Back to Journals » International Journal of Nanomedicine » Volume 12
A pH-responsive prodrug delivery system of 10-HCPT for controlled release and tumor targeting
Authors Liu Y, Li D, Guo X, Xu H, Li Z, Zhang Y, Song C, Fan R, Tang X, Zhang Z
Received 27 October 2016
Accepted for publication 8 January 2017
Published 22 March 2017 Volume 2017:12 Pages 2227—2242
DOI https://doi.org/10.2147/IJN.S125849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yang Liu,1–3 Dan Li,2 Xinhong Guo,2 Haiwei Xu,2 Zhi Li,2 Yanling Zhang,2 Chuanjun Song,4 Ruhan Fan,2 Xing Tang,1,* Zhenzhong Zhang2,*
1Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, 2Department of Pharmaceutics, School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, 3Department of Pharmaceutics, Collaborative Innovation Center of New Drug Research and Safety Evaluation, Henan, Zhengzhou, 4Department of Organic Chemistry, College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: We synthesized a pH-responsive conjugate of 10-hydroxycamptothecin-thiosemicarbazide-linear polyethylene glycol 2000 (PEG2000). The conjugate was confirmed by matrix-assisted laser desorption time of flight mass spectrometry, 1H NMR, and 13C NMR. The water solubility of the prodrug was increased by over 3,000 times; much longer body circulation time, higher tumor-targeting ability, and reduced toxicity were observed, compared with commercial 10-HCPT injection. The linker contains a pH-sensitive hydrazone bond, which breaks under low pH conditions in the tumor microenvironment. The conjugates showed good stability in phosphate-buffered saline (pH 7.4) and rat plasma. This amphiphilic conjugate could self-assemble into nanosized micelles of 80–100 nm. Cytotoxicity assay results indicate significantly higher efficacy of the conjugate (IC50 [half maximal inhibitory concentration] =0.117 µM on SW180 cells) than 10-HCPT solution (IC50 =0.241 µM on SW480 cells). Cellular uptake analysis suggested its rapid internalization and nuclear transport. Pharmacokinetic analysis of the conjugates demonstrated that the conjugate circulated for a longer time in the blood circulation system (T2/1 =10.516±1.158 h) than did 10-HCPT solution (T2/1 =1.859±1.385 h), and that it also enhanced the targeting and mean residence time (MRT0–inf =39.873±4.549 h) in the tumor site, compared with 10-HCPT (MRT0–inf =9.247±1.026 h). Finally, the conjugate demonstrated an increased tumor growth inhibition effect (TIR =82.66%±7.175%) in vivo and lower side effects than 10-HCPT (TIR =63.85%±5.233%). This prodrug holds great promise in improving therapeutic efficacy and overcoming multidrug resistance.
Keywords: 10-hydroxycamptothecin, conjugate, controlled release, prodrug, pH-responsive, hydrazone
Introduction
10-Hydroxycamptothecin (10-HCPT) has a broad spectrum of anticancer activity in vitro and in vivo.1 Its mechanism of therapeutic action is based on targeting the nuclear enzyme topoisomerase I by stabilizing a cleavable complex to inhibit DNA S-phase replication and RNA transcription.2 However, the clinical use of 10-HCPT has been hampered by its poor water solubility and chemically unstable lactone ring (E-ring).2 Moreover, it is accompanied by severe side effects, such as myelosuppression and hematuria, which also limit its clinical application. Combination with a macromolecule as a prodrug to achieve better targeting and controlled release is one of the most promising strategies. Polymer–drug prodrugs, which are obtained by covalent linkage of active drugs to water-soluble macromolecular carriers, have been investigated in a number of studies.3–5 These prodrugs lead to the improvement of drug water solubility and chemical stability,6 a much longer systemic circulation time or sustained release effect,7 passive tumor targeting through an enhanced permeability and retention effect,8 promotion of drug uptake into cells via endocytosis,9 decreased toxicity,10 better availability at the tumor site, and antimultidrug resistance.11 Apart from introducing cell-targeting biomolecules for specific delivery, stimuli-responsive drug systems based on different internal environments in the human body can also enable controlled drug delivery and targeted therapeutics.12 Owing to rapid proliferation-induced glucose consumption and lactic acid accumulation, the pH in tumor tissues is frequently 0.5–1.0 units less than that in healthy tissues,13 and endosomes and lysosomes show lower pH values (4.5–6.5) than do normal extracellular matrices and blood (pH =7.4). As an important signal, an acid-sensitive chemical bond between drug and polymer can be considered an ideal trigger for selectively releasing drugs in tumor tissues or directly within tumor cells. Of all pH-responsive bonds, the most important are hydrazone, acetal,14–16 and cis-aconityl.17–20 As the preferred pH-responsive chemical bond, hydrazone has been extensively studied, owing to its acute response in drug delivery behavior.
We designed a pH-responsive conjugate with a hydrazone bond as a linker to provide triggered release of 10-HCPT after cellular internalization in acidic endosomes and lysosomes (Figure 1). The blue part is the lactone functionality in the E-ring, which is not only essential for anticancer activity but also confers a degree of instability in aqueous solutions.21 Therefore, we did not choose this site to avoid the destruction of lactone functionality. The yellow part is an ester bond, which is stable under physiological pH conditions and degrades under high pH conditions. The red part is a hydrazone bond, which degrades under low pH conditions.6 The green part is a 2,000 Da MPEG (methoxy polyethylene glycol) group, which is highly hydrophilic and which can greatly improve the water solubility of 10-HCPT by >3,000 times.22 Owing to its high molecular weight, the conjugate circulates in the body for much longer than free 10-HCPT.23 After entering cells through endocytosis, the hydrazone bond degrades and the drug is released to enter the cell nucleus.
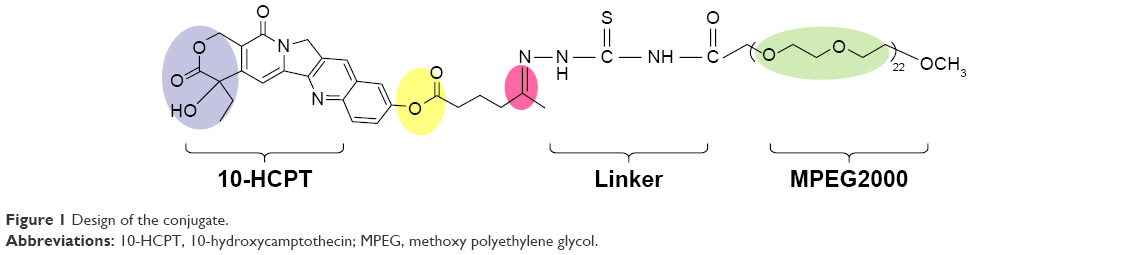 | Figure 1 Design of the conjugate. |
Multidrug resistance is the major obstacle that troubles almost all of drug delivery systems.24,25 According to the literature, macromolecules can bypass the P-glycoprotein efflux system and promote intracellular accumulation of drugs.26,27 Unlike small molecule drugs, which diffuse passively across the plasma membrane, polymer–drug conjugates are liberated from within the lysosomal compartment and subsequently eliminated from the cytoplasm. Consequently, the interaction and recognition effects of the P-glycoprotein efflux pump toward such macromolecules are minimized.28–30 Furthermore, the pH responsivity of conjugates tends to trigger effective drug release in acid tumor cells and concentrates the intracellular drug to a sufficiently high level,31,32 see Figure 2. The final conjugate was synthesized in four steps (Figure 3). In vitro release, cytotoxicity, and endocytosis of the conjugate were monitored. Finally, in vivo pharmacokinetics, biodistribution, and antitumor efficacy were evaluated.
