for continued efficacy.
Food and Drug Administration (FDA)
FDA includes pharmacogenetic PK data in drug labels (ie, prescribing information), however, specific dose change recommendations are not provided, except for pantoprazole.78 Specifically, the FDA recommends a dosage reduction in children who are CYP2C19 PMs taking pantoprazole, although the magnitude of dose reduction is not specified. There is no dose adjustment recommended in the pantoprazole product label for adult IMs or PMs.
The FDA Table of Pharmacogenetic Associations reported higher systemic concentrations in IMs or PMs taking pantoprazole, dexlansoprazole, or omeprazole; and higher systemic concentrations in PMs taking esomeprazole, lansoprazole, or rabeprazole.79 As evidence continues to emerge on the gene-drug relationship of CYP2C19 and PPIs, updates to this table will be necessary to accurately reflect the association. Specifically, including that IMs may also have higher systemic concentrations of lansoprazole, and an inclusion of decreased systemic PPI concentrations seen in RMs and UMs.
As mentioned earlier, the FDA-approved drug labels (ie, prescribing information), highlight the adverse effects associated with PPIs in the “Warnings and Precautions” section. These adverse effect warnings are relevant when considering long-term PPI therapy, especially in IMs and PMs.
Special Populations
Both age and PGx status of patients can be instrumental in optimizing dosing. Specifically, when determining optimal PPI dosing, consideration of CYP2C19 phenotype and age range may be beneficial in improving PPI treatment response. For example, a recent pharmacokinetic study on the influence of CYP2C19 polymorphisms in elderly participants receiving omeprazole concluded that aging had a significant effect on non-PM phenotypes.80 In the NM and IM groups of the elderly, overall omeprazole exposure increased 4.2-fold and 3.7-fold, respectively, compared to the corresponding phenotype groups in young adults. Na et al also observed an increase in half-life for elderly in the NM (1.8-fold) and IM (1.9-fold) groups, compared to young adult NMs and IMs, however, no difference in half-life was observed in the elderly PM group compared to young adult PMs. Implementing a personalized pharmacotherapy approach with different standards for older versus young adults can achieve better effects.
PPI use in children is common and continues to increase.16 Pediatric manifestations of GERD are often harder to identify as typical symptoms of heartburn and regurgitation are not easily evaluated in younger patients. Anti-reflux surgery (ARS) is one of the most common major surgeries in pediatric patients with uncontrolled GERD symptoms despite pharmacological treatment. A recent study identified children with confirmed GERD who underwent ARS and found that the CYP2C19 RM and UM phenotypes were a significant predictor of ARS compared to the non-ARS controls.33 PMs were underrepresented in the ARS population, supporting the association of rapid PPI metabolism with those expressing one or two gain of function alleles. This study implies that surgery may be avoided with more aggressive PPI dosing for RMs and UMs, therefore, there is potential that preemptive CYP2C19 testing would be beneficial in this population.
Also in the pediatric population, emerging data have shown associations between PPI therapy and increased upper respiratory tract infections and asthma exacerbation, specifically in individuals expressing one or more no-function allele(s) (ie, IM and PM phenotypes).10,81,82
CYP2C19 PGx Testing
In practice, patients may or may not present with prior CYP2C19 pharmacogenetic results and clinicians are faced with a decision on whether to pursue testing. PGx testing prior to initiating therapy may improve the likelihood of treatment success and decrease the burden of potential adverse effects.
Only PGx tests conducted by Clinical Laboratory Improvement Amendments (CLIA)-certified laboratories should be used to inform clinical decisions. The Association for Molecular Pathology (AMP) PGx Working Group recommends that, at a minimum, Tier 1 CYP2C19 alleles (ie,*2, *3 and *17) be included in clinical CYP2C19 genotyping tests.83 These alleles are most common across the ethnic populations encountered in the US and are known to alter function. Tier 2 CYP2C19 variant alleles (ie, *4A, *4B, *5, *6, *7, *8, *9, *10 and *35) are also recommended in clinical genotyping tests, however they are considered optional to include.
CYP2C19 Implementation Experience at Our Institution
Implementation of CYP2C19 pharmacogenetic testing at the University of Florida (UF) was possible with close collaboration between the GI faculty in clinical practice and the multidisciplinary faculty within the precision medicine program (PMP).84 The implementation began with gastroenterologists expressing that readily available clinical recommendations for PPI dosing based on CYP2C19 genotype would be helpful in their practice. The PMP, led by pharmacogenetic-specialized pharmacists, provided education and support to the GI clinicians. Specifically, in-person education was done with a slide set sent out to all faculty afterwards. Key information was provided on how to order tests, contact information for questions, reviewing results, and dosing recommendations based on CYP2C19 phenotype. This investment in time and effort by our PGx pharmacists enhanced GI physician participation at our institution.
Implementation of CYP2C19 testing in PPI-treated GI patients (who primarily had GERD) made clear that in addition to the potential clinical utility of using CYP2C19 genotype to guide therapy decisions with PPIs, the prevalence of PGx medication prescribing is higher than previously thought. This could lead to additional clinical benefits for the patient over their course of care, particularly if they were tested with a multi-pharmacogene panel and not just for CYP2C19. Based on data from our institution, 70.2% of patients with GERD are prescribed other medications that can be guided by PGx panel-based testing within 12 months post-genotyping,84 causing us to conclude that panel-based testing provides more clinical information compared to single-gene testing, relative to costs and efficiencies associated with panel-based testing. Not only can CYP2C19 genotype be of clinical utility for several other medications, but use of a panel with additional pharmacogenes expands the future utility of the data.
