Back to Journals » International Journal of Nanomedicine » Volume 15
A pH-Sensitive Prodrug Nanocarrier Based on Diosgenin for Doxorubicin Delivery to Efficiently Inhibit Tumor Metastasis
Authors Wei Z, Wang H, Xin G, Zeng Z, Li S, Ming Y, Zhang X, Xing Z ![]() , Li L, Li Y, Zhang B, Zhang J, Niu H, Huang W
, Li L, Li Y, Zhang B, Zhang J, Niu H, Huang W
Received 20 February 2020
Accepted for publication 17 July 2020
Published 4 September 2020 Volume 2020:15 Pages 6545—6560
DOI https://doi.org/10.2147/IJN.S250549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Zeliang Wei,1,2 Haibo Wang,3 Guang Xin,1 Zhi Zeng,1 Shiyi Li,1 Yue Ming,1 Xiaoyu Zhang,1 Zhihua Xing,1 Li Li,2 Youping Li,1 Boli Zhang,1,4 Junhua Zhang,4 Hai Niu,1,5 Wen Huang1
1Laboratory of Ethnopharmacology, West China Medical School, West China Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Department of Dermatology, West China Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 3Textile Institute, College of Light Industry, Textile and Food Engineering, Sichuan University, Chengdu, People’s Republic of China; 4Tianjin University of Traditional Chinese Medicine, Tianjin, People’s Republic of China; 5College of Mathematics, Sichuan University, Chengdu, Sichuan, People’s Republic of China
Correspondence: Wen Huang; Hai Niu Email [email protected]; [email protected]
Background: The metastasis, one of the biggest barriers in cancer therapy, is the leading cause of tumor deterioration and recurrence. The anti.-metastasis has been considered as a feasible strategy for clinical cancer management. It is well known that diosgenin could inhibit tumor metastasis and doxorubicin (DOX) could induce tumor apoptosis. However, their efficient delivery remains challenging.
Purpose: To address these issues, a novel pH-sensitive polymer-prodrug based on diosgenin nanoparticles (NPs) platform was developed to enhance the efficiency of DOX delivery (DOX/NPs) for synergistic therapy of cutaneous melanoma, the most lethal form of skin cancer with high malignancy, early metastasis and high mortality.
Methods and Results: The inhibitory effect of DOX/NPs on tumor proliferation and migration was superior to that of NPs or free DOX. What is more, DOX/NPs could combine mitochondria-associated metastasis and apoptosis with unique internalization pathway of carrier to fight tumors. In addition, biodistribution experiments proved that DOX/NPs could efficiently accumulate in tumor sites through enhancing permeation and retention (EPR) effect compared with free DOX. Importantly, the data from in vivo experiment revealed that DOX/NPs without heart toxicity significantly inhibited tumor metastasis by exerting synergistic therapeutic effect, and reduced tumor volume and weight by inducing apoptosis.
Conclusion: The nanocarrier DOX/NPs with satisfying pharmaceutical characteristics based on the establishment of two different functional agents is a promising strategy for synergistically enhancing effects of cancer therapy.
Keywords: anti-metastasis, antitumor, self-assembly, codelivery, doxorubicin
Introduction
Cutaneous melanoma is one of the most malignant and fastest growing forms of human cancers to affect younger populations.1,2 Despite advances in surgery and chemotherapy, nearly 80% of patients still die from metastatic melanoma.3–5 Moreover, metastasis is highly resistant to traditional chemotherapies.6,7 Accumulated clinical research also manifest that once the tumor metastasizes, single drug chemotherapy is much less effective.8 Therefore, it is extremely urgent to develop a novel treatment strategy for inhibiting the tumor metastasis.
Diosgenin, extracted from Dioscorea zingiberensis, is a typical natural product.9 It displays various pharmacological activities such as immunity-regulating, anti-inflammation, antithrombus as well as antitumor.10–12 More important, extensive studies reveal that diosgenin and its derivative could inhibit tumor cell metastasis in lung cancer, breast cancer, and melanoma. Meanwhile, numerous research studies also indicate that diosgenin might be a promising agent to prevent the DOX-induced cardiotoxicity in vivo.13 Unfortunately, the conventional chemotherapy using small molecular size doxorubicin (DOX) as well as diosgenin still exhibits several limitations, including poor intracellular penetration, rapid in vivo clearance, low efficiency of accumulation at tumor site, and adverse effects for normal tissues.14–17 The recent studies report that diosgenin could potentiate the apoptotic effect of DOX and paclitaxel in hepatocellular carcinoma cells.18 And the polymeric nanoparticles based on diosgenin and 10-hydroxycamptothecin could efficiently inhibit B16F10 tumor cell growth.19 Furthermore, potential nanoparticle drug delivery systems including short peptides and macromolecule polymers, have been widely used for codelivery of poorly soluble drugs to inhibit tumor growth, because they allow for well flexibility, high drug loading, good biocompatibility and controlled release.20–22 However, to our knowledge, the developing high potent codelivery system of diosgenin with chemotherapeutic agents including the trackable DOX for fighting metastasis, one of the most knotty questions in cancer therapy, is few involved. Therefore, an effective and safe codelivery system of the combined administration of DOX and diosgenin to synergistically enhance antimetastasis as well as antiproliferation activities is highly desirable.
It is generally known that prodrug nanocarriers with functional characteristics including stimuli-responsiveness and long-circulating, play an important role for releasing the drug effectively in the pathological position.23,24 Prodrug nanocarriers, which are composed of chemotherapeutic drugs with polymers, could efficiently and selectively release drugs in response to dysfunctional environment conditions for chemotherapeutics of tumor metastasis.25,26 In recent decades, more and more researchers focus on drug delivery targeted by using a multifunctional prodrug nanocarrier. Due to the pH-sensitive characteristic, nanocarriers could stimuli-responsively control drug release in the tumor microenvironment via the intracellular lower pH signal.27 The size of nanocarrier is also another key factor for drug delivery efficiency.28 It is well known that poly(ethylene glycol) (PEG), which is widely used to modify the small molecule or macromolecular drug, could dissolve very insoluble small molecule compounds, prolong circulation time, enhance permeability and retention (EPR) effects, and alter parent drug biodistribution.29–31 However, the platform with satisfying pharmacological characteristics for synergistic antimetastasis and anti-apoptosis is highly required in tumor therapy.