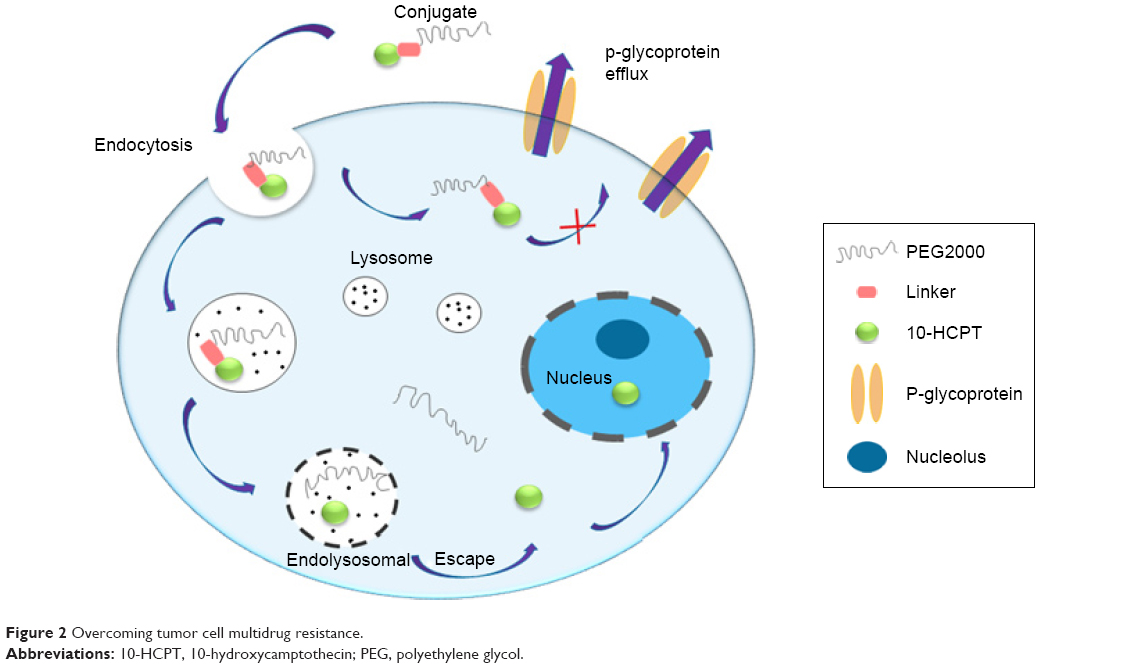 | Figure 2 Overcoming tumor cell multidrug resistance. |
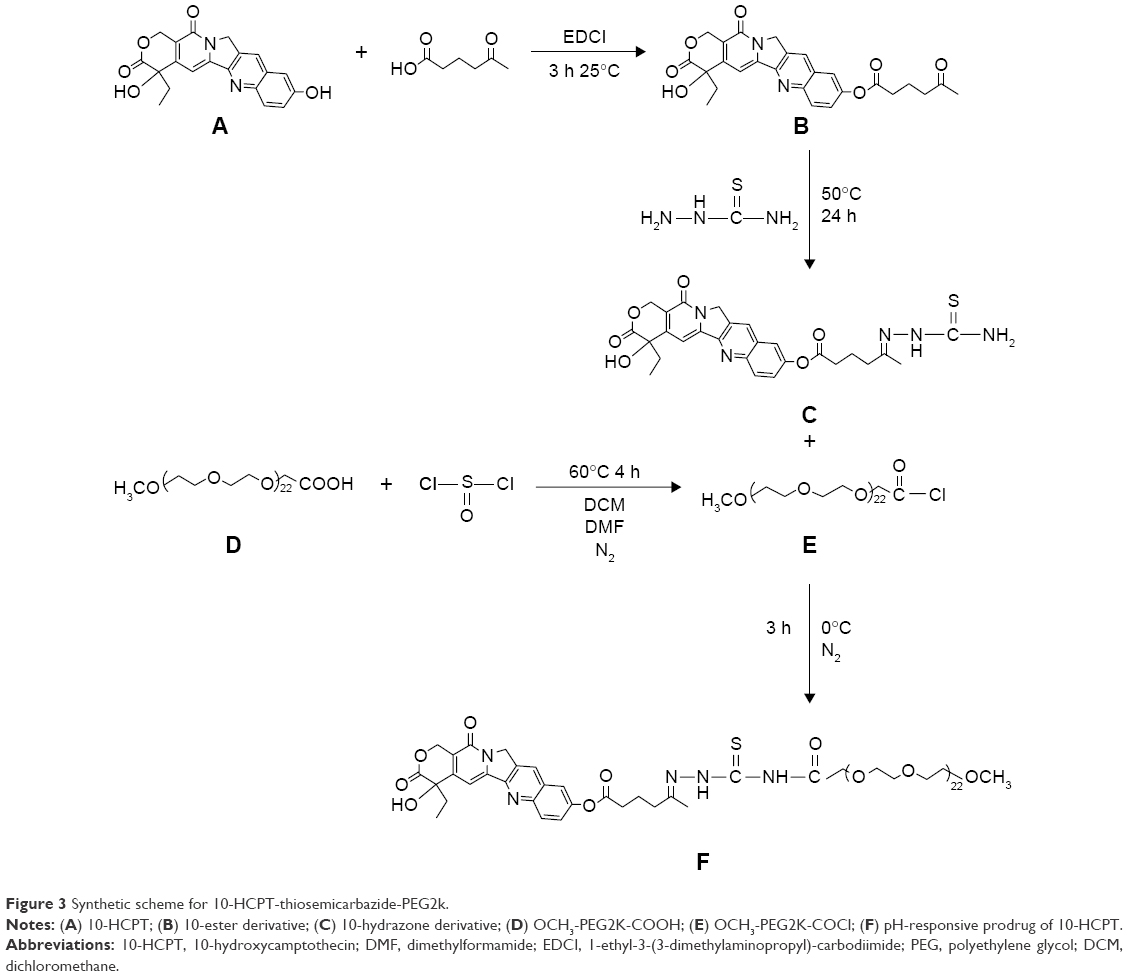 | Figure 3 Synthetic scheme for 10-HCPT-thiosemicarbazide-PEG2k. |
Materials and methods
Materials
10-HCPT, EDCI (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide), and MTT (methylthiazolyldiphenyl-tetrazolium bromide) were obtained from Sigma Chemical Co. (St Louis, MO, USA). 5-carbonyl caproic acid and dichloromethane were obtained from Shanghai Topbiochem Ltd. (Shanghai, People’s Republic of China). Anhydrous N, N-dimethylformamide (DMF), thiosemicarbazide, and sulfoxide chloride were obtained from Aladdin Industrial Corporation (Shanghai, People’s Republic of China). MeO-PEG2000–COOH (molecular weight ~2,000 Da) was obtained from Xi’an Ruixi Biological Technology Co. Ltd. (Xi’an, People’s Republic of China). Sephadex LH-20 was obtained from Shanghai Yuanye Bio-Technology Co. Ltd. (Shanghai, People’s Republic of China). All other chemicals were of analytical grade and were used without further purification.
Cells and animals
Cell lines MCF-7, HepG2, and SW480 were obtained from ATCC and cultured in Roswell Park Memorial Institute 1640 or L-15, supplemented with 10% inactivated fetal bovine serum in a humidified atmosphere (5% CO2) at 37°C. BALB/c mice (4–5 weeks old, female) were purchased from Aiermaite Inc. (Suzhou, People’s Republic of China). Tumor-bearing mice were housed under a 12 h light/12 h dark cycle and were given free access to water and food.
Synthesis of CPT-5-carbonyl caproic acid ester (compound B)
A solution of compound A (1.00 g, 2.7 mmol) and 5-carbonyl caproic acid (1.07 g, 8.1 mmol) in pyridine (10 mL) was added to EDCI (2.06 g, 10.8 mmol) at 25°C, and stirred for 3 h. Thin layer chromatography was used to detect the reaction progress. The mixture was added to dichloromethane (50 mL) and H2O (50 mL) and stirred for 30 min; the organic layer was separated, dried, and concentrated to obtain a residue, which was purified by silica gel column (dichloromethane: MeOH =10:1) to produce compound B (1.25 g, 96%) as a yellow solid.
Synthesis of CPT-5-carbonyl caproic acid-thiosemicarbazide hydrazone (compound C)
Compound B (377 mg, 2.1 mmol) was dissolved in 100 mL of anhydrous dichloromethane at room temperature.33 Thiosemicarbazide was dissolved in 50 mL of anhydrous methanol at room temperature. The two solutions were mixed together and stirred for 24 h at 50°C. Subsequently, the mixture was allowed to come to room temperature. A white precipitate was produced, which was filtered and washed with 100 mL dichloromethane and 100 mL methanol, in turn. The solvent was evaporated under reduced pressure.34
Synthesis of MeO-PEG2000–COOCl (compound E)
Linear MeO-PEG2000–COOH was dissolved in 2 mL anhydrous DMF at room temperature; to it was added 100 mL of anhydrous dichloromethane at room temperature and then 5 mL sulfoxide chloride. The reaction was stirred for 5 h at 50°C under N2. The solvent and excess sulfoxide chloride were removed under reduced pressure. Compound E was kept at 4°C under N2.
Synthesis of 10-HCPT-thiosemicarbazide-PEG2000-MeO conjugate (compound F)
Compound E (205 mg, 0.1 mmol) was dissolved in 50 mL anhydrous DMF and to it were added compound C (55 mg, 0.1 mmol) drop by drop at 0°C under N2.35 K2CO3 was added to the reaction to maintain a basic pH. After stirring for 3 h in an ice-bath, the solvent was evaporated under reduced pressure to concentrate the mixture. The product was purified using size-exclusion chromatography (LH-20 Sephadex). A Sephadex LH-20 column (3.0×20 cm) was packed using the gravity method. The product was applied to the column as a DMF solution at room temperature. Elution was performed using dichloromethane and methanol (1:1). Each 5 mL of eluent was collected in a tube and inspected using thin layer chromatography. The pure fractions of compound F were pooled and the solvent was evaporated under reduced pressure. The purified product was washed thrice by dichloromethane and methanol (1:1) to remove remaining impurities and then dried under vacuum overnight. The conjugate was characterized using proton nuclear magnetic resonance (1H NMR; Bruker, 400 MHz), carbon nuclear magnetic resonance (13C NMR), and matrix-assisted laser desorption time of flight mass spectrometry (MALDI-TOF MS). The instrument was operated in the linear positive ion mode at 15 mV (sum =850 mV). Data processing used was Kratos PC Axima CFR plus V 2.4.1.