As further support for running a panel-based versus single gene tests, the costs for the two approaches are now similar, based on our experience. Reimbursement data from our institution from January 2019 to December 2021 revealed a higher reimbursement rate for panel-based tests compared to a CYP2C19 test alone among those with commercial insurance (74% vs 42%, respectively). Specifically, for insurance claims submitted for GI diagnoses, we found 76% of claims submitted received partial or full reimbursement for the panel PGx tests as opposed to 53% of claims submitted for a single CYP2C19 gene test. Of note, our data showed a significantly higher reimbursement rate for Medicare insurance compared to commercial insurance.85
Drug-Drug-Gene Interactions
Along with CYP2C19 genotype, drug-drug-gene interactions (DDGI) may also be an important factor to consider with PPIs and could be important for personalization of therapy. There are few published studies that evaluate drug interactions with CYP2C19 after PPI administration, but based on study findings from Zhang et al46 genotype detection and drug interactions are helpful determinants of optimal PPI dosing in order to avoid treatment failure and PPI-associated adverse effects. Co-administration of PPIs (except rabeprazole) with strong CYP2C19 inhibitors (ie, fluvoxamine, fluconazole, fluoxetine)86 led to markedly increased PPI plasma concentrations for non-PMs, suggesting that a dose increases in NMs, RMs, and potentially UMs, may not be needed to achieve adequate acid suppression.16,34 Alternatively, strong CYP2C19 inducers (ie, rifampin, St. John’s wort) can lead to decreased systemic exposure and treatment failure, suggesting the need for a PPI dose increase in NMs, IMs, and potentially PMs.87 Particularly in patients for whom a CYP2C19 inducer cannot be avoided (eg treatment with rifampin), use of rabeprazole is likely to be the preferred PPI since the magnitude of induction (and thus dose changes needed for the other drugs) would be difficult to predict. Additionally, although many clinical studies have explored the DDGI potential of pantoprazole, they found none to be clinically relevant.88–90
In patients with normal CYP2C19 activity, CYP3A4 inhibitors (ie, ketoconazole, fluconazole, clarithromycin) have minimal effect on PPI metabolism. Clinically relevant alterations in PPI metabolism are seen for those with CYP2C19 deficiency (ie, IMs and PMs) who shift from CYP2C19 as the primary metabolic pathway to primarily CYP3A4 pathway.34,91 In CYP2C19 IMs and PMs, CYP3A4 inhibitors may markedly increase PPI concentrations; therefore, increasing the risk for adverse effects with chronic dosing (greater than 12 weeks).16,91
Omeprazole and esomeprazole are weak autoinhibitors of CYP2C197 which can influence the effectiveness of concurrently administered CYP2C19-metabolized prodrugs (ie, clopidogrel) and increase drug concentrations of CYP2C19 inactivated drugs (ie, diazepam, warfarin, phenytoin, voriconazole, SSRIs) which may raise the risk of side effects.34 However, further studies demonstrating the clinical consequence of these DDGIs are needed. In addition, the FDA issued a warning of reduced effectiveness of clopidogrel in individuals with impaired CYP2C19 function and advised avoiding its concomitant use with omeprazole or esomeprazole.92,93 It is recommended to instead use an alternative PPI with weaker CYP2C19 inhibition, such as pantoprazole.88–90
Consideration of CYP2C19 Metabolism in Drug Development
The relationship between PPI metabolism and CYP2C19 polymorphism has had a significant role in drug development. Modifications to areas in the omeprazole structure which are prone to CYP2C19 metabolism (via o-demethylation and hydroxylation) have been made in order to reduce drug interactions and PK variability affected by pharmacogenetics. For example, the methyl group on omeprazole’s oxygen moiety was substituted with a difluoromethyl group (ie, pantoprazole) to reduce CYP2C19 O-demethylation, therefore, pantoprazole is less prone metabolism via CYP2C19 compared to omeprazole. In addition, esomeprazole is the S-enantiomer of omeprazole (a racemic mixture). With less dependence on CYP2C19 metabolism due to stereoselectivity, esomeprazole offers an advantage of less interindividual variability of PK parameters and improved therapeutic effect.94–96
Although approved in other countries, ilaprazole and an imidazopyridine-derived PPI (ie tenatoprazole) are not approved for clinical use in the US and are currently undergoing preclinical and clinical evaluation. Ilaprazole has shown an extended plasma half-life (ie, 3.6 hours) and metabolism not significantly influenced by CYP2C19 genetic polymorphism.97 Likewise, tenatoprazole offers a theoretical advantage over its benzimidazole cousins, as its prolonged half-life (~7-fold higher than other PPIs) may aid in consistent acid suppression in the daytime as well as night-time; specifically reducing nocturnal gastric acidification in healthy, H. pylori negative patients.98,99 A recent PK study investigated the metabolism of tenatoprazole via CYP2C19 and CYP3A4 pathways in human liver microsomes,100 whereas previous PK studies investigated metabolism in vitro. Le et al identified that the R-isomer of tenatoprazole is metabolized predominantly by CYP2C19, whereas the S-isomer is metabolized via CYP2C19 and CYP3A4.100 Since CYP3A4 may compensate for decreased metabolism via CYP2C19 pathway (ie IMs and PMs) the authors of the aforementioned study proposed isolating and administering just the S-isomer as it may lead to uniform PK parameters regardless of genotype, however, further investigation is needed to prove the benefit over a racemic mixture and R-isomer.