In this work, we established a novel pH-sensitive prodrug nanocarrier as a drug delivery system for codelivery of DOX and diosgenin in order to efficiently suppress tumor metastasis and proliferation (Scheme 1). The drug carrier was a diosgenin derivative prodrug nanoparticles (NPs) with Schiff-based bond, which could self-assemble in aqueous media, and load DOX without any surface coating. The characterization of DOX/NPs was measured by transmission electron microscope (TEM) and dynamic light scattering (DLS). Effects of DOX/NPs on cellular viability, uptake, apoptosis, and mitochondrial function were also assessed in vitro. Meanwhile, the tumor targeting ability and DOX/NPs biodistribution were detected on B16F10 tumor xenograft models using near infrared (NIR) imaging. Afterwards, the efficacy of DOX/NPs on inhibiting tumor metastasis and proliferation were estimated in vivo by H&E staining and immunohistochemistry. Finally, the pathological analysis of organs was used to evaluate the safety of DOX/NPs. The obtained data demonstrate that DOX/NPs with significant antimetastasis and antiproliferation is potential for synergistic therapy of cancer. This is the first study to report a prodrug nanocarrier combining diosgenin with DOX for inhibiting tumor metastasis, which is a great challenge for cancer treatment, especially preventing tumor deterioration and recurrence.
 |
Scheme 1. After intravenous injection, self-assembly DOX/NPs were taken up by cells via endocytosis, and DOX/NPs released DOX and diosgenin to synergistically enhance anti-metastasis and anti-proliferation by triggering mitochondrial dysfunction. |
Materials and Methods
Materials
Doxorubicin hydrochloride (DOX·HCl) was purchased from Meilun Biological Technology Co., Ltd (Liaoning, China). Triethylamine (TEA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Hoechst 33258, dimethyl sulfoxide (DMSO) and BCA protein assay kit were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Annexin V-FITC apoptosis detection kit was purchased from Sino Biological, Inc. (Beijing, China). The detection kits of ATP, ROS, Mito-sox and JC-1, were purchased from Beyotime Institute of Biotechnology (Shanghai, China). Unless stated otherwise, the other reagents were purchased from Sigma-Aldrich.
Culture medium (RPIM 1640 medium), FBS were purchased from Gibco Life Technologies (Thermo Fisher Scientific, Waltham, MA, USA). Mouse anti-GAPDH antibody, rabbit anti-Bcl-2 antibody and rabbit anti-Bax antibody were purchased from Abcam (Burlingame, CA, USA). Cleaved caspase-3 rabbit mAb was purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Goat anti-mouse IgG and goat anti-rabbit IgG were purchased from Beijing Biosynthesis Biotechnology Co., Ltd (Beijing, China).
Cell Culture
Murine melanoma B16F10 cell line was received from the Lab of Transplant Engineering and Immunology, West China Hospital, and was approved for use in experimental research by the Ethics Committee of West China Center of Medical Sciences, Sichuan University. The cells were cultured in RPMI-1640 medium with 10% of heat-inactivated FBS, streptomycin (100 mg/mL) and penicillin (100 U/mL). Cells were sustained at 37°C under a humidified atmosphere with 5% CO2.
Animals
Male BALB/c mice (four weeks old) with body weights ranging from 18–22 g were housed in a temperature, humidity, and illumination regulated environment (22±2°C, 50±10% and 12 h light–dark cycle). They were fed with standard animal water and food. The experimental protocol for all methods and experiments was approved by the Ethics Committee of West China Center, Sichuan University. The animal welfare guidelines of the Institutional Animal Care and Welfare Committee of West China Hospital, Sichuan University were strictly followed.
Synthesis of NPs
The prodrug nanocarrier was synthesized as previously described.32 Briefly, methoxy-PEG-4-formylbenzoic acid (MPEG-CBA) and diosgenin amino caproic acid ester were synthesized by our laboratory, and the products were purified by recrystallization for next reaction. Then MPEG-CBA (6.5 g) and diosgenin amino caproic acid ester (1.5 g) were added to 150 mL tetrahydrofuran (THF) solution with stirring at 40°C for 12 h. Afterwards, the mixture was evaporated with a vacuum pump, and products were dissolved in methanol and purified in anhydrous ethyl ether. Finally, the impurities were removed by filtration, and the product was recovered by precipitation in isopropanol, dried in the vacuum drying machine at 40°C for subsequent experiments.
Preparation of DOX/NPs
The DOX/NPs were prepared in a simple self-assembly approach between these two molecules as previously reported.33 In brief, DOX·HCl (4 mg) and NPs (10 mg) were dissolved in 1 mL DMSO. And 100 μL TEA was added into mixed solution. Furthermore, 1 mL of this solution was slowly injected into 4 mL double-distilled water (ddH2O) with stirring. The mixture was stirred at room temperature for two hours and dialyzed in ddH2O overnight using a dialysis bag. NPs self-assemble into nanoparticles in the same way.
Characterization of DOX/NPs
The morphology and particle size of DOX/NPs were examined by TEM (H-600-4, Hitachi, Tolyo, Japan).34 Briefly, samples were placed on a copper grid, stained with phosphotungstic acid solution (1%, w/v), and operated with an acceleration voltage of 75 kV at room temperature. The change of size distribution was measured to evaluate the pH-responsive property and stability of DOX/NPs in different pH values or temperatures by a DLS instrument (LB-500, Horibk, Japan).
The amount of DOX encapsulated into nanoparticles was calculated according to a colorimetric method. The lyophilized DOX/NPs were dissolved in DMSO, the fluorescence intensity was measured at 495/593 nm by a F-7000 fluorescence spectrometer (Hitachi). Various concentrations DOX solutions were measured to generate a calibration curve for calculating drug concentration. The drug loading content (DLC) and loading efficiency (DLE) was calculated using the following formula:


Drug Release Test
The drug release profiles were detected by a dialysis method.35 DOX released from DOX/NPs due to hydrolytic degradation in PBS with different pH (7.4 or 5.0). Two milliliters of DOX/NPs solution was sealed in a dialysis bag and soaked in 20 mL PBS at 37°C in the dark. Different samples were withdrawn at predetermined time points, and DOX concentration was analyzed by the fluorescence spectrometer. The concentration of diosgenin releasing from DOX/NPs was calculated in the same way by the HPLC (Shimadzu LC20A, Japan).
In vitro Cytotoxicity Study
The cytotoxicities of drugs were measured by the MTT assay on B16F10 cells. Cells were seeded into 96-well culture plates and incubated with NPs, DOX and DOX/NPs with different concentrations for 24 and 48 h. DOX and DOX/NPs with gradient concentrations of DOX ranged from 0.01 to 5.0 μg/mL, and the gradient concentrations of NPs ranged from 1 to 100 μg/mL. After that, 100 μL MTT (5 mg/mL in medium) was injected to each well and incubated for four hours. Afterwards, 100 μL DMSO was added and incubated for 10 min at room temperature. The optical density (OD) of each well was measured by a SpectraMax microplate reader (Molecular Devices, San Jose, CA, USA) at 492 nm. Cytotoxicity was calculated as percentage relative to the untreated control group. We evaluated the synergistic effects between DOX and diosgenin in the DOX/NPs by applying the combination index (CI) using the following formula:

whereby DOXcombined and diosgenincombinedis the IC50 value of the drugs used in the combination treatment, and DOXsingle or diosgeninsingle is the single drug IC50 value. When the value of CI lower than one denotes a synergistic effect, a value is equal to one an additive effect is indicated, and a value larger than one indicates an antagonistic effect.