Fluorescence properties of compound F
Compound F was dissolved in methanol at a concentration of 5 μg/mL and scanned for its fluorescence spectrum from 300 to 900 nm in a fluorescence spectrophotometer (F-4600, Hitachi High-Technologies Corporation, Japan) to get its maximum fluorescence excitation and absorption wavelength.
Preparation of coumarin-6 incorporated micelles
Twenty milligram of compound F was dissolved in 4 mL water. The solution was added with 1.0 mL of coumarin-6 ethanol solution at the concentration of 60 μg/mL, and then was ultrasonicated for 3 min in ice-bath by a probe-type ultrasonicator (JY92-2D, Ningbo Scentz Biotechnology Co., Ltd., People’s Republic of China). The solution was dialyzed against an excess amount of distilled water with a dialysis bag (MWCO 2000) overnight, filtering through a 0.22 μm pore-sized microporous membrane and the particle size, size distribution, and zeta potential of micelles were determined by dynamic light scattering. The morphology was observed by transmission electron microscopy.
In vitro drug release from conjugates
Drug release from conjugates in phosphate-buffered saline (PBS)
The conjugates were dissolved in a series of PBS solutions (0.01 mol/L) at pH 3.0, 4.0, 5.0, 6.0, and 7.4, to give a final concentration of 0.5 mg/mL. The buffer solutions were incubated at 37°C for 48 h and 0.1 mL portions of each solution were withdrawn and centrifuged at 12,000 rpm for 5 min at 0, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h and the supernatants were analyzed by high performance liquid chromatography–mass spectrum (UPLC/MS) (Agilent Technologies 6460 Triple Quad, Palo Alto, CA, USA; ZORBAX Eclipse XDB-C18 Rapid Resolution HD 2.1×100 mm, 1.8 μm, Agilent). The multiple reactions monitoring mode was used to confirm the precursor ions and select product ions. Full-scan spectra produced a predominant peak of (MH)+ at M/Z 477.0. Collision-induced dissociation of the (MH)+ ion produced a major fragment at M/Z 433.1. Therefore, the transition pair 477.0/433.1 was selected.7
Drug release from conjugates in plasma
Solutions of compound F (5.0 mg/mL) and camptothecin with a final concentration of 1.0 μg/mL as an internal standard were added to 5 mL human serum to give a final concentration of 0.5 mg/mL and incubated at 37°C. At intervals of 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h, 100 μL samples were withdrawn. After precipitating the protein with 300 μL acetonitrile, the mixture was vortexed and centrifuged at 12,000 rpm for 5 min; 300 μL of the supernatant was evaporated to dryness under N2 at 40°C. The dry extracts were dissolved in 100 μL of acetonitrile for UPLC/MS analysis. The 10-HCPT was then further diluted with human serum to make a set of standards ranging from 0.5 to 1,000 ng/mL and tested quantitatively by the internal standard method. Test conditions were the same as described in the “Fluorescence properties of compound F” section.
In vitro cytotoxicity
Cell lines were seeded in 96-well plates at a concentration of 5,000 cells/well and allowed to attach overnight. Cells were then exposed to various concentrations of test compounds in quintuplicate for 48 h under 5% CO2 at 37°C. Next, MTT was added and the culture was incubated for an additional 4 h and the absorbance was measured using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 490 nm. The results were also expressed as IC50 (the compound concentration required for 50% growth inhibition of tumor cells).36 All experiments were performed three times.
In vitro endocytosis
To analyze the endocytosis effect of the conjugate, MCF-7 cells were seeded at 8,000 cells per well in 24-well plates. After 24 h culture, cells were treated with compound F and 10-HCPT solution for 1, 2, or 4 h under 5% CO2 at 37°C. The cells were washed three times with PBS and then fixed in cold ethanol for 10 min at room temperature. A laser scanning confocal fluorescence microscope (Olympus IX81 microscope, Olympus corporation, Japan) was used to visualize the cellular uptake and intracellular distribution of coumarin-6 incorporated micelles.
In vivo pharmacokinetics and biodistribution
All animal research were performed following the protocol approved by the ethics committee of Henan Laboratory Animal Center and followed the guidelines of the Regulations for the administration of affairs concerning experimental animals.
Tumor-bearing nude mice were randomly divided into two groups (n=6). Mice were administered a single dose of 10-HCPT (5 mg/kg) and compound F solution (equal to 10-HCPT 5 mg/kg) intravenously. After 10, 20, 30 min, 1, 2, 4, 8, 12, 24, and 48 h of injection, blood samples were collected from the orbital vein, then centrifuged to obtain plasma samples. The mice were then killed to obtain tissue samples. The organs (heart, liver, spleen, lung, kidney, brain, and tumor) were removed and washed with physiological solution (0.9% NaCl), weighed and stored at −20°C. Plasma samples were also stored at −20°C until analysis. For the analysis, 150 μL of plasma was mixed with 50 μL of 0.1 N NaOH for 20 min in a water bath at 37°C, allowing hydrolysis of the conjugate. After this, 0.1 N HCl (50 μL) was added, followed by 20 μL camptothecin in acetonitrile (10 μg/mL) as internal standard and 600 μL acetonitrile. After vortexing for 2 min, the mixture was sonicated for 5 min and centrifuged at 10,000 rpm for 5 min. The clear supernatant was dried under nitrogen at 40°C, reconstituted by 100 μL acetonitrile and centrifuged at 12,000 rpm for 10 min before analysis.23,37,38 Tissues were homogenized in saline. Samples of 200 μL of tissue homogenate were analyzed using the same processing steps as for the plasma samples. The concentrations of 10-HCPT from each tissue homogenate and plasma were measured using UPLC/MS. Pharmacokinetic data were analyzed using Drug and Statistics (DAS) software version 3.0 (Mathematical Pharmacology Professional Committee of China, Shanghai, People’s Republic of China).
In vivo antitumor efficacy
In vivo antitumor efficacy of compound F was assessed using tumor-bearing nude mouse models. Tumor treatment was started when the tumor volume reached ~50–100 mm3. Tumor-bearing mice were divided into three groups (n=8) in such a way as to minimize weight and tumor size differences among the groups: the control group received saline; one treatment group received 10-HCPT solution, which is a commercial injection (5.0 mg/kg); the other treatment group received compound F solution (35.1 mg/kg, equivalent to 5.0 mg/kg 10-HCPT). The mice received a dose of 5 mg/kg once a day through tail vein injection for 14 days. The tumor volume was measured every 2 days and calculated as a × b2/2, with a the largest and b the smallest diameter. Moreover, the toxicity of these regimens was determined by monitoring animal behavior and weight loss.
Statistical analysis
All statistical tests were performed using Statistical Package for Social Science, version 13.0 (SPSS Inc., Chicago, IL, USA). A minimal P=0.05 was used as the significance level for all tests. One-way analysis of variance and Tukey’s test were performed on the uptake data. All data are reported as mean ± standard deviation unless otherwise noted.