A novel PPI, azeloprazole (Z-215), is primarily metabolized via CYP3A4. A study conducted by Kinoshita et al showed that at weeks 4 and 8, 3 doses of Z-215 (10 mg, 20 mg, and 40 mg) were just as effective for the initial treatment of reflux esophagitis as 10 mg dose of rabeprazole.101 High endoscopic healing rates for reflux esophagitis, were observed with Z-215 administration, regardless of CYP2C19 genotype.
PPIs are the mainstay therapy for GERD and have some unmet needs such as nocturnal acid breakthrough (NAB) (ie, intragastric pH less than 4.0 lasting for greater than 1 hour during the overnight period) and CYP2C19 phenotype-related variability. Tegoprazan (ie, a novel potassium-competitive acid blocker) has shown benefits in improving night symptoms and does not depend on CYP2C19, which makes it a potential therapy for patients experiencing GERD nightly symptoms.102 Recently, tegoprazan 25 mg was determined to be non-inferior to lansoprazole 15 mg in maintenance of healing of GERD (LA grade A and grade B).103 Tegoprazan also appeared to have a similar safety profile to lansoprazole. In Japan, another novel potassium-competitive acid blocker vonoprazan (TAK-438), is clinically available to treat acid-related diseases. Jenkins et al, demonstrated TAK-438 has potent, rapid acid inhibition and its pharmacokinetics were unaffected by CYP2C19 genotype as it is primarily metabolized via CYP3A4 enzyme.104 The study also showed TAK-438 might have advantages over existing acid-suppressing drugs, such as prevention of NAB, reduction in night-time gastric acidity, and the ability to dose at any time regardless of meals.
Future Perspectives
Genetic variability in CYP2C19 across continental populations is very common and confers a range of metabolism phenotypes that have been clearly shown in the literature to influence pharmacokinetics and clinical responses to PPIs in adults, and special populations. Substantial additional data support differential clinical outcomes based on CYP2C19 genotype and inferred CYP2C19 phenotype, and guidance is available from pharmacogenetic expert groups and the FDA. Despite these facts, CYP2C19 genotype-guided PPI therapy has not seen widespread clinical uptake. The North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) treatment guidelines support empiric PPI dose increase for refractory symptomatic GERD prior to esophageal pH testing, yet continue to exclude genetic factors related to PPI metabolism in clinical practice guidelines.105
To address this issue, an increased awareness of the relationship between CYP2C19 variability and PPI metabolism, as well as the widespread availability and relatively low cost of CYP2C19 genotyping is needed, especially among frontline clinicians. This need can be met through large scale educational efforts. To date, many clinical pharmacogenomic implementations have been developed and/or are currently led by pharmacists, so this group is a logical starting point to increase awareness and advocate for practice change. Pharmacists can serve as champions to educate gastroenterologists within their institutions. There is also a dearth of published literature describing successful CYP2C19-PPI implementations, with only a single such publication10 identified by the authors. Additional literature is needed describing challenges, lessons learned, and shared experiences surrounding CYP2C19 testing with PPIs from clinical practice leaders in this area. One journal is publishing a series of articles on various successful clinical implementations106,107 that should include experience with CYP2C19-guided PPI therapy.
As described above, emerging evidence suggests dose increases are appropriate for patients with GERD who are CYP2C19 NM or RM. CPIC guidelines are updated regularly, and it would not be surprising if the GERD indication is added into the updated recommendations. Within the clinical guidelines, it would be helpful to establish common nomenclature to define GERD. Specifically, including language for treating GERD symptoms prior to esophagogastroduodenoscopy will provide clarity as to whether the recommendations apply to this population. Further, including LA grade into recommendations may also be of value as these patients have been diagnosed with GERD. It will also provide additional clarity to clinicians when CPIC guidelines are updated to provide guidance for genotype-guided dosing of PPIs in CYP2C19 NMs and RMs for GERD-related indications. Finally, CYP2C19 genotyping to guide PPI use should be addressed in gastroenterology guidelines. With the widespread use, and overuse, of PPIs, genetic data can be an invaluable tool to optimize efficacy and avoid adverse effects of PPIs.
Until CYP2C19 genotyping is addressed in gastroenterology guidelines, we advocate for the incorporation of CYP2C19 genotype data into the currently available guidelines, as illustrated in Figure 2. This hybrid application of decision making based on clinical and genetic factors holds promise for optimizing patient outcomes while additional evidence is accumulated and awareness of this important issue increases.
 |
Figure 2 Recommendations for PGx testing and PGx-guided PPI dosing for patients with GERD symptoms despite PPI treatment (Algorithm based on CPIC guidelines in conjunction with ACG guidelines). Note: *If PGx testing is performed, at minimum, ensure Tier 1 CYP2C19 alleles (ie, *2, *3, *17) are included in genotyping assays. |
Disclosure
Drs Johnson, Wiisanen, Cicali and Eken were supported in part through NIH grant U01 HG007269.The authors report no other conflicts of interest in this work.