Wound Healing Assay
To observe the cell migration, B16F10 cells were seeded on flame-dried coverslips at a density of 1×105 cells per well in a six-well culture plate. The cells were scratched with a sterile plastic pipette tip (10 μL) and rinsed three times with PBS. Scratched B16F10 cells were incubated with NPs (13.3 μg/mL), DOX or DOX/NPs with concentration of DOX at 5 μg/mL (the same below) for 24 h. The cell migration to the wound area was recorded, and the wound area of each group was analyzed using ImageJ(NIH, Bethesda, MD, USA). At last, the rate of migration was calculated as a percentage by quantifying the change of wound area after treatment.
In vitro Cellular Uptake
To observe the cellular uptake, B16F10 cells were seeded on flame-dried coverslips at a density of 1×105 cells per well in a six-well culture plate. After incubation overnight, the cells were cultured with fresh media containing DOX/NPs or DOX·HCl with concentration of DOX at 5 μg/mL for two, four, and six hours. The cells were rinsed with PBS three times and stained with Hoechst 33258 for 10 min at 37°C. After staining, the cellular uptake behavior was scanned by a fluorescence microscope (Zeiss, Oberkochen, Germany).
Measurement of Intracellular Mitochondrial Function
To assess the effect of drugs on mitochondrial function in cells, ATP level, mitochondrial membrane potential and ROS level were examined as previously described. In brief, B16F10 cells were treated with NPs (13.3 μg/mL), DOX (5 μg/mL) or DOX/NPs (5 μg/mL) for 48 h. After washing twice with RPMI-1640 medium, cells were incubated with 5 μM DCFH-DA, mito-sox or ATP detection reagent for 30 min at 37°C. Then, the fluorescence intensity in different wavelengths were detected by a multimode microplate reader (BioTek, Synergy IMX, USA) referring to instructions. Finally, the results were calculated according to ratios of fluorescence intensity to protein concentration. The JC-1 probe was employed to measure the mitochondrial membrane potential of cells after treatment with drugs for 48 h. The cells were cultured with JC-1 probe (2 mg/L) at 37°C for 20 min. Then the fluorescence images were photographed by a fluorescence microscope (Zeiss, Oberkochen, Germany).
Western Blot Analysis
B16F10 cells were seeded into a six-well culture plate at a density of 1×106 cells per well and treated with NPs (13.3 μg/mL), DOX·HCl (5 μg/mL) or DOX/NPs (5 μg/mL) for 48 h. The cells were harvested by scraping them out with lysis buffer, and supernatants of lysate samples were collected by centrifuging at 12,000 g for 10 min at 4°C. The protein contents were assayed by a BCA protein assay kit. Afterwards, the equal amount (20 μg) of protein was separated by SDS-PAGE gel and then electrotransferred to polyvinylidene difluoride (PVDF) membranes. Furthermore, membranes were blocked in nonfat powdered milk (5% skimmed milk in TBST, w/v) at room temperature for one hour. Then the membranes were incubated with primary antibodies overnight at 4°C. After incubating with secondary antibodies, the protein bands were detected by Immobilon-Western Chemiluminescence HRP substrate (EMD Millipore, Billerica, MA, USA).
In vivo Imaging
The biodistribution of DOX/NPs was detected by in vivo NIR imaging. A NIR fluorophore, indocyanine green (ICG), was encapsulated into the nanoparticles. The process for encapsulating ICG in the nanoparticles was the same as that for encapsulation of DOX. The tumor xenograft models were established by subcutaneous injection of B16F10 cells into the male BALB/c mice (six to eight weeks old), and the mice were treated with the ICG-loaded DOX/NPs or free DOX via tail-vein injection. Whole body fluorescence images were taken at 3, 6, 12, and 24 h after injection using a Spectral AMI X (Spectral Instruments Imaging, USA) IVIS instrument with a 780 nm excitation wavelength. Images of various organs, including heart, liver, spleen, lung, kidney and tumor were also collected after sacrifice of the mice at 12 h postinjection.
In vivo Antitumor Assay
To investigate antitumor growth efficiency of drugs in vivo, a subcutaneous tumor model was employed. Male BALB/c mice (six to eight weeks old) were subcutaneously injected with 2×106 B16F10 cells in 100 μL of medium. The mice were randomly divided into different treatment groups and raised for a week. When tumor volume reached an acceptable size, mice were given NPs (13.3 mg/kg), DOX·HCl (5 mg/kg) or DOX/NPs (5 mg/kg) via tail-vein injection every two days, and saline was administered intravenously as control group. During that period, the body weight, tumor volume and survival rate of mice were measured. After treatment for 21 days, the mice were sacrificed.
In vivo Lung Metastasis Assay
To investigate antimetastasis efficiency of drugs in vivo, a lung metastasis model was established. Male BALB/c mice (six to eight weeks old) were injected intravenously with 1×106 B16F10 cells via the tail vein, and then randomly divided into several groups. The mice were given NPs (13.3 mg/kg), DOX·HCl (5 mg/kg) or DOX/NPs (5 mg/kg) via tail-vein injection every three days, and saline was administered intravenously as normal control group and model group. After treatment for 21 days, the mice were sacrificed. Black dots on lung surface and weight of lung were measured.
Histology and Immunohistochemistry
The organs and tumor tissue of the mice in the melanoma tumor xenograft model and lung metastasis model were collected after the mice were sacrificed. The dissected tissue samples were fixed in 10% formaldehyde solution, wrapped with paraffin wax and cut into slices at 5 μm. Tissue sections were stained with H&E.
In addition, fixed and embedded tumor and lung tissue samples were sectioned at 5 μm and mounted on poly-lysine-coated slides for immunohistochemistry. Tris–EDTA buffer (pH 9.0) was used for antigen retrieval. Then the sections were soaked with 3% hydrogen peroxide for 20 min to inhibit endogenous peroxides, and incubated with primary antibodies for caspase-3 overnight at 4°C. After that, the secondary antibody was employed at 37°C for 30 min and diaminobenzidine was used as the chromogenic substrate. Finally, sections were counterstained with H&E. Negative controls were processed in the same way, but primary antibody was replaced by PBS.
Statistical Analysis
All statistical analyses were performed using the statistical software package SPSS 19.0 (IBM Corporation, Armonk, NY, USA). Data were expressed as the means ±SD. The statistically significant difference was determined using a two-sample t-test or a one-way ANOVA. The statistical significance of difference was considered for P<0.05.