Results
Characterization of compound B
The ester formation was confirmed by 1H NMR (400 MHz), 13C NMR (100 MHz), and high resolution mass spectrometry (ESI, Agilent 6540; Figure 4). The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublet, dt = double triplet. The 1H NMR spectra and 13C NMR spectra were recorded in CDCl3-d6. The following signals were obtained:
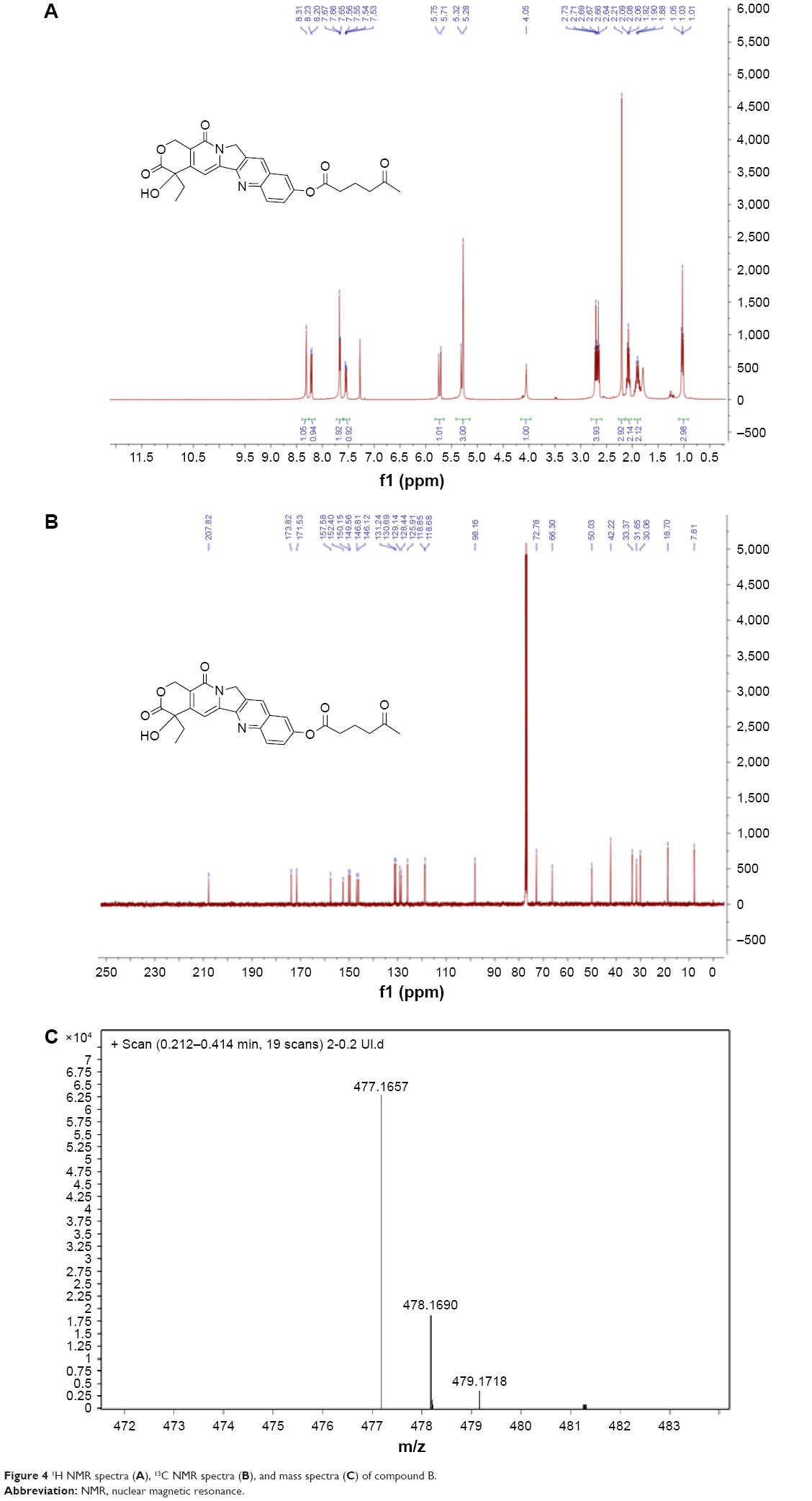 | Figure 4 1H NMR spectra (A), 13C NMR spectra (B), and mass spectra (C) of compound B. |
1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.22 (d, J =9.2 Hz, 1H), 7.72–7.61 (m, 2H), 7.53 (dt, J =35.3, 17.7 Hz, 1H), 5.71 (t, J =17.7 Hz, 1H), 5.30 (d, J =15.5 Hz, 3H), 4.17–3.96 (m, 1H), 2.68 (dt, J =19.8, 7.1 Hz, 4H), 2.22 (d, J =9.8 Hz, 3H), 2.08 (dt, J =12.0, 6.1 Hz, 2H), 2.01–1.85 (m, 2H), 1.03 (t, J =7.4 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 207.82, 173.82, 171.53, 157.58, 152.40, 150.15, 149.56, 146.81, 146.12, 131.24, 130.69, 129.14, 128.44, 125.91, 118.85, 118.68, 98.16, 72.78, 66.30, 50.03, 42.22, 33.37, 31.65, 30.06, 18.70, 7.84.
Characterization of compound C
The hydrazone formation was confirmed by 1H NMR (400 MHz), 13C NMR (100 MHz), and high resolution mass spectrometry (ESI, Agilent 6540; Figure 5). 1H NMR spectra and 13C NMR spectra were recorded in dimethyl sulfoxide (DMSO)-d6. The following signals were obtained:
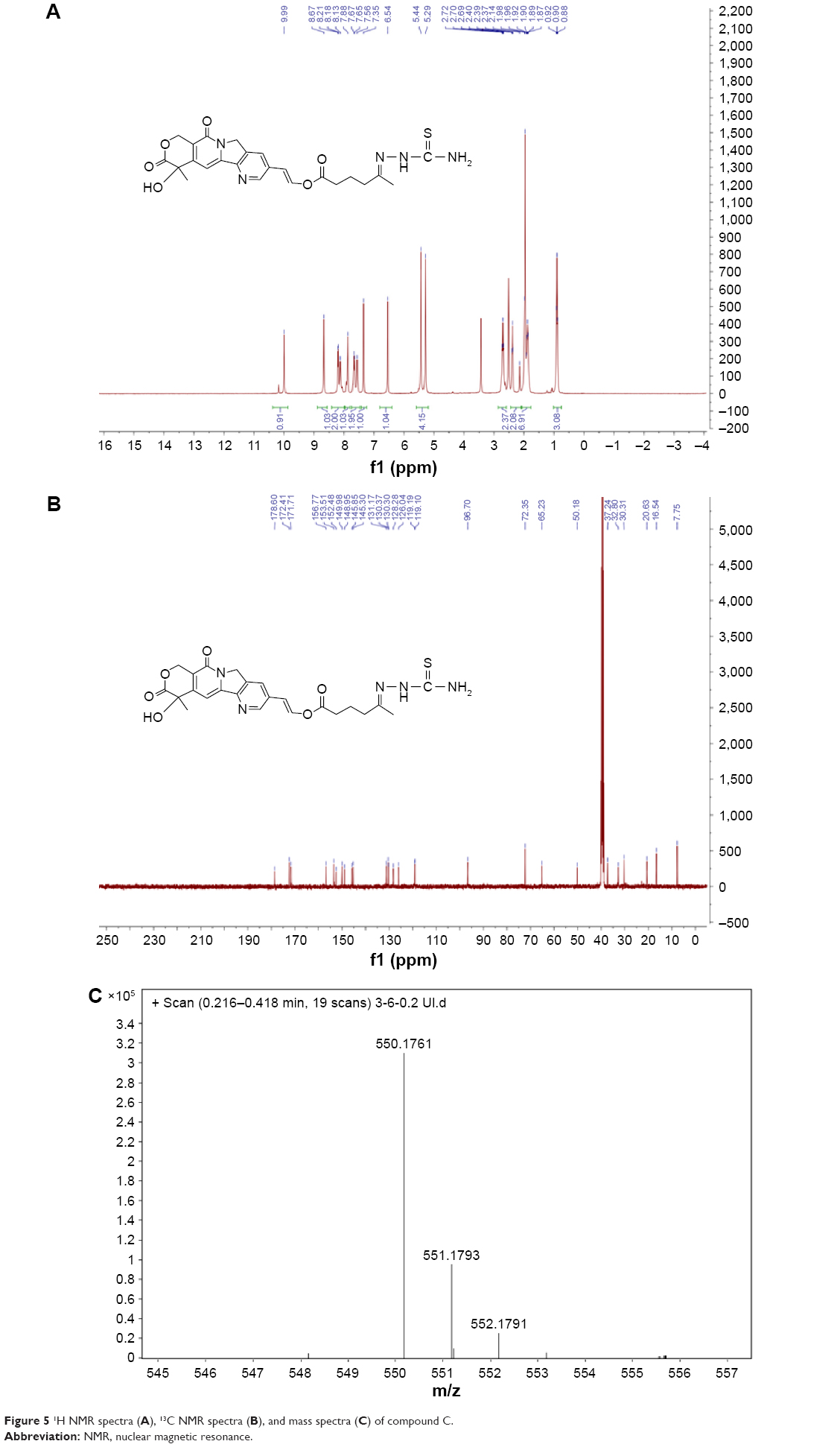 | Figure 5 1H NMR spectra (A), 13C NMR spectra (B), and mass spectra (C) of compound C. |
1H NMR (400 MHz, DMSO) δ 9.99 (s, 1H), 8.67 (s, 1H), 8.40–7.99 (m, 2H), 7.98–7.23 (m, 4H), 6.54 (s, 1H), 5.36 (d, J =60.0 Hz, 4H), 2.70 (t, J =6.7 Hz, 2H), 2.32 (dd, J =55.8, 49.1 Hz, 2H), 1.92 (dt, J =13.7, 7.7 Hz, 7H), 0.90 (t, J =6.8 Hz, 3H).