References
1. Peery AF, Crockett SD, Murphy CC, et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2021. Gastroenterology. 2022;162(2):621–644. doi:10.1053/j.gastro.2021.10.017
2. Mathews SC, Izmailyan S, Brito FA, Yamal JM, Mikhail O, Revere FL. Prevalence and financial burden of digestive diseases in a commercially insured population. Clin Gastroenterol Hepatol. 2022;20(7):1480–1487.e1487. doi:10.1016/j.cgh.2021.06.047
3. Medical Expenditure Panel Survey (MEPS). Agency for Healthcare Research and Quality (AHRQ). ClinCalc DrugStats Database version 2022.08. Rockville, MD; 2013–2020.
4. Katz PO, Dunbar KB, Schnoll-Sussman FH, Greer KB, Yadlapati R, Spechler SJ. ACG clinical guideline for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2022;117(1):27–56. doi:10.14309/ajg.0000000000001538
5. Katzka DA, Pandolfino JE, Kahrilas PJ. Phenotypes of gastroesophageal reflux disease: where Rome, Lyon, and Montreal meet. Clin Gastroenterol Hepatol. 2020;18(4):767–776. doi:10.1016/j.cgh.2019.07.015
6. Yadlapati R, Gyawali CP, Pandolfino JE; Participants CGCC. AGA clinical practice update on the personalized approach to the evaluation and management of GERD: expert review. Clin Gastroenterol Hepatol. 2022;20(5):984–994.e981. doi:10.1016/j.cgh.2022.01.025
7. Armstrong D, Bennett JR, Blum AL, et al. The endoscopic assessment of esophagitis: a progress report on observer agreement. Gastroenterology. 1996;111(1):85–92. doi:10.1053/gast.1996.v111.pm8698230
8. Gyawali CP, Fass R. Management of gastroesophageal reflux disease. Gastroenterology. 2018;154(2):302–318. doi:10.1053/j.gastro.2017.07.049
9. Gyawali CP, Kahrilas PJ, Savarino E, et al. Modern diagnosis of GERD: the Lyon consensus. Gut. 2018;67(7):1351–1362. doi:10.1136/gutjnl-2017-314722
10. Cicali EJ, Blake K, Gong Y, et al. Novel implementation of genotype-guided proton pump inhibitor medication therapy in children: a pilot, randomized, multisite pragmatic trial. Clin Transl Sci. 2019;12(2):172–179. doi:10.1111/cts.12589
11. Ichikawa H, Sugimoto M, Sugimoto K, Andoh A, Furuta T. Rapid metabolizer genotype of CYP2C19 is a risk factor of being refractory to proton pump inhibitor therapy for reflux esophagitis. J Gastroenterol Hepatol. 2016;31(4):716–726. doi:10.1111/jgh.13233
12. Tang M, Blake KV, Lima JJ, et al. Genotype tailored treatment of mild symptomatic acid reflux in children with uncontrolled asthma (GenARA): rationale and methods. Contemp Clin Trials. 2019;78:27–33. doi:10.1016/j.cct.2019.01.009
13. Sabet S, McGhee JE. New guidance on cytochrome P450 2C19 phenotype-based use of proton pump inhibitors. J Pediatr Gastroenterol Nutr. 2021;72(5):697–699. doi:10.1097/MPG.0000000000003082
14. Franciosi JP, Mougey EB, Williams A, et al. Association between CYP2C19*17 alleles and pH probe testing outcomes in children with symptomatic gastroesophageal reflux. J Clin Pharmacol. 2018;58(1):89–96. doi:10.1002/jcph.977
15. Caudle KE, Dunnenberger HM, Freimuth RR, et al. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet Med. 2017;19(2):215–223. doi:10.1038/gim.2016.87
16. Lima JJ, Thomas CD, Barbarino J, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2C19 and proton pump inhibitor dosing. Clin Pharmacol Ther. 2021;109(6):1417–1423. doi:10.1002/cpt.2015
17. PharmGKB. Gene-specific information tables for CYP2C19. Available from: https://www.pharmgkb.org/page/cyp2c19RefMaterials.
18. Scott SA, Sangkuhl K, Shuldiner AR, et al. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 19. Pharmacogenet Genomics. 2012;22(2):159–165. doi:10.1097/FPC.0b013e32834d4962
19. Sim SC, Risinger C, Dahl ML, et al. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther. 2006;79(1):103–113. doi:10.1016/j.clpt.2005.10.002
20. Ionova Y, Ashenhurst J, Zhan J, et al. CYP2C19 allele frequencies in over 2.2 million direct-to-consumer genetics research participants and the potential implication for prescriptions in a large health system. Clin Transl Sci. 2020;13(6):1298–1306. doi:10.1111/cts.12830
21. Clinical Pharmacogenetics Implementation Consortium (CPIC). CYP2C19 frequency table. Available from: https://files.cpicpgx.org/data/report/current/frequency/CYP2C19_frequency_table.xlsx.