Results and Discussion
Characterization of DOX/NPs
The NPs formulation was prepared by our laboratory as described in our previous publication, which could self-assemble to form stable nanoparticles in aqueous solution, because of its amphiphilic structure with good uniformity (Figure S1A and B).32 And the hydrophobic DOX was successfully loaded into the NPs through interaction between DOX and hydrophobic part. These DOX/NPs were verified to have an average diameter of 165.4±5.4 nm by DLS, as shown in Figure 1A. The morphology of the DOX/NPs was further observed by TEM. The TEM image with homogenous spherical shapes indicated that the amphiphilic DOX/NPs indeed formed nanoparticles (Figure 1B), which were able to passively target to tumor tissues via the EPR effect. Furthermore, the stability of DOX/NPs was evaluated by detecting the change of particle size in a simulative physiological condition with different temperatures. It was found that the DOX/NPs were quite stable in aqueous solution, and the particle size was an insignificant change in different temperatures over 72 h (Figures 1C and S2). These results illustrate that DOX and NPs were successfully self-assembled together and stable under physiological conditions.
 |
Figure 1 Characterization of DOX/NPs. (A) The size distribution of NPs and DOX/NPs. (B) TEM images of DOX/NPs. (C) The size changes of DOX/NPs in different temperatures and times. (D) The cumulative DOX release profiles of DOX/NPs in physiological and acidic conditions at 37°C. The scale bar of the TEM is 200 nm. The data represent mean ±SD. n=3–4. |
Controllable Drug Release of DOX/NPs
Since DOX was encapsulated as the hydrophobic anticancer drug, we studied the release of DOX from DOX/NPs in the buffer solutions simulating in vivo situations at pH 7.4 and 5.0. According to the previously provided calculation methods in the Materials and Methods section, the drug loading content (DLC) and loading efficiency (DLE) of DOX/NPs were 13.0 and 37.5%, respectively. The DOX release was controlled by the cleavage of NPs Schiff-base bond.36,37 As shown in Figure 1D, the release rates of DOX at 24 and 48 h is 31.4 and 36.2% at pH 7.4, respectively. In contrast, the acidic pH significantly accelerated the release of DOX. The release rates of DOX significantly increased to 67.8 and 79.8% at 24 and 48 h at pH 5.0. The release profile of diosgenin derivative was similar to that of DOX (Figure S3). Compared with reported programs, NPs/DOX could more feasibly release drugs to exert therapeutic effects under weak acidic conditions because of the pH-responsive characteristic.19,38 Also, the experimental phenomenon was consistent with previous reports that the pH-responsive drug delivery systems play an important role in the drug release behavior, while the enhanced solubility of DOX at pH 5.0 has a weak effect.39–41 These results demonstrate that acidic condition in tumor or late endosome could facilitate the release of DOX from the DOX/NPs, thus reduce the damage to normal cells and organs.
Cytotoxicity and Antimigration of DOX/NPs
We next investigated antitumor activity of DOX/NPs compared with that of free DOX and NPs in B16F10 cells by the MTT assay in vitro. Figure 2A and B showed the viability of cells treated with free DOX and DOX/NPs at different equivalent concentrations ranging from 0.01 to 5 μg/mL of DOX for 24 and 48 h. Dose-dependent cytotoxicity was observed for both free DOX and DOX/NPs. Meanwhile, DOX/NPs exhibited a weaker cell inhibition than free DOX, especially at low concentrations for 24 h. However, DOX/NPs showed better cell inhibition than free DOX for 48 h, because of the controlled release from NPs and different cellular uptake pathway of DOX. The IC50 of free DOX for 24 and 48 h incubation were 0.32 and 0.18 μg/mL, while that of DOX/NPs for 24 and 48 h were 0.54 and 0.14 μg/mL. The IC50 of NPs were 30.98 and 20.36 μg/mL for 24 and 48 h, respectively (Figure 2C). Moreover, imagines of colony formation assay proved that the size of the colony formation after cells incubated with NPs, DOX or DOX/NPs was significantly smaller for 24 and 48 h. At the same time, the number of B16F10 cells treated with DOX/NPs significantly decreased compared with free DOX for 48 h (Figures 2D and S4). Meanwhile, effect of DOX/NPs on antimigration was assayed by the wound healing assay. As shown in Figure 2E and F, after treatment with DOX/NPs for 24 h, cell migration was obviously inhibited compared with the control group treated without drugs. Furthermore, the group treated with DOX/NPs exhibited better effect on antimigration than that from the group treated with DOX or NPs. The results indicate that the DOX/NPs have excellent potential for antimigration and antiproliferation on B16F10 cells, and improves antitumor efficiency in time course.
 |
Figure 2 Effect of free DOX, NPs and DOX/NPs on cell viability and migration. (A) for 24 and (B) 48 h. (C) Cytotoxicity of NPs on B16F10 cells for 24 and 48 h. (D) Colony formation after drugs treatment via crystal violet staining. (E) Quantification of wound area in free DOX, NPs and DOX/NPs treated B16F10 cells. (F) Wound healing assay in B16F10 cells treated with free DOX, NPs or DOX/NPs for 24 h (magnification ×100). The data represent mean ±SD. n=3–4. *p<0.05 vs control, **p<0.05 vs DOX or NPs. |
In addition, combination index (CI) values were calculated to explore the relationship between diosgenin and DOX. The CI values of DOX and diosgenin in NPs/DOX was 0.78 for B16F10 cells for 48 h. A value of CI less than, equal to, or greater than one is considered as indicating synergistic, additive, or antagonistic effects, respectively. The results demonstrate that a synergistic and efficient effect of NPs/DOX on inhibiting tumor growth could be accomplished by the combination of two anticancer drugs, DOX and diosgenin.
Effect of DOX/NPs on Mitochondrial Function
Mitochondria are the major site of ATP and ROS production, as well as being responsible for vital cellular functions and tumor metastasis, which is an energy-dependent process.42,43 The ROS overload is the key cause to induce the dysfunction and structural deterioration of the mitochondria.44 To further evaluate the effect of NPs and DOX/NPs on mitochondrial function, B16F10 cells were incubated with NPs or DOX/NPs for 48 h. The results showed a decrease in intracellular ATP level of B16F10 cells treated with NPs or DOX/NPs (Figure 3A). When the mitochondrial dysfunction occurs, membrane potential may decrease significantly. We detected the change of MMP by using JC-1. And colors change from red to green as MMP decreased from confocal microscopic images. Therefore, the ratio of red to green fluorescence illustrated the polarization of the MMP after treating cells with NPs or DOX/NPs (Figure 3B). In addition, an increase in intracellular ROS level was also confirmed after cells were treated with NPs or DOX/NPs (Figure 3C). Mitochondria are considered as the major site for production of ROS, we found that NPs or DOX/NPs promoted mitochondrial ROS accumulation in B16F10 cells by flow cytometry (Figure 3D and E). The data from the mitochondrial function suggest that NPs or DOX/NPs could inhibit tumor metastasis through impairing mitochondrial function.
