")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
A Novel Variant and a Missense Variant Identified in the DKC1 Gene in Three Chinese Familieswith Dyskeratosis Congenita
Authors Yuan C , Deng D, Yang J, Liu S, Qian Q, Chen M, Zhou S , Li Y, Li M
Received 5 May 2022
Accepted for publication 19 August 2022
Published 9 September 2022 Volume 2022:15 Pages 1837—1845
DOI https://doi.org/10.2147/CCID.S371794
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Chunyu Yuan,1,* Dongmei Deng,2,* Jianqiu Yang,1 Simeng Liu,3 Qihong Qian,4 Min Chen,1 Shengru Zhou,1 Yujiang Li,5 Min Li1,4
1Department of Dermatology, Dushu Lake Hospital Affiliated to Soochow University (Medical Center of Soochow University, Suzhou Dushu Lake Hospital), Suzhou Ctiy, Jiangsu, People’s Republic of China; 2Health Management Center, The Fifth People’s Hospital of Hainan Province, Haikou Ctiy, Hainan, People’s Republic of China; 3Department of Dermatology, Aerospace Center Hospital, Beijing Ctiy, People’s Republic of China; 4Department of Dermatology, The First Affiliated Hospital of Soochow University, Suzhou Ctiy, Jiangsu, People’s Republic of China; 5Department of Dermatology, Sanmenxia Central Hospital, Sanmenxia Ctiy, Henan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yujiang Li, Department of Dermatology, Sanmenxia Central Hospital, Middle Road of Yaoshan, Hubin District, Sanmenxia, Henan, 472000, People’s Republic of China, Tel +86-13525857005, Email [email protected] Min Li, Department of Dermatology, the First Affiliated Hospital of Soochow University, No. 188 Shizi Road, Gusu District, Suzhou, Jiangsu, 215006, People’s Republic of China, Tel +86-18936140383, Email [email protected]
Purpose: Dyskeratosis congenita (DC) is an inherited telomere biology disorder characterized clinically by mucocutaneous triad of reticulate hyperpigmentation, nail changes and oral leukoplakia. Bone marrow failure, pulmonary fibrosis and malignancies are the mainly life-threatening causes. There are X-linked recessive, autosomal dominant and autosomal recessive patterns of DC. DKC1 is the most common pathogenic mutation gene responsible for X-linked DC, and it encodes a protein, dyskerin, which is a component of telomerase holoenzyme complex essential for telomere maintenance. Patients with DC have very short telomeres, but the precise pathogenic mechanism remains unclear. This study aimed to identify the causative mutations in the DKC1 gene in three Chinese families with the X-linked form of DC.
Patients and Methods: Three Chinese families with DC were included in this study. Whole exome sequencing and Sanger sequencing were performed to clarify the mutation of DKC1 gene. Measurement of relative telomere length through qPCR. Predictions of protein structure and function were performed using bioinformatics tools, including I-TASSER, Polyphen-2 and SIFT.
Results: There were four males with DC and a female carrier in three Chinese pedigrees. The novel mutation c.92A>C (p. Q31P) and the missense mutation c.1058C>T (p. A353V) in DKC1 were identified. Both mutations locally changed the structure of dyskerin. Variant Q31P and A353V were predicted to have “deleterious” and “natural” effects on the function of dyskerin, respectively.
Conclusion: The novel variant and missense variant detected in the DKC1 gene improve our understanding of DC and broaden the mutation spectrum of the DKC1 gene.