22. Furuta T, Sugimoto M, Kodaira C, et al. CYP2C19 genotype is associated with symptomatic recurrence of GERD during maintenance therapy with low-dose lansoprazole. Eur J Clin Pharmacol. 2009;65(7):693–698. doi:10.1007/s00228-009-0628-5
23. Hu XP, Xu JM, Hu YM, Mei Q, Xu XH. Effects of CYP2C19 genetic polymorphism on the pharmacokinetics and pharmacodynamics of omeprazole in Chinese people. J Clin Pharm Ther. 2007;32(5):517–524. doi:10.1111/j.1365-2710.2007.00851.x
24. Hunfeld NG, Mathot RA, Touw DJ, et al. Effect of CYP2C19*2 and *17 mutations on pharmacodynamics and kinetics of proton pump inhibitors in Caucasians. Br J Clin Pharmacol. 2008;65(5):752–760. doi:10.1111/j.1365-2125.2007.03094.x
25. Lou HY, Chang CC, Sheu MT, Chen YC, Ho HO. Optimal dose regimens of esomeprazole for gastric acid suppression with minimal influence of the CYP2C19 polymorphism. Eur J Clin Pharmacol. 2009;65(1):55–64. doi:10.1007/s00228-008-0552-0
26. Hunfeld NG, Touw DJ, Mathot RA, et al. A comparison of the acid-inhibitory effects of esomeprazole and pantoprazole in relation to pharmacokinetics and CYP2C19 polymorphism. Aliment Pharmacol Ther. 2010;31(1):150–159. doi:10.1111/j.1365-2036.2009.04150.x
27. Gawrońska-Szklarz B, Adamiak-Giera U, Wyska E, et al. CYP2C19 polymorphism affects single-dose pharmacokinetics of oral pantoprazole in healthy volunteers. Eur J Clin Pharmacol. 2012;68(9):1267–1274. doi:10.1007/s00228-012-1252-3
28. Sahara S, Sugimoto M, Uotani T, et al. Twice-daily dosing of esomeprazole effectively inhibits acid secretion in CYP2C19 rapid metabolisers compared with twice-daily omeprazole, rabeprazole or lansoprazole. Aliment Pharmacol Ther. 2013;38(9):1129–1137. doi:10.1111/apt.12492
29. Payan M, Rouini MR, Tajik N, Ghahremani MH, Tahvilian R. Hydroxylation index of omeprazole in relation to CYP2C19 polymorphism and sex in a healthy Iranian population. Daru. 2014;22(1):81. doi:10.1186/s40199-014-0081-6
30. Furuta T, Shirai N, Watanabe F, et al. Effect of cytochrome P4502C19 genotypic differences on cure rates for gastroesophageal reflux disease by lansoprazole. Clin Pharmacol Ther. 2002;72(4):453–460. doi:10.1067/mcp.2002.127637
31. Chen WY, Chang WL, Tsai YC, Cheng HC, Lu CC, Sheu BS. Double-dosed pantoprazole accelerates the sustained symptomatic response in overweight and obese patients with reflux esophagitis in Los Angeles grades A and B. Am J Gastroenterol. 2010;105(5):1046–1052. doi:10.1038/ajg.2009.632
32. Sheu BS, Cheng HC, Yeh YC, Chang WL. CYP2C19 genotypes determine the efficacy of on-demand therapy of pantoprazole for reflux esophagitis as Los-Angeles grades C and D. J Gastroenterol Hepatol. 2012;27(1):104–109. doi:10.1111/j.1440-1746.2011.06848.x
33. Franciosi JP, Mougey EB, Williams A, et al. Association between CYP2C19 extensive metabolizer phenotype and childhood anti-reflux surgery following failed proton pump inhibitor medication treatment. Eur J Pediatr. 2018;177(1):69–77. doi:10.1007/s00431-017-3051-4
34. El Rouby N, Lima JJ, Johnson JA. Proton pump inhibitors: from CYP2C19 pharmacogenetics to precision medicine. Expert Opin Drug Metab Toxicol. 2018;14(4):447–460. doi:10.1080/17425255.2018.1461835
35. Shirai N, Furuta T, Xiao F, et al. Comparison of lansoprazole and famotidine for gastric acid inhibition during the daytime and night-time in different CYP2C19 genotype groups. Aliment Pharmacol Ther. 2002;16(4):837–846. doi:10.1046/j.1365-2036.2002.01229.x
36. Ieiri I, Kishimoto Y, Okochi H, et al. Comparison of the kinetic disposition of and serum gastrin change by lansoprazole versus rabeprazole during an 8-day dosing scheme in relation to CYP2C19 polymorphism. Eur J Clin Pharmacol. 2001;57(6–7):485–492. doi:10.1007/s002280100342
37. Adachi K, Katsube T, Kawamura A, et al. CYP2C19 genotype status and intragastric pH during dosing with lansoprazole or rabeprazole. Aliment Pharmacol Ther. 2000;14(10):1259–1266. doi:10.1046/j.1365-2036.2000.00840.x