13C NMR (101 MHz, DMSO) δ 178.60, 172.41, 171.71, 156.77, 153.51, 152.48, 149.98, 148.95, 145.85, 145.30, 131.17, 130.37, 130.30, 128.28, 126.04, 119.19, 119.10, 96.70, 72.35, 65.23, 50.18, 37.24, 32.80, 30.31, 20.63, 16.54, 7.75.
Characterization of compound F
The conjugate was confirmed by 1H NMR, 13C NMR, and MALDI-TOF MS (Figure 6). The 1H NMR spectra and 13C NMR spectra were recorded in DMSO-d6. The following signals were obtained:
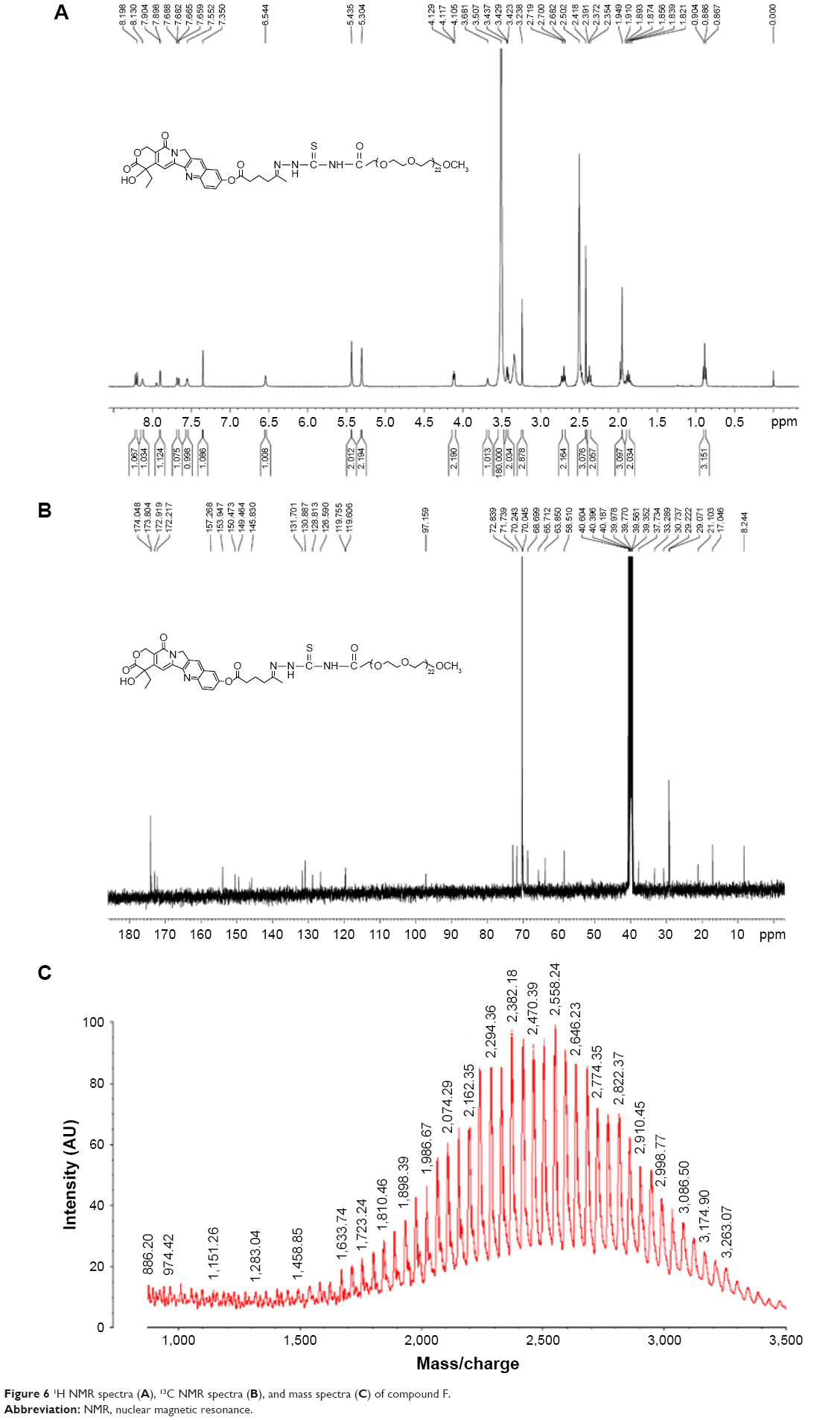 | Figure 6 1H NMR spectra (A), 13C NMR spectra (B), and mass spectra (C) of compound F. |
1H NMR (Bruker, 400 MHz) δ 0.89 CH3 (t), δ 1.87 CH2 (m), δ 1.95 CH3 (s), δ 2.37 CH2 (t), δ 2.42 CH3 (s), δ 2.70 CH2 (t), δ 3.23 CH2 (s), δ 3.43 CH2 (t), δ 3.50 (CH2CH2)45 (s), δ 3.68 OH (s), δ 4.11 CH2 (t), δ 5.30 CH2 (s), δ 5.44 CH2 (s), δ 6.54 CH (s), δ 7.35 CH (s), δ 7.55 NH (s), δ 7.67 CH (dd), δ 7.90 CH (d), δ 8.13 NH (s), δ 8.21 CH (d).
13C NMR (Bruker, 100 MHz) 8.2, 17.0, 21.1, 29.1, 29.2, 30.7, 33.3, 37.7, 58.5, 63.8, 65.7, 68.7, 70.0, 70.2, 71.7, 72.8, 97.1, 119.6, 119.8, 126.6, 128.8, 130.9, 131.7, 145.8, 149.5, 150.5, 153.9, 157.3, 172.2, 172.9, 173.8, 174.0.
A comparison of MALDI spectra of unreacted polyethylene glycol (PEG) (MW 2,026) with those of the final bio conjugate revealed a shift of 531 Da in the single mass peaks, indicating that the polymer conjugate carried exact molecule. Additionally, no sub-distribution of unreacted PEG was observed in the mass spectra of the camptothecin bioconjugate confirming that the conjugate solutions contained no free amino-PEG. The yield of the final conjugate was 32.4%.
Fluorescence properties of compound F
The fluorescence spectra of compound F are shown in Figure 7. We can see that the maximum excitation wavelength of compound F is 365 nm, and the maximum emission wave-length of compound F is 472 nm.
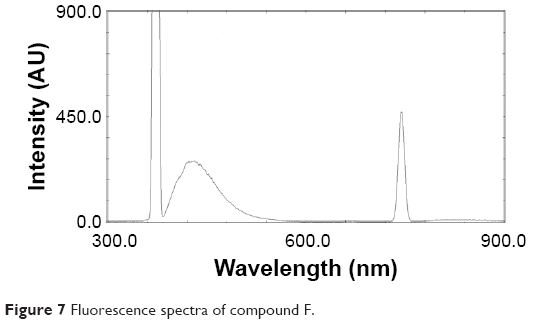 | Figure 7 Fluorescence spectra of compound F. |
Characterization of compound F self-assembly micelles
It was found that conjugates can self-assemble into micelles at the concentration of 7.5 mg/mL. Transmission electron microscopy images revealed their spherical shapes and particle size, and zeta potential was measured using dynamic light scattering (Figure 8). The particle size of micelles was ~80–100 nm and zeta potential was about −0.23 mV.