Keywords: dyskeratosis congenita, mucocutaneous triad, DKC1 gene, mutation
Introduction
Dyskeratosis congenita (DC) is a rare inherited disease, also known as Zinsser–Cole–Engman syndrome, with an estimated prevalence of 1/1,000,000. Three hereditary patterns have been recognized: X-linked recessive (XLR, OMIM #305000) (accounting for about 30% of the cases), autosomal dominant (AD, OMIM #127550) and autosomal recessive (AR, OMIM #224230).1 It occurs predominantly in males and clinically presents with a mucocutaneous triad of reticulate hyperpigmentation, nail changes and oral leukoplakia.2,3 In addition, hair and teeth involvements, palm-plantar hyperkeratosis and hyperhidrosis and other cutaneous manifestations are reported in DC.2 A variety of other abnormalities of digestive system, cardiovascular system and bones and so on are also described in DC patients.2,3 The cardinal and life-threatening extracutaneous manifestations are bone marrow failure, pulmonary disorders and malignancies, accounting for 60–70%, 15–18% and 8–10% of the deaths in DC, respectively.2 It is clear that a patient with DC has a high risk of cancers, such as squamous cell carcinoma and lymphoma.4 In general, the prognosis of DC is poor and the mean age of death is the third to fifth decade of life.2,4 Actually, the phenotype is variable where some patients with severe multi-system involvement in early life and others with mild presentations.4
DC is now primarily regarded as a telomere biology disorder of defective telomere maintenance.4 There are 19 mutated genes reported in association with DC and they shorten the length of telomere directly or indirectly.2 DKC1 (OMIM #300126) is the most common pathogenic mutation gene where X-linked recessive pattern is noticeable, maps to Xq28 and encodes a highly conserved protein called dyskerin.1,5 It participates in premessenger RNA splicing events and pseudouridylation of ribosomal RNA and spliceosomal small nuclear RNAs.2 Furthermore, as a part of telomerase holoenzyme complex, dyskerin also interacts with a particular H/ACA RNA, the telomerase RNA component (TERC, or hTR in humans), regulating the accumulation of hTR. Consequently, dyskerin is crucial to the activity of telomerase in elongation of telomeres.1,2,4 Likewise, mutations in TERC (OMIM #602322) and telomerase reverse transcriptase, TERT (OMIM #613989) have been found in AD-DC patients.1,5 It is also been identified that AR-DC is a result of mutations in NHP2, NOP10, TERT, USB1, TCAB1, CTC1, and RTEL1.1,2,5
Collectively, DC has a wide spectrum of clinical characteristics and genetic heterogeneity. Here, we reported three Chinese DC families associated with DKC1 gene mutations, and identified a novel mutation c.92A>C (p. Q31P) and a missense mutation c.1058C>T (p. A353V) in the DKC1 gene.
Patients and Methods
Patients
Family 1 was a three-generation Chinese DC pedigree with two affected males (Figure 1A). The proband (III2) was a 23-year-old man presented with skin pigmentation for more than 10 years. Physical examination showed mottled and reticulate hyperpigmentation of the face, neck, trunk and upper arms (Figure 1B–D), leukoplakia on the dorsum of the tongue (Figure 1E) and mild fingernail changes (Figure 1F) without systemic involvement. His elder brother in this family had similar clinical features.
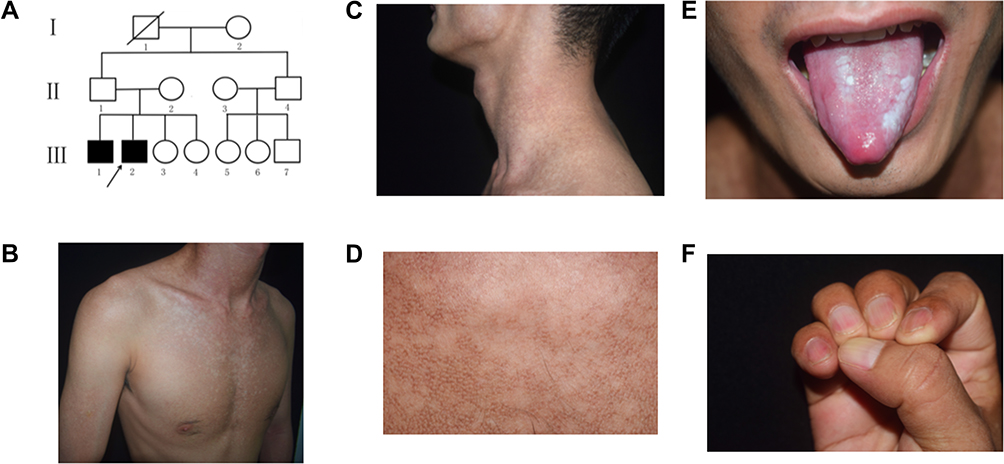 |
Figure 1 Pedigree and clinical features of the proband in family 1. (A) The pedigree of the family. (B and C) The mottled and reticulate pigmentation on the chest and neck. (D) Detail of reticulate hyperpigmentation of the neck. (E) Oral leukoplakia. (F) Mild longitudinal ridges, onychoschizia and brown lines of fingernails. The black arrow indicated the proband. |
Family 2 was a three-generation Chinese DC lineage with one affected male and one carrier (Figure 2A). The proband (III2) was a 17-year-old boy with the complaint of skin hyperpigmentation and nail changes for about 10 years. The reticulate hyperpigmentation was initially from the ears and then gradually spread to face, neck and limbs (Figure 2B). White nails, atrophy and longitudinal ridges were found in 20 nails (