38. Sugimoto M, Shirai N, Nishino M, et al. Comparison of acid inhibition with standard dosages of proton pump inhibitors in relation to CYP2C19 genotype in Japanese. Eur J Clin Pharmacol. 2014;70(9):1073–1078. doi:10.1007/s00228-014-1713-y
39. Howden CW, Metz DC, Hunt B, et al. Dose-response evaluation of the antisecretory effect of continuous infusion intravenous lansoprazole regimens over 48 h. Aliment Pharmacol Ther. 2006;23(7):975–984. doi:10.1111/j.1365-2036.2006.02849.x
40. Wang Y, Zhang H, Meng L, et al. Influence of CYP2C19 on the relationship between pharmacokinetics and intragastric pH of omeprazole administered by successive intravenous infusions in Chinese healthy volunteers. Eur J Clin Pharmacol. 2010;66(6):563–569. doi:10.1007/s00228-010-0821-6
41. Furuta T, Ohashi K, Kosuge K, et al. CYP2C19 genotype status and effect of omeprazole on intragastric pH in humans. Clin Pharmacol Ther. 1999;65(5):552–561. doi:10.1016/S0009-9236(99)70075-5
42. Li ZS, Zhan XB, Xu GM, Cheng NN, Liao Z. Effect of esomeprazole and rabeprazole on intragastric pH in healthy Chinese: an open, randomized crossover trial. J Gastroenterol Hepatol. 2007;22(6):815–820. doi:10.1111/j.1440-1746.2006.04709.x
43. Shimatani T, Inoue M, Kuroiwa T, Horikawa Y, Mieno H, Nakamura M. Effect of omeprazole 10 mg on intragastric pH in three different CYP2C19 genotypes, compared with omeprazole 20 mg and lafutidine 20 mg, a new H2-receptor antagonist. Aliment Pharmacol Ther. 2003;18(11–12):1149–1157. doi:10.1046/j.1365-2036.2003.01804.x
44. Kee PS, Maggo SDS, Kennedy MA, et al. Omeprazole treatment failure in gastroesophageal reflux disease and genetic variation at the CYP2C locus. Front Genet. 2022;13:869160. doi:10.3389/fgene.2022.869160
45. Bråten LS, Haslemo T, Jukic MM, et al. A novel CYP2C-haplotype associated with ultrarapid metabolism of escitalopram. Clin Pharmacol Ther. 2021;110(3):786–793. doi:10.1002/cpt.2233
46. Zhang HJ, Zhang XH, Liu J, et al. Effects of genetic polymorphisms on the pharmacokinetics and pharmacodynamics of proton pump inhibitors. Pharmacol Res. 2020;152:104606. doi:10.1016/j.phrs.2019.104606
47. Li H, Meng L, Liu F, Wei JF, Wang YQ. H+/K+-ATPase inhibitors: a patent review. Expert Opin Ther Pat. 2013;23(1):99–111. doi:10.1517/13543776.2013.741121
48. Sun LN, Cao Y, Li YQ, et al. Impact of gastric H+/K+-ATPase rs2733743 on the Intragastric pH-values of dexlansoprazole injection in Chinese subjects. Front Pharmacol. 2017;8:670. doi:10.3389/fphar.2017.00670
49. Kodaira C, Sugimoto M, Nishino M, et al. Effect of MDR1 C3435T polymorphism on lansoprazole in healthy Japanese subjects. Eur J Clin Pharmacol. 2009;65(6):593–600. doi:10.1007/s00228-009-0625-8
50. Thurber KM, Otto AO, Stricker SL. Proton pump inhibitors: understanding the associated risks and benefits of long-term use. Am J Health Syst Pharm. 2023;80(8):487–494. doi:10.1093/ajhp/zxad009
51. Targownik LE, Fisher DA, Saini SD. AGA clinical practice update on de-prescribing of proton pump inhibitors: expert review. Gastroenterology. 2022;162(4):1334–1342. doi:10.1053/j.gastro.2021.12.247
52. Protonix (Pantoprazole) [package insert]. Philadelphia, PA: Pfizer Inc.; 2020.
53. Prilosec (Omeprazole) [package insert]. Wilmington DE: AstraZeneca LP; 2020.
54. Prevacid (Lansoprazole) [package insert]. Warren, NJ: Takeda Pharmaceuticals America, Inc.; 2020.
55. Nexium (Esomeprazole) [package insert]. Wilmington DE: AstraZeneca LP; 2021.
56. Dexilant (Dexlansoprazole) [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2020.
57. Food and Drug Administration. FDA drug safety communication: possible increased risk of fractures of the hip, wrist, and spine with the use of proton pump inhibitors; 2010.
58. Food and Drug Administration. FDA drug safety communication: Clostridium difficile associated diarrhea can be associated with stomach acid drugs known as proton pump inhibitors (PPIs). Food and Drug Administration; 2012.
59. Food and Drug Administration. FDA drug safety communication: low magnesium levels can be associated with long-term use of proton pump inhibitor drugs (PPIs); 2011.