 |
Figure 3 Effect of NPs and DOX/NPs on mitochondrial function. (A) The relative ATP levels indicated by protein content after NPs or DOX/NPs treatment for 48 h. (B) The representative images of mitochondrial membrane potential determined by confocal microscopy. The cells with green-positive and red-negative fluorescence were counted as depolarized cells. (C) The relative fluorescent intensity of intracellular ROS level. (D) The relative fluorescent intensity and (E) flow cytometry of MitoROS levels. The data represent mean ±SD. n=3–4. *p<0.05 vs control, **p<0.01 vs NPs. |
In addition, since Bcl-2 and the caspase family were involved in mitochondria-associated apoptosis,45,46 we researched their change after B16F10 cells were treated with DOX or DOX/NPs. Western blot protein expression of Bax (proapoptotic protein), Bcl-2 (antiapoptotic protein) and cleaved caspase-3 in the cells were performed and quantified. As shown in Figure S5A–C, expression of Bax and cleaved caspase-3 were in the order PBS <DOX<DOX/NPs, but expression of Bcl-2, an antitumor factor, was in the opposite order. Diosgenin induces tumor cell apoptosis by activating caspase-3 via a mitochondrial-related death pathway.47,48 Moreover, efficiency of DOX/NPs on inducing apoptosis was superior to free DOX or NPs (Figure S6), which took full advantage of the synergism of two chemotherapy agents. These results demonstrate that DOX/NPs inhibit Bcl-2 expression, activates Bax and caspase-3 resulting in mitochondria-mediated apoptosis.
Cellular Uptake and Cell Imaging of DOX/NPs
The cellular uptake and intracellular drug release behaviour of DOX/NPs were investigated by fluorescence microscope in B16F10 melanoma cells. Since DOX itself has intrinsic red fluorescence, it was used directly to study cellular uptake and location of the prodrug nanocarrier in B16F10 cells monitored by fluorescence microscopy and flow cytometry. As shown in Figure 4A and B, the red fluorescence in the cells gradually intensified accompanyied with treatment time extending from two to six hours. When incubating B16F10 cells with free DOX for two hours, the intense red fluorescent signal was internalized fast, and observed mainly accumulated in the cellular nuclei. In comparison, the red fluorescence signal was observed in the cytoplasm after cells were treated with DOX/NPs for two hours, and weaker than that of group treated with free DOX for two hours. As the time increases from two to six hours, red fluorescence of DOX gradually moved into the nuclei and covered the blue fluorescence of nuclei stain. But the fluorescent signal was still clearly seen in the cytoplasm in most cells for up to six hours. These findings provided the impressive evidence that free DOX internalization in cell nuclei was a relatively fast process, while DOX/NPs might take more time for cellular uptake, and the subsequent transference from cytoplasm to nucleus of DOX.
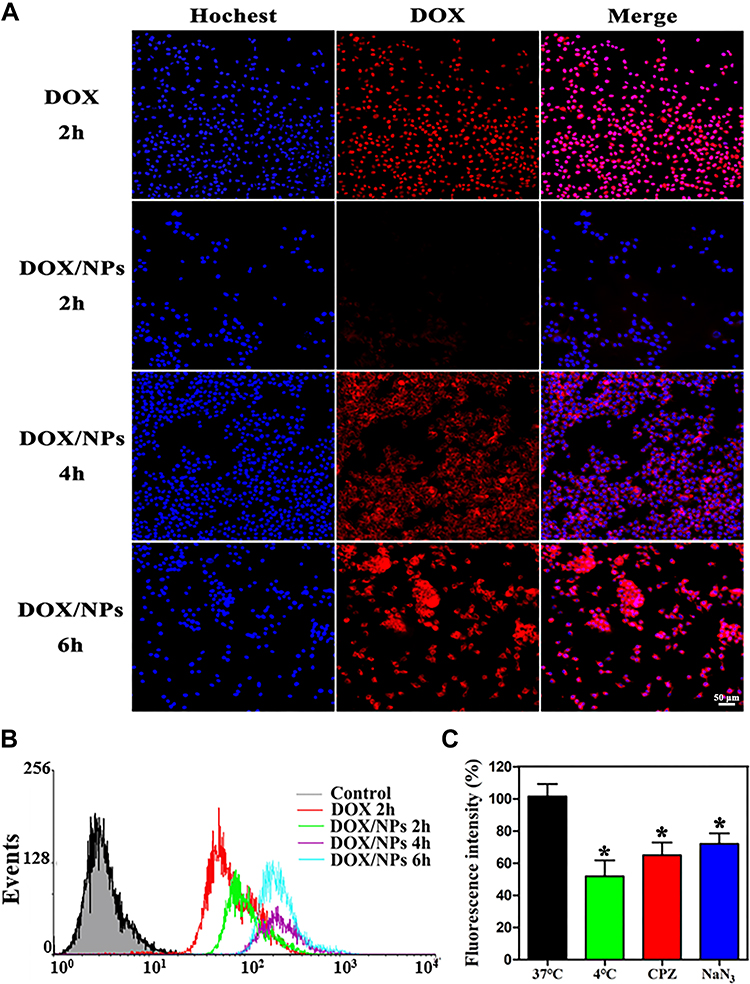 |
Figure 4 Cellular uptake and cell imaging of DOX/NPs. (A) Fluorescence microscopy images of intracellular uptake of DOX (red) in B16F10 cells after treatment with DOX·HCl for two hours, DOX/NPs for two, four, and six hours. Nuclei are labeled by Hoechst staining (blue). (B) Intracellular uptake of DOX detected by flow cytometry for two, four, and six hours. (C) Uptake of DOX/NPs was monitored by keeping the cells at 37°C pretreating them with 30 μg/mL CPZ and 120 mM NaN3 or 4°C by confocal microscopy. The data represent mean ±SD. n=3–4. *p<0.05 vs group at 37°C. |
It was reported that nanoparticles entered into cells through endocytosis pathway, resulting in the difference of fluorescence distribution.49 Endocytosis could be blocked by endocytosis inhibitor chlorpromazine (CPZ), the energy-depleting agent NaN3 as well as low temperature, because endocytosis is an energy dependent process. As shown in Figure 4C, compared to B16F10 cells, the cells treated with DOX/NPs at 4°C, and CPZ+DOX/NPs or NaN3+DOX/NPs at 37°C exhibited only 51.9, 65.3, and 72.2% of red fluorescent signal internalization, respectively. The reduction of red fluorescent signals confirm the unique internalization manner of DOX/NPs via endocytosis, contributing to reverse the resistance to DOX of cells.50,51 These results indicate that DOX/NPs entered into B16F10 cells via endocytosis pathway.