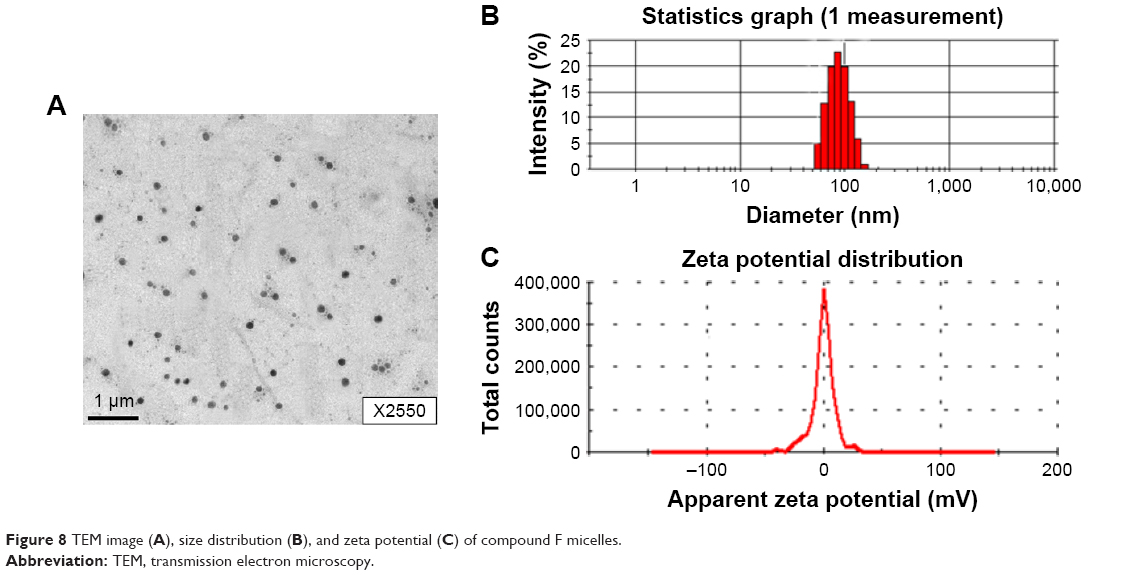 | Figure 8 TEM image (A), size distribution (B), and zeta potential (C) of compound F micelles. |
Drug release from conjugate
From the results, we can see that compound B has stronger in vitro cell cytotoxicity than 10-HCPT. After entering tumor cells, the hydrazone bond in compound F is to rupture to generate compound B and play efficacy so the release of compound B is important. As shown in Figure 9, the release of compound B from conjugate was significantly pH-dependent. Specifically, compound B was released rapidly from the conjugate for pH <6.0. After 1 h, the amounts of compound B released from the conjugate in solutions of pH 3.0, 4.0, 5.0, and 6.0 at 37°C were 87%, 79%, 72%, and 31%, respectively; however, in the solution of pH 7.4 and in plasma the releases were 2% and 5%, respectively. The less the pH values were, the more quickly the drug was released. After 4 h, as shown, the amount of compound B released under pH 5.0 at 37°C was almost 100%; however, in solutions of pH 6.0, 7.4 PBS, and plasma, the releases were 47%, 8%, and 15%, respectively. In addition, the amounts that compound B released from the conjugate in pH 7.4 PBS or plasma after 48 h were all <30%. These results indicate that in blood and at physiological pH levels, the conjugate is relatively stable; however, in the acidic microenvironment of tumor cells, particularly in lysosomes, compound B would be released quickly. This means that the drug delivery system exerts a better antitumor effect and while exerting a smaller toxicity on normal tissues.
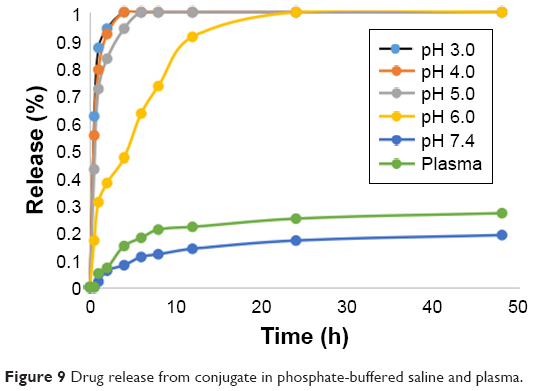 | Figure 9 Drug release from conjugate in phosphate-buffered saline and plasma. |
In vitro cytotoxicity
The antitumor activities of 10-HCPT and compound F were tested on three cancer cell lines using MTT assay. The results showed that compound F had the strongest cytotoxicity for the three cell lines. Compound B had stronger cytotoxicity than 10-HCPT or compound C, which probably because its molecular polarity was reduced and the ability to pass through the cell membrane was improved or presumably because its molecular structure has higher topoisomerase I affinity and led to higher cytotoxicity. Compound C had stronger cytotoxicity than 10-HCPT. This probably because after entering cells the hydrazone bond in its molecule ruptured to generate compound C and produce stronger cytotoxicity. When conjugated with PEG, the cytotoxicity of 10-HCPT was much enhanced for the three cell lines (Figure 10). In contrast, PEG has no inhibitory effect. This is presumably because tumor cells lack an exocytosis pathway for macromolecules, which promotes the gathering of drug in cells to achieve a higher concentration. 5-Carbonyl caproic acid has almost no cytotoxicity for the three cell lines.
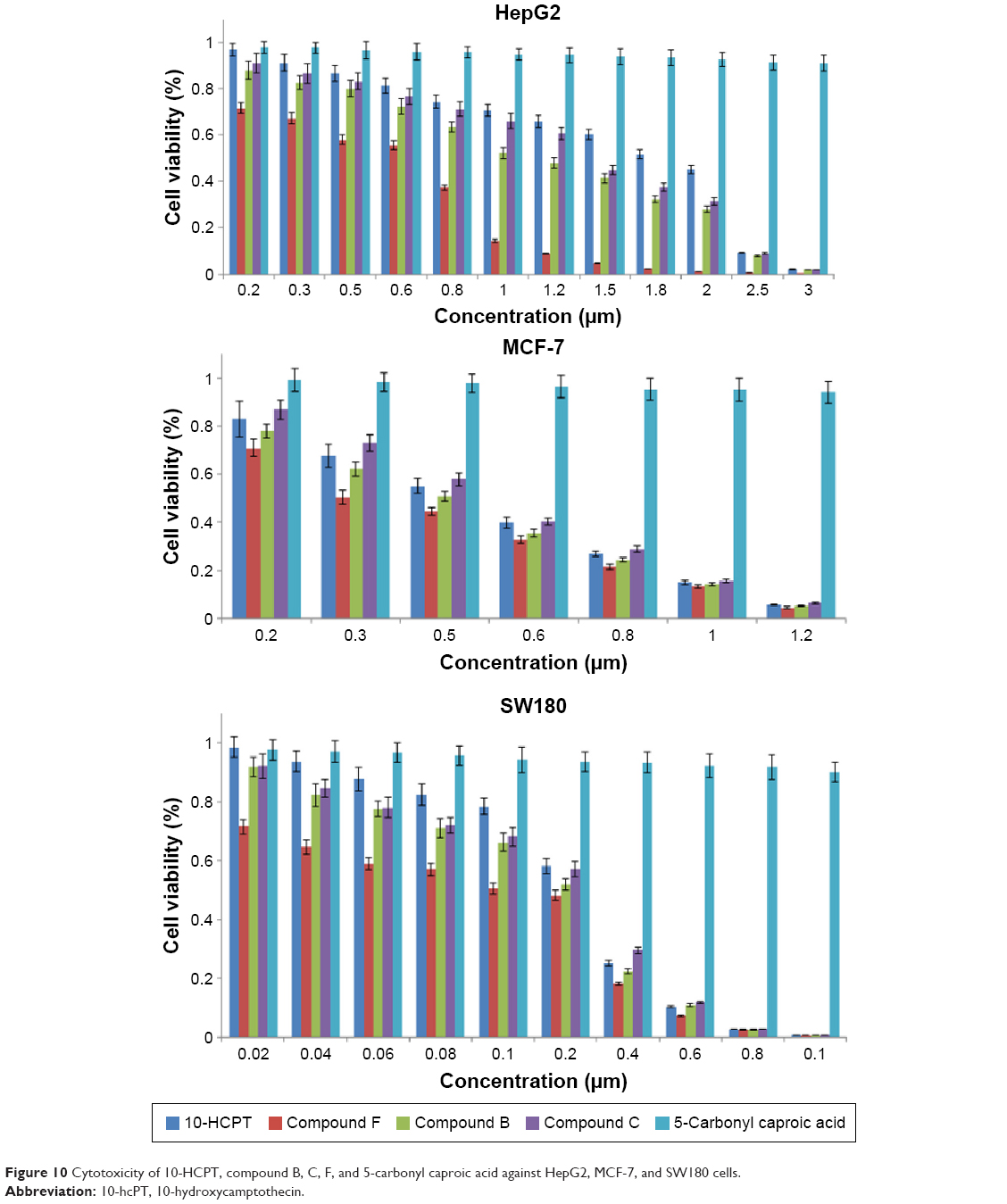 | Figure 10 Cytotoxicity of 10-HCPT, compound B, C, F, and 5-carbonyl caproic acid against HepG2, MCF-7, and SW180 cells. |
In vitro endocytosis
Endocytosis was examined using MCF-7 cells. Coumarin-6 was used as fluorescent marker by being incorporated into compound F mice. In vitro release of coumarin-6 from micelles was negligible (<1%) after 24 h (data not shown). This guaranteed that the fluorescence detected was attributed to coumarin-6 incorporated micelles.
As shown in Figure 11, the fluorescent signals were detected from tumor cells that were treated with micelles for 1 h, and became stronger as the incubation time increased. In addition, fluorescence of coumarin-6 was observed in the nuclei of MCF-7 cells after 1 h, suggesting the rapid internalization and nuclear transport of compound F micelles.