60. Chen Y, Liu B, Glass K, Du W, Banks E, Kirk M. Use of proton pump inhibitors and the risk of hospitalization for infectious gastroenteritis. PLoS One. 2016;11(12):e0168618. doi:10.1371/journal.pone.0168618
61. Hassing RJ, Verbon A, de Visser H, Hofman A, Stricker BH. Proton pump inhibitors and gastroenteritis. Eur J Epidemiol. 2016;31(10):1057–1063. doi:10.1007/s10654-016-0136-8
62. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrol. 2013;14:150. doi:10.1186/1471-2369-14-150
63. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837–844. doi:10.1038/ki.2014.74
64. Antoniou T, Macdonald EM, Hollands S, et al. Proton pump inhibitors and the risk of acute kidney injury in older patients: a population-based cohort study. CMAJ Open. 2015;3(2):E166–E171. doi:10.9778/cmajo.20140074
65. Chen G, Ning LJ, Qin Y, Zhao B, Mei D, Li XM. Acute kidney injury following the use of different proton pump inhibitor regimens: a real-world analysis of post-marketing surveillance data. J Gastroenterol Hepatol. 2021;36(1):156–162. doi:10.1111/jgh.15151
66. Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol. 2016;27(10):3153–3163. doi:10.1681/ASN.2015121377
67. Arora P, Gupta A, Golzy M, et al. Proton pump inhibitors are associated with increased risk of development of chronic kidney disease. BMC Nephrol. 2016;17(1):112. doi:10.1186/s12882-016-0325-4
68. Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238–246. doi:10.1001/jamainternmed.2015.7193
69. Lambert AA, Lam JO, Paik JJ, Ugarte-Gil C, Drummond MB, Crowell TA. Risk of community-acquired pneumonia with outpatient proton-pump inhibitor therapy: a systematic review and meta-analysis. PLoS One. 2015;10(6):e0128004. doi:10.1371/journal.pone.0128004
70. Xie Y, Bowe B, Li T, Xian H, Yan Y, Al-Aly Z. Risk of death among users of proton pump inhibitors: a longitudinal observational cohort study of United States veterans. BMJ Open. 2017;7(6):e015735. doi:10.1136/bmjopen-2016-015735
71. Toubasi AA, AbuAnzeh RB, Khraisat BR, Al-Sayegh TN, AlRyalat SA. Proton pump inhibitors: current use and the risk of coronavirus infectious disease 2019 development and its related mortality. meta-analysis. Arch Med Res. 2021;52(6):656–659. doi:10.1016/j.arcmed.2021.03.004
72. Furuta T, Shirai N, Xiao F, Ohashi K, Ishizaki T. Effect of high-dose lansoprazole on intragastic pH in subjects who are homozygous extensive metabolizers of cytochrome P4502C19. Clin Pharmacol Ther. 2001;70(5):484–492. doi:10.1067/mcp.2001.119721
73. Wang WH, Huang JQ, Zheng GF, et al. Head-to-head comparison of H2-receptor antagonists and proton pump inhibitors in the treatment of erosive esophagitis: a meta-analysis. World J Gastroenterol. 2005;11(26):4067–4077. doi:10.3748/wjg.v11.i26.4067
74. El-Serag HB. Epidemiology of non-erosive reflux disease. Digestion. 2008;78(Suppl 1):6–10. doi:10.1159/000151249
75. Inadomi JM, Jamal R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology. 2001;121(5):1095–1100. doi:10.1053/gast.2001.28649
76. Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: from bench to byte-an update of guidelines. Clin Pharmacol Ther. 2011;89(5):662–673. doi:10.1038/clpt.2011.34
77. KNMP. RDPA. Dutch Pharmacogenetics Working Group (DPWG). Pharmacogenetic guidelines [Internet]. Netherlands. Dutch Pharmacogenetics Working Group Recommendations; 2020. Available from: https://www.knmp.nl/media/1058.
78. Food and Drug Administration. FDA table of pharmacogenomic biomarkers in drug labeling; 2022.
79. Food and Drug Administration. FDA table of pharmacogenetics association; 2022.
80. Na JY, Jeon I, Yoon J, et al. Influence of CYP2C19 polymorphisms on the pharmacokinetics of omeprazole in elderly subjects. Clin Pharmacol Drug Dev. 2021;10(12):1469–1477. doi:10.1002/cpdd.966
81. Lima JJ, Lang JE, Mougey EB, et al. Association of CYP2C19 polymorphisms and lansoprazole-associated respiratory adverse effects in children. J Pediatr. 2013;163(3):686–691. doi:10.1016/j.jpeds.2013.03.017
82. Bernal CJ, Aka I, Carroll RJ, et al. CYP2C19 phenotype and risk of proton pump inhibitor-associated infections. Pediatrics. 2019;144(6):e20190857. doi:10.1542/peds.2019-0857
83. Pratt VM, Del Tredici AL, Hachad H, et al. Recommendations for clinical CYP2C19 genotyping allele selection: a report of the association for molecular pathology. J Mol Diagn. 2018;20(3):269–276. doi:10.1016/j.jmoldx.2018.01.011
84. Elchynski AL, Cicali EJ, Ferrer Del Busto MC, et al. Determining the potential clinical value of panel-based pharmacogenetic testing in patients with chronic pain or gastroesophageal reflux disease. Pharmacogenomics J. 2021;21(6):657–663. doi:10.1038/s41397-021-00244-6
85. Lemke L. Reimbursement of Pharmacogenetic Tests at UF Health. Denver, CO: Clinical Pharmacogenetics Implementation Consortium; 2022.