In vivo Imaging and Biodistribution of DOX/NPs
The biodistribution of DOX/NPs or free DOX after intravenous injection into the B16F10 cells tumor-bearing BALB/C mice were detected using the in vivo imaging technique. DOX/NPs were labelled with ICG. As shown in Figure S7, DOX/NPs revealed good tumor accumulation because of the EPR effect. The strong fluorescence signal of ICG was observed in the tumor site at three hours after intravenous injection, and the stronger fluorescence signal was monitored at 12 h and attenuated gradually with prolonged time. The fluorescence signals in the tumor continued to 24 h and the fluorescence intensity was still strong at the tumor site. These data indicated that intravenously injected DOX/NPs had a passive tumor targeting ability as a result of the EPR effect, and the high contrast of animal imaging exhibited excellent potential of DOX/NPs for cancer diagnosis.31,52 To further quantitatively compare the biodistribution of free DOX with DOX/NPs, the mice were sacrificed after 12 h postinjection, and the major organs including heart, liver, spleen, lung and kidney, as well as tumor were excised. The ex vivo fluorescent images further indicated the high accumulation of DOX/NPs was superior to free DOX in the tumor site. In addition, compared to free DOX, the DOX/NPs significantly reduced the accumulation of drug in the liver and kidneys (Figure 5A and B). Altogether, the high accumulation of DOX/NPs in the tumor could contribute to the EPR effect and successful surface modification with PEG, which is feasible for escaping the reticuloendothelial system (RES) organs.53
 |
Figure 5 Biodistribution of free DOX and DOX/NPs measured by the fluorescence of DOX in major organs and tumor tissues. (A) Biodistribution of free DOX in major organs and tumor tissues at 12 h after intravenous injection. (B) Biodistribution of DOX/NPs in major organs and tumor tissues at 12 h after intravenous injection. |
In Vivo Anticancer Activity of DOX/NPs
To evaluate the synergistic antitumor effect of prodrug nanocarrier, the anticancer effect of NPs, DOX and DOX/NPs was further investigated in B16F10 tumor-bearing BALB/c mice. As shown in Figure 6A and B, the average tumor weight and volume were measured during the experiment for evaluating antitumor efficacy. The tumor weight and volume grew rapidly in the saline treated group over 21 days (Figure S8). In contrast, NPs and free DOX groups showed moderate antitumor efficiency, while DOX/NPs displayed the significantly better tumor inhibition efficacy than NPs and free DOX, which corresponded with the results of in vitro cytotoxicity and cell apoptosis experiments, demonstrating a synergetic therapeutic effect of DOX/NP formulation. Moreover, the body weight and survival rate of each group was measured to evaluate the side effects (Figure 6C). Only mice treated with DOX lost about 20% weight and had a 30% survival rate at the end of the experiment (Figure 6D), suggesting the NPs and DOX/NPs have insignificant systemic toxicity.
 |
Figure 6 In vivo antitumor efficacy of free DOX, NPs and DOX/NPs. (A and B) Tumor growth curves. (C) Body weights of mice. (D) Survival rate of mice. (E) H&E staining of tumor sections and heart sections harvested from each treatment group. (F) Immunohistochemical staining of caspase-3 in tumor sections from each treatment group. The data represent mean ±SD. n=6–8. *p<0.05 vs saline, **p<0.01 vs NPs and DOX. |
We processed representative tumors and organs harvested from various groups on day 21 for histopathological analysis. H&E staining showed that the group treated with DOX/NPs revealed lowest cell density and most apoptosis in the tumor tissue compared with groups treated with PBS, NPs or DOX (Figure 6E), confirming that proliferation of tumor is prevented effectively in the group treated with DOX/NPs. Meanwhile, H&E assays of heart tissue showed that little heart toxicity in the DOX group, but no obvious damage in other groups and other organs (Figures 6E and S9). In addition, immunohistochemistry study showed that the DOX/NPs treated group induced a higher cleaved caspase-3 expression in tumor tissue than other groups (Figure 6F). These results indicate that the synergetic therapeutic DOX/NPs could significantly enhance antitumor efficiency by inducing apoptosis, and reduce heart toxicity of DOX in vivo.
In Vivo Antimetastasis Efficacy of DOX/NPs
Metastasis is the primary cause leading to tumor deterioration and recurrence in melanoma patients.54 We found that migration could be inhibited by affecting mitochondrial function in cell experiments. Next, we determined the antimetastatic spread effect of DOX/NPs in vivo by inoculating B16F10 cells intravenously into BALB/c mice. As shown in Figure 7A–C, the lung weight and the number of lung metastatic nodules obviously decreased in the group treated with DOX/NPs over 21 days, but the groups treated with NPs or DOX exhibited a weaker inhibitory effect. The increased lung weight in the model group was probably as a result of the metastatic burden of the tumors. In addition, mice treated with DOX/NPs exhibited the fewest and smallest thrombi in the pulmonary vasculature and lungs among those found in the mice of other groups (Figure 7D and E). And that, the body weight of the mice in the group treated with DOX/NPs showed no obvious change compared with the control group treated with saline (Figure 7F). However, it might be noted that the body weight of mice treated with DOX decreased, which was consistent with our data from anticancer experiments in vivo. These results demonstrate that the synergetic therapeutic DOX/NPs could significantly inhibit B16F10 tumor metastasis by suppressing tumor cell proliferation. The results are consistent with the data from in vitro experiments, further confirming that DOX/NPs have potential to become an alternative strategy of efficient and synergistic therapy for antimetastasis.
 |
Figure 7 In vivo antimetastasis efficacy of free DOX, NPs and DOX/NPs. (A) Lung morphologies from each treatment group. (B) The weight of lungs of mice in each treatment group. (C) The number of visible colonies in the lung of each treatment group. (D) Quantification of thrombi formation within lung tissue. (E) H&E staining of lung sections harvested from each treatment group. Black arrows indicate thrombi formation induced by tumor. (F) The body weight of mice. The data represent mean ±SD. n=6–8. *p<0.05 vs control, **p<0.01 vs control, **p<0.05 vs model, ***p< 0.05 vs NPs and DOX, ****p<0.01 vs NPs and DOX. |
Conclusion
In summary, we successfully constructed a pH-sensitive prodrug nanocarrier DOX/NPs with satisfying pharmaceutical characteristics as drug delivery system to codeliver the DOX/diosgenin combination to efficient and synergistic antimetastasis and anti-apoptosis. The DOX/NPs could self-assemble into nanoparticles, exhibit homogenous spherical shapes with high loading efficiency, and further lead to significantly enhanced drug release at tumor intracellular pH. Compared with free DOX or NPs, DOX/NPs possess great synergistic effects on tumor migration and growth inhibition by inducing mitochondria-associated metastasis and apoptosis in B16F10 cancer cells, which is the most lethal form of skin cancer with early metastasis and high mortality. Furthermore, DOX/NPs could penetrate deep of into the tumor tissues as well as increase tumor targeting by biodistribution imaging in vivo. More importantly, DOX/NPs show better effective antimetastasis behavior than DOX and NPs in the lung metastasis, which is the most intractable question in controlling tumor deterioration and recurrence. The established DOX/NPs nanocarrier also reduce side effects on normal tissues compared with free DOX. It is concluded that the established codelivery system of the combination of DOX and diosgenin, which could effectively facilitate an efficient and synergistic antimetastasis and antiproliferation, provides a potential strategy for cancer treatment, especially in avoiding tumor deterioration and recurrence.