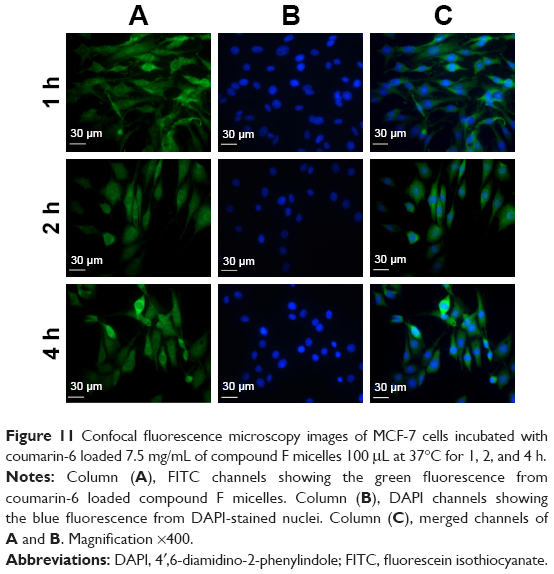 | Figure 11 Confocal fluorescence microscopy images of MCF-7 cells incubated with coumarin-6 loaded 7.5 mg/mL of compound F micelles 100 μL at 37°C for 1, 2, and 4 h. |
In vivo pharmacokinetics and biodistribution
The concentrations of compound F in the plasma and tissues of heart, liver, spleen, lung, kidney, brain, and tumor were measured after intravenous injection of compound F solution. The results are presented in Table 1. A value of P<0.05 was considered statistically significant, using SPSS.
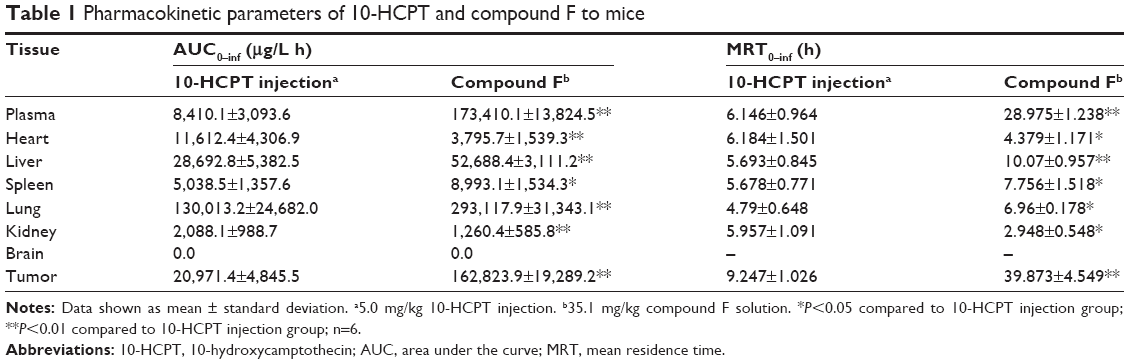 | Table 1 Pharmacokinetic parameters of 10-HCPT and compound F to mice |
As can be seen, the total area under the curve (AUC) and mean residence time (MRT) of compound F were significantly higher than those of 10-HCPT solution in plasma (Figure 12). It is suggested that compound F could circulate for a much longer time in the blood circulation system than 10-HCPT solution, which would result in their higher affinity to the tumor cells or extracellular spaces. Additionally, compared with 10-HCPT solution, the MRT0–inf values of compound F were 4.31-fold in tumors. The AUC0–inf values of compound F in tumor were 7.76-fold higher than those of 10-HCPT solution. These results indicated that a prolonged residence time supports reversible binding of the drug to topoisomerase I, which improved antitumor efficacy. Compound F also exhibited a higher accumulation in the liver, spleen, and lung. Drug levels in the heart and kidney were closely related to the inherent cardiac and renal toxicity. Therefore, compound F could reduce the corresponding toxic effects.
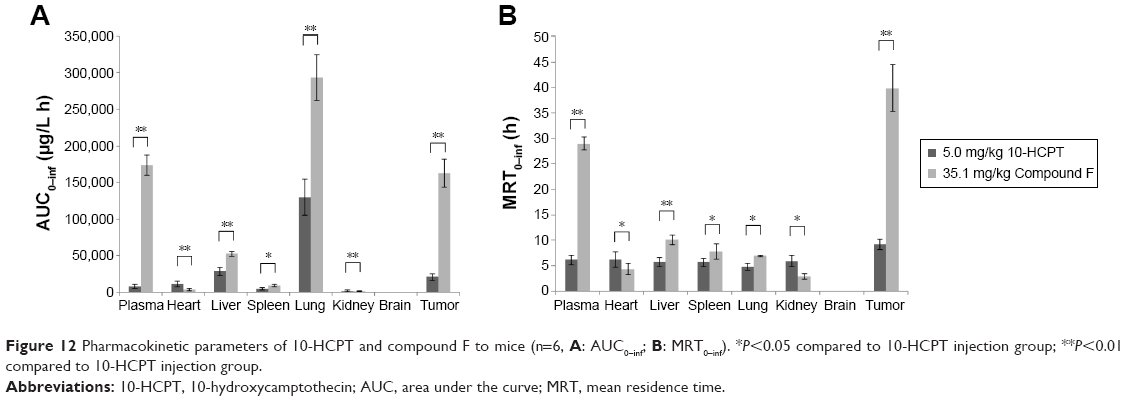 | Figure 12 Pharmacokinetic parameters of 10-HCPT and compound F to mice (n=6, A: AUC0–inf; B: MRT0–inf). *P<0.05 compared to 10-HCPT injection group; **P<0.01 compared to 10-HCPT injection group. |
In vivo antitumor efficacy
To provide in vivo evidence for the antitumor potential of compound F, antitumor efficacy was evaluated using an in vivo nude mouse model. As shown in Figure 13, the tumor volume of the group treated with compound F solution was significantly smaller than that of the group treated with either saline or 10-HCPT solution (P<0.05). Moreover, the strongest antitumor efficacy was observed in this group. An important consideration is potential toxicity, which is commonly assessed by analyzing the effect on animal behavior and body weight change. Body weight changes are shown in Figure 14. As can be seen, compound F did not cause significant loss of body weight, whereas 10-HCPT solution exhibited significant body weight loss (P<0.05), which means that compound F has lower toxicity than 10-HCPT.
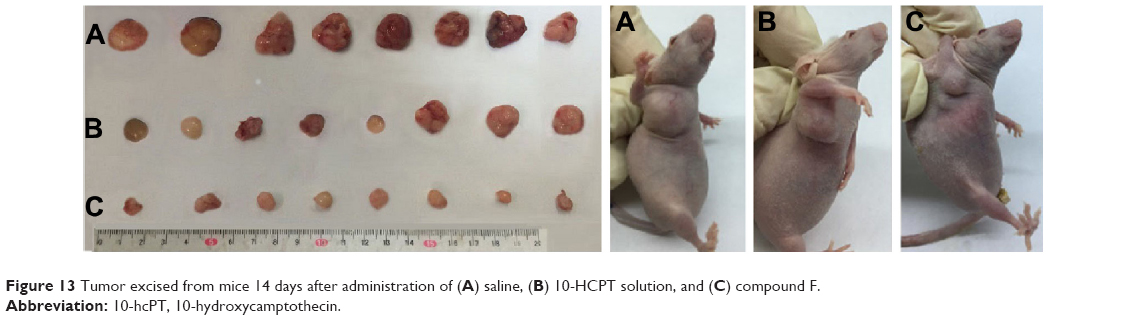 | Figure 13 Tumor excised from mice 14 days after administration of (A) saline, (B) 10-HCPT solution, and (C) compound F. |
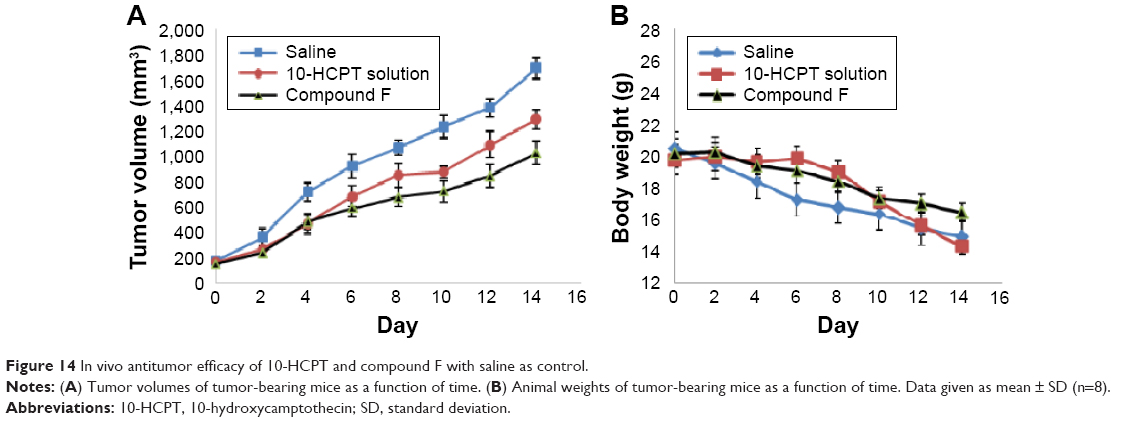 | Figure 14 In vivo antitumor efficacy of 10-HCPT and compound F with saline as control. |
Conclusion
This article proposes a new-targeted conjugate for delivering HCPT specifically to tumors. The molecular weight of PEG affects the yield of reaction. When using PEG2000, the reaction yield was <40%; however, when using PEG3400, the yield was <10% and unstable. When using PEG5000, the yield is too low to obtain a pure product. Since our main purpose is to improve drug solubility, we intentionally selected PEG2000; prolonged circulation and good tumor localization were observed.