86. Administration UFD. Drug development and drug interactions: table of substrates, inhibitors and inducers; 2022.
87. Blume H, Donath F, Warnke A, Schug BS. Pharmacokinetic drug interaction profiles of proton pump inhibitors. Drug Saf. 2006;29(9):769–784. doi:10.2165/00002018-200629090-00002
88. Desta Z, Zhao X, Shin JG, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet. 2002;41(12):913–958. doi:10.2165/00003088-200241120-00002
89. Calabresi L, Pazzucconi F, Ferrara S, Di Paolo A, Tacca MD, Sirtori C. Pharmacokinetic interactions between omeprazole/pantoprazole and clarithromycin in health volunteers. Pharmacol Res. 2004;49(5):493–499. doi:10.1016/j.phrs.2003.10.010
90. Cheer SM, Prakash A, Faulds D, Lamb HM. Pantoprazole: an update of its pharmacological properties and therapeutic use in the management of acid-related disorders. Drugs. 2003;63(1):101–133. doi:10.2165/00003495-200363010-00006
91. Wedemeyer RS, Blume H. Pharmacokinetic drug interaction profiles of proton pump inhibitors: an update. Drug Saf. 2014;37(4):201–211. doi:10.1007/s40264-014-0144-0
92. Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022;112(5):959–967. doi:10.1002/cpt.2526
93. Plavix (Clopidogrel) [Prescribing Information]. Bristol-myers squibb/sanofi; 2020.
94. Andersson T, Hassan-Alin M, Hasselgren G, Röhss K, Weidolf L. Pharmacokinetic studies with esomeprazole, the (S)-isomer of omeprazole. Clin Pharmacokinet. 2001;40(6):411–426. doi:10.2165/00003088-200140060-00003
95. Marom H, Agranat I. Racemization of the gastrointestinal antisecretory chiral drug esomeprazole magnesium via the pyramidal inversion mechanism: a theoretical study. Chirality. 2010;22(9):798–807. doi:10.1002/chir.20839
96. Jana K, Bandyopadhyay T, Ganguly B. Stereoselective metabolism of omeprazole by cytochrome P450 2C19 and 3A4: mechanistic insights from DFT study. J Phys Chem B. 2018;122(22):5765–5775. doi:10.1021/acs.jpcb.8b01179
97. Shin JS, Lee JY, Cho KH, et al. The pharmacokinetics, pharmacodynamics and safety of oral doses of ilaprazole 10, 20 and 40 mg and esomeprazole 40 mg in healthy subjects: a randomised, open-label crossover study. Aliment Pharmacol Ther. 2014;40(5):548–561.
98. Galmiche JP, Bruley Des Varannes S, Ducrotté P, et al. Tenatoprazole, a novel proton pump inhibitor with a prolonged plasma half-life: effects on intragastric pH and comparison with esomeprazole in healthy volunteers. Aliment Pharmacol Ther. 2004;19(6):655–662. doi:10.1111/j.1365-2036.2004.01893.x
99. Hunt RH, Armstrong D, James C, et al. Effect on intragastric pH of a PPI with a prolonged plasma half-life: comparison between tenatoprazole and esomeprazole on the duration of acid suppression in healthy male volunteers. Am J Gastroenterol. 2005;100(9):1949–1956. doi:10.1111/j.1572-0241.2005.41956.x
100. Le TK, Park YJ, Cha GS, Oktavia FARH, Kim DH, Yun CH. Roles of human liver cytochrome P450 enzymes in tenatoprazole metabolism. Pharmaceutics. 2022;15(1):23.
101. Kinoshita Y, Kusano M, Iwakiri K, Fujishiro M, Tachikawa N, Haruma K. Efficacy and safety profile of Z-215 (azeloprazole sodium), a proton pump inhibitor, compared with rabeprazole sodium in patients with reflux esophagitis: a Phase II, multicenter, randomized, double-blind, comparative study. Curr Ther Res Clin Exp. 2018;88:26–34. doi:10.1016/j.curtheres.2018.03.004
102. Yang E, Kim S, Kim B, et al. Night-time gastric acid suppression by tegoprazan compared to vonoprazan or esomeprazole. Br J Clin Pharmacol. 2022;88(7):3288–3296. doi:10.1111/bcp.15268
103. Cho YK, Kim JH, Kim HS, et al. Randomised clinical trial: comparison of tegoprazan and lansoprazole as maintenance therapy for healed mild erosive oesophagitis. Aliment Pharmacol Ther. 2023;57(1):72–80. doi:10.1111/apt.17255
104. Jenkins H, Sakurai Y, Nishimura A, et al. Randomised clinical trial: safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK-438 (vonoprazan), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment Pharmacol Ther. 2015;41(7):636–648. doi:10.1111/apt.13121
105. Lightdale JR, Gremse DA, Heitlinger LA. Section on gastroenterology Hp, and nutrition. Gastroesophageal reflux: management guidance for the pediatrician. Pediatrics. 2013;131(5):e1684–e1695. doi:10.1542/peds.2013-0421
106. Ramsey LB, Prows CA, Chidambaran V, et al. Implementation of CYP2D6-guided opioid therapy at Cincinnati Children’s Hospital Medical Center. Am J Health Syst Pharm. 2023. doi:10.1093/ajhp/zxad025
107. Gammal RS, Fieg E. Pharmacist and genetic counselor collaboration in pharmacogenomics. Am J Health Syst Pharm. 2022;79(18):1516–1520. doi:10.1093/ajhp/zxac168
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Proton Pump Inhibitors Prescribing Among Hospitalized Patients: A Cross-Sectional Study
Abukhalil AD, Ali O, Saad A, Falana H, Al-Shami N, Naseef HA, Rabba A
International Journal of General Medicine 2023, 16:141-150
Published Date: 11 January 2023
Awareness and Predictors of the Use of Bioinformatics in Genome Research in Saudi Arabia
Alomair L, Abolfotouh MA
International Journal of General Medicine 2023, 16:3413-3425
Published Date: 11 August 2023