Acknowledgment
This work was financed by Grant-in-aid for scientific research from the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant no. 2019ZX09201005-005-001 and 2019ZX09201005-005-004), Sichuan Science and Technology Department Emergency Project (Grant no. 2020YFS0554), the National Natural Science Foundation of China (Grant no. 81673710, 81973580), West China Innovative Chinese Medicine and Health Products Research Academician Workstation of Academician Zhang Boli and Academician Zhu Beiwei, West China Hospital, Sichuan University (Grant no. HXYS19001 and HXYS19002), the National Science Foundation for Young Scientists of China (Grant no. 81803866), the Project funded by China Postdoctoral Science Foundation (Grant no. 2019M663533, 2019T120854) and the Post-Doctor Research Project, West China Hospital, Sichuan University (Grant no. 2018HXBH061, 2019HXBH024).
The authors would like to thank Song Lei (Department of Pathology, West China Hospital, Sichuan University) for providing transmission electron microscope and experimental data analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Miller AJ, Mihm MC. Mechanisms of disease - melanoma. N Engl J Med. 2006;355(1):51–65. doi:10.1056/NEJMra052166
2. Evans MS, Madhunapantula SRV, Robertson GP, Drabick JJ. Current and future trials of targeted therapies in cutaneous melanoma. Adv Exp Med Biol. 2013;779:223–225.
3. Orgaz JL, Sanz-Moreno V. Emerging molecular targets in melanoma invasion and metastasis. Pigment Cell Melanoma Res. 2013;26(1):39–57. doi:10.1111/pcmr.12041
4. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6(6):449–458. doi:10.1038/nrc1886
5. Ortega A. A new role for gaba: inhibition of tumor cell migration. Trends Pharmacol Sci. 2003;24(4):151–154. doi:10.1016/S0165-6147(03)00052-X
6. Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol. 2008;5(4):206–219. doi:10.1038/ncponc1066
7. Martin OA, Anderson RL, Narayan K, Macmanus MP. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat Rev Clin Oncol. 2017;14(1):32–44. doi:10.1038/nrclinonc.2016.128
8. Chen CT, Yamaguchi H, Lee HJ, et al. Dual targeting of tumor angiogenesis and chemotherapy by endostatin-cytosine deaminase-uracil phosphoribosyl transferase. Mol Cancer Ther. 2011;10(8):1327–1336. doi:10.1158/1535-7163.MCT-10-1117
9. Qin Y, Wu X, Huang W, et al. Acute toxicity and sub-chronic toxicity of steroidal saponins from Dioscorea Zingiberensis C.H.Wright in rodents. J Ethnopharmacol. 2009;126(3):543–550. doi:10.1016/j.jep.2009.08.047
10. Chen Y, Tang YM, Yu SL, et al. Advances in the pharmacological activities and mechanisms of diosgenin. Chin J Nat Med. 2015;13(8):578–587. doi:10.1016/S1875-5364(15)30053-4
11. He Z, Chen H, Li G, et al. Diosgenin inhibits the migration of human breast cancer MDA-MB-231 cells by suppressing Vav2 activity. Phytomedicine. 2014;21(6):871–876. doi:10.1016/j.phymed.2014.02.002
12. Bhuvanalakshmi G, Rangappa KS, Dharmarajan A, et al. Breast cancer stem-like cells are inhibited by diosgenin, a steroidal saponin, by the attenuation of the Wnt β-catenin signaling via the Wnt antagonist secreted frizzled related protein-4. Front Pharmacol. 2017;8:124. doi:10.3389/fphar.2017.00124
13. Chen CT, Wang ZH, Hsu CC, Lin HH, Chen JH. In vivo protective effects of diosgenin against doxorubicin-induced cardiotoxicity. Nutrients. 2015;7(6):4938–4954. doi:10.3390/nu7064938
14. Zhang R, Xing R, Jiao T, et al. Carrier-free, chemophotodynamic dual nanodrugs via self-assembly for synergistic antitumor therapy. ACS Appl Mater Interfaces. 2016;8(21):13262–13269. doi:10.1021/acsami.6b02416
15. Glasgow MDK, Chougule MB. Recent developments in active tumor targeted multifunctional nanoparticles for combination chemotherapy in cancer treatment and imaging. J Biomed Nanotechnol. 2015;11(11):1859–1898. doi:10.1166/jbn.2015.2145
16. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016;17(1):20–37. doi:10.1038/nrc.2016.108
17. Sun N, Zhao C, Cheng R, et al. Cargo-free nano-medicine with pH-sensitivity for co-delivery of dox conjugated prodrug with sn38 to synergistically eradicate breast cancer stem cells. Mol Pharm. 2018;15(8):3343–3355. doi:10.1021/acs.molpharmaceut.8b00367
18. Li F, Fernandez PP, Rajendran P, Hui KM, Sethi G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett. 2010;292(2):197–207. doi:10.1016/j.canlet.2009.12.003
19. Li C, Dai J, Zheng D, et al. An efficient prodrug-based nanoscale delivery platform constructed by water soluble eight-arm-polyethylene glycol-diosgenin conjugate. Mat Sci Eng C Mater. 2019;98:153–160. doi:10.1016/j.msec.2018.12.078
20. Reithofer MR, Chan KH, Lakshmanan A, Dang HL, Hauser CAE. Ligation of anti-cancer drugs to self-assembling ultrashort peptides by click chemistry for localized therapy. Chem Sci. 2014;5(2):625–630. doi:10.1039/C3SC51930A
21. Chan KH, Lee WH, Ni M, Loo YH. C-Terminal residue of ultrashort peptides impacts on molecular self-assembly, hydrogelation, and interaction with small-molecule drugs. Sci Rep. 2018;8:17127. doi:10.1038/s41598-018-35431-2
22. Rashed HM, Shamma RN, Basalious EB. Contribution of both olfactory and systemic pathways for brain targeting of nimodipine-loaded lipo-pluronics micelles: in vitro characterization and in vivo biodistribution study after intranasal and intravenous delivery. Drug Deliv. 2017;24(1):181–187. doi:10.1080/10717544.2016.1236848
23. Yang Z, Luo H, Cao Z, et al. Dual-targeting hybrid nanoparticles for the delivery of SN38 to Her2 and CD44 overexpressed human gastric cancer. Nanoscale. 2016;8(22):11543–11558. doi:10.1039/C6NR01749E
24. Han H, Wang H, Chen Y, et al. Theranostic reduction-sensitive gemcitabine prodrug micelles for near-infrared imaging and pancreatic cancer therapy. Nanoscale. 2016;8(1):283–291. doi:10.1039/C5NR06734K
25. Cheetham AG, Chakroun RW, Ma W, Cui H. Self-assembling prodrugs. Chem Soc Rev. 2017;46(21):6638–6663. doi:10.1039/C7CS00521K