Compounds B and F showed blue fluorescence while 10-HCPT showed yellow fluorescence and compound C showed violet fluorescence under 360 nm excitation, indicating that neither 10-HCPT nor compound C was taken in by the cells. It can therefore be concluded that the ester bond in the linker is stable during the endocytosis process.
A pH-responsive ternary conjugate was successfully synthesized for tumor-targeted delivery of 10-HCPT. It could significantly facilitate specific uptake by cancer cells via specific endocytosis and induce cell-dependent nuclear translocation. In addition, compound F showed a prolonged circulation time and preferential accumulation in tumors, resulting in enhanced therapeutic efficacy with better safety and lower toxicity. Overall, compound F could be a promising drug delivery system for cancer chemotherapy.
Acknowledgments
This work was supported by the major national scientific research projects (2015CB932103) and by the National Natural Sciences Foundation of China (No 81573364).
Disclosure
The authors report no conflicts of interest in this work.
References
Bailly C. Homocamptothecins: potent topoisomerase I inhibitors and promising anticancer drugs. Crit Rev Oncol Hematol. 2003;45(1):91–108. | ||
Kehrer DF, Soepenberg O, Loos WJ, Verweij J, Sparreboom A. Modulation of camptothecin analogs in the treatment of cancer: a review. Anticancer Drugs. 2001;12(2):89–105. | ||
Liu R, Li D, He B, et al. Anti-tumor drug delivery of pH sensitive poly(ethylene glycol)-poly(L-histidine)-poly(L-lactide) nanoparticles. J Control Rel. 2011;152(1):49–56. | ||
Wu XL, Kim JH, Koo H, et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconjug Chem. 2010;21(2):208–213. | ||
Na Liu, Bingqiang Li, Chu Gong, Yuan Liu, Yanming Wang, Guolin Wu. A pH- and thermo-responsive poly(amino acid)-based drug delivery system. Colloids Surf B Biointerfaces. 2015;136:562–569. | ||
Pang X, Jiang Y, Xiao Q, Leung AW, Hua H, Xu C. pH-responsive polymer–drug conjugates: design and progress. J Control Release. 2016;222:116–129. | ||
Li G, Li Y, Tang Y, et al. Hydroxyethyl starch conjugates for improving the stability, pharmacokinetic behavior and antitumor activity of 10-hydroxy camptothecin. Int J Pharm. 2014;471(1–2):234–244. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Sheng Y, You Y, Chen Y. Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int J Pharm. 2016;512(1):1–13. | ||
Chen W, Meng F, Cheng R, Zhong Z. pH-Sensitive degradable polymersomes for triggered release of anticancer drugs: a comparative study with micelles. J Control Release. 2010;142(1):40–46. | ||
Zhou Z, Murdoch WJ, Shen Y. A linear polyethylenimine (LPEI) drug conjugate with reversible charge to overcome multidrug resistance in cancer cells. Polymer. 2015;76:150–158. | ||
Ma Y, Fan X, Li L. pH-sensitive polymeric micelles formed by doxorubicin conjugated prodrugs for co-delivery of doxorubicin and paclitaxel. Carbohydr Polym. 2016;137:19–29. | ||
Volk T, Jähde E, Fortmeyer HP, Glüsenkamp KH, Rajewsky MF. pH in human tumour xenografts: effect of intravenous administration of glucose. Br J Cancer. 1993;68(3):492–500. | ||
Gu Y, Zhong Y, Meng F, Cheng R, Deng C, Zhong Z. Acetal-linked paclitaxel prodrug micellar nanoparticles as a versatile and potent platform for cancer therapy. Biomacromolecules. 2013;14(8):2772–2780. | ||
Zou J, Jafr G, Themistou E, et al. pH-sensitive brush polymer-drug conjugates by ring-opening metathesis copolymerization. Chem Commun (Camb). 2011;47(15):4493–4495. | ||
Gillies ER, Goodwin AP, Fréchet JM. Acetals as pH-sensitive linkages for drug delivery. Bioconjug Chem. 2004;15(6):1254–1263. | ||
Park JH, Cho YW, Son YJ, et al. Preparation and characterization of self-assembled nanoparticles based on glycol chitosan bearing adriamycin. Colloid Polym Sci. 2006;284(7):763–770. | ||
Hu FQ, Liu LN, Du YZ, Yuan H. Synthesis and antitumor activity of doxorubicin conjugated stearic acid-g-chitosan oligosaccharide polymeric micelles. Biomaterials. 2009;30(36):6955–6963. | ||
Yoo HS, Lee EA, Park TG. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J Control Release. 2002;82(1):17–27. | ||
Zhu S, Hong M, Tang G, et al. Partly PEGylated polyamidoamine dendrimer for tumor-selective targeting of doxorubicin: the effects of PEGylation degree and drug conjugation style. Biomaterials. 2010;31(6):1360–1371. | ||
Paranjpe PV, Chen Y, Kholodovych V, Welsh W, Stein S, Sinko PJ. Tumor-targeted bioconjugate based delivery of camptothecin: design, synthesis and in vitro evaluation. J Control Release. 2004;100(2):275–292. | ||
Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev. 2003;55(2):217–250. | ||
Hong M, Zhu S, Jiang Y, et al. Novel anti-tumor strategy: PEG-hydroxycamptothecin conjugate loaded transferrin-PEG-nanoparticles. J Control Release. 2010;141(1):22–29. | ||
Lage H. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 2008;65(20):3145–3167. | ||
Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer–drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–1213. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymericmicelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Davis ME, Chen ZG, Shin DM, Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Omelyanenko V, Kopečková P, Gentry C, Kopecek J. Targetable HPMA copolymer-adriamycin conjugates. Recognition, internalization, and subcellular fate. J Control Release. 1998;53(1–3):25–37. | ||
Minko T, Kopečková P, Kopeček J. Efficacy of the chemotherapeutic action of HPMA copolymer-bound doxorubicin in a solid tumor model of ovarian carcinoma. Int J Cancer. 2000;86(1):108–117. | ||
Kopeček J. Polymer-drug conjugates: origins, progress to date and future directions. Adv Drug Deliv Rev. 2013;65(1):49–59. | ||
He Q, Gao Y, Zhang L, et al. A pH-responsive mesoporous silica nanoparticles-based multi-drug delivery system for overcoming multidrug resistance. Biomaterials. 2011;32(30):7711–7720. | ||
Lee ES, Na K, Bae YH. Super pH-sensitive multifunctional polymeric micelle. Nano Lett. 2005;5(2):325–329. | ||
Sarmiento GP, Rouge PD, Fabian L, et al. Efficient synthesis of chiral Δ2-1,3,4-thiadiazolines from α-pinene and verbenone. Tetrahedron: Asymmetry. 2011;22(20–22):1924–1929. | ||
Nami N, Mighani H, Kia N, Sadatfaraji H. Synthesis of Some Polyamides from Bis(thiosemicarbazone)acenaphthenequinone. Chem Sci Trans. 2013;2(S1):S267–S275. | ||
Ghaemy M, Mighani H, Alizadeh R. Synthesis and characterization of Schiff-base containing polyamides. Chin J Polym Sci. 2011;29(2):149–155. | ||
Minko T, Paranjpe PV, Qiu B, et al. Enhancing the anticancer efficacy of camptothecin using biotinylated poly(ethylene glycol) conjugates in sensitive and multi-drug resistant human ovarian carcinoma cells. Cancer Chemother Pharmacol. 2002;50(2):143–150. | ||
Goldwirt L, Lemaitre F, Zahra N, Farinotti R, Fernandez C. A new UPLC-MS/MS method for the determination of irinotecan and 7-ethyl-10-hydroxycamptothecin (SN-38) in mice: application to plasma and brain pharmacokinetics. J Pharm Biomed Anal. 2012;66:325–333. | ||
Khan S, Ahmad A, Guo W, Wang YF, Abu-Qare A, Ahmad I. A simple and sensitive LC/MS/MS assay for 7-ethyl-10-hydroxycamptothecin (SN-38) in mouse plasma and tissues: application to pharmacokinetic study of liposome entrapped SN-38(LE-SN38). J Pharm Biomed Anal. 2005;37(1):135–142. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.