26. Wang H, Agarwal P, Zhao S, et al. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials. 2015;72:74–89. doi:10.1016/j.biomaterials.2015.08.048
27. Pang X, Jiang Y, Xiao Q, Leung AW, Hua H, Xu C. pH-responsive polymer–drug conjugates: design and progress. J Control Release. 2016;222:116–129. doi:10.1016/j.jconrel.2015.12.024
28. Kobayashi K, Wei J, Iida R, Ijiro K, Niikura K. Surface engineering of nanoparticles for therapeutic applications. Polym J. 2014;46(8):460–468. doi:10.1038/pj.2014.40
29. Yang Z, Sun N, Cheng R, et al. pH multistage responsive micellar system with charge-switch and peg layer detachment for co-delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. Biomaterials. 2017;147:53–67. doi:10.1016/j.biomaterials.2017.09.013
30. Rao W, Wang H, Han JF, et al. Chitosan-decorated doxorubicin-encapsulated nanoparticle targets and eliminates tumor reinitiating cancer stem-like cells. Acs Nano. 2015;9(6):5725–5740. doi:10.1021/nn506928p
31. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. Acs Nano. 2014;8(6):5696–5706. doi:10.1021/nn500299p
32. Wei ZL, Xin G, Wang HB, et al. The diosgenin prodrug nanoparticles with ph-responsive as a drug delivery system uniquely prevents thrombosis without increased bleeding risk. Nanomed Nanotechnol. 2018;14(3):673–684. doi:10.1016/j.nano.2017.12.019
33. Zhang Y, He J, Cao D, Zhang M, Ni P. Galactosylated reduction and ph dual-responsive triblock terpolymer Gal-PEEP-a-PCL-ss-PDMAEMA: a multifunctional carrier for the targeted and simultaneous delivery of doxorubicin and DNA. Polym Chem. 2014;5(17):5124–5138. doi:10.1039/C4PY00538D
34. Zeng Z, Wei ZL, Ma LM, et al. pH-responsive nanoparticles based on ibuprofen prodrug as drug carriers for inhibition of primary tumor growth and metastasis. J Mater Chem B. 2017;5(33):6860–6868. doi:10.1039/C7TB01288H
35. Chi TN, Tran TH, Amiji M, Lu X, Kasi RM. Redox-sensitive nanoparticles from amphiphilic cholesterol-based block copolymers for enhanced tumor intracellular release of doxorubicin. Nanomed Nanotechnol. 2015;11(8):2071–2082. doi:10.1016/j.nano.2015.06.011
36. Song N, Ding M, Pan Z, et al. Construction of targeting-clickable and tumor-cleavable polyurethane nanomicelles for multifunctional intracellular drug delivery. Biomacromolecules. 2013;14(12):4407–4419. doi:10.1021/bm401342t
37. Gu JX, Cheng WP, Liu JG, et al. pH-triggered reversible “stealth” polycationic micelles. Biomacromolecules. 2008;9(1):255–262. doi:10.1021/bm701084w
38. Ilkar Erdagi S, Yildiz U. Diosgenin-conjugated PCL–MPEG polymeric nanoparticles for the co-delivery of anticancer drugs: design, optimization, in vitro drug release and evaluation of anticancer activity. New J Chem. 2019;43(17):6622–6635. doi:10.1039/C9NJ00659A
39. Li F, Chen WL, You BG, et al. Enhanced cellular internalization and on-demand intracellular release of doxorubicin by stepwise pH-/reduction-responsive nanoparticles. ACS Appl Mater Interfaces. 2016;8(47):32146–32158. doi:10.1021/acsami.6b09604
40. He YJ, Su ZG, Xue LJ, Xu H, Zhang C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release. 2016;229:80–92. doi:10.1016/j.jconrel.2016.03.001
41. Zhao KD, Li D, Xu WG, et al. Targeted hydroxyethyl starch prodrug for inhibiting the growth and metastasis of prostate cancer. Biomaterials. 2017;116:82–94. doi:10.1016/j.biomaterials.2016.11.030
42. Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212(2):167–178. doi:10.1016/j.taap.2006.01.003
43. Yu TZ, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. PNAS. 2006;103(8):2653–2658. doi:10.1073/pnas.0511154103
44. Xin G, Wei ZL, Ji CJ, et al. Xanthohumol isolated from humulus lupulus prevents thrombosis without increased bleeding risk by inhibiting platelet activation and mtDNA release. Free Radical Biol Med. 2017;108:247–257. doi:10.1016/j.freeradbiomed.2017.02.018
45. Cory S, Adams JM. The bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi:10.1038/nrc883
46. He Y, Li X, Ma J, Ni G, Yang G, Zhou S. Programmable codelivery of doxorubicin and apatinib using an implantable hierarchical-structured fiber device for overcoming cancer multidrug resistance. Small. 2019;15(8):e1804397. doi:10.1002/smll.201804397
47. Lepage C, Liagre B, Cook-Moreau J, Pinon A, Beneytout JL. Cyclooxygenase-2 and 5-lipoxygenase pathways in diosgenin-induced apoptosis in HT-29 and HCT-116 colon cancer cells. Int J Oncol. 2010;36(5):1183–1191. doi:10.3892/ijo_00000601
48. Liagre B, Bertrand J, Leger DY, Beneytout JL. Diosgenin, a plant steroid, induces apoptosis in cox-2 deficient k562 cells with activation of the p38 MAP kinase signalling and inhibition of NF-kappaB binding. Int J Mol Med. 2005;16(6):1095–1101.
49. Zhu SJ, Qian LL, Hong MH, et al. RGD-modified PEG-PAMAM-DOX conjugate: in vitro and in vivo targeting to both tumor neovascular endothelial cells and tumor cells. Adv Mater. 2011;23(12):H84–H89. doi:10.1002/adma.201003944
50. Yu C, Zhou M, Zhang X, Wei W, Chen X, Zhang X. Smart doxorubicin nanoparticles with high drug payload for enhanced chemotherapy against drug resistance and cancer diagnosis. Nanoscale. 2015;7(13):5683–5690. doi:10.1039/C5NR00290G
51. Ni WF, Li ZY, Liu ZQ, et al. Dual-targeting nanoparticles: codelivery of curcumin and 5-fluorouracil for synergistic treatment of hepatocarcinoma. J Pharm Sci. 2019;108(3):1284–1295. doi:10.1016/j.xphs.2018.10.042
52. Petros RA, Desimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi:10.1038/nrd2591
53. Sun J, Jiang L, Lin Y, et al. Enhanced anticancer efficacy of paclitaxel through multistage tumor-targeting liposomes modified with RGD and KLA peptides. Int J Nanomed. 2017;12:1517–1537. doi:10.2147/IJN.S122859
54. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19(11):1450–1464. doi:10.1038/nm.3391
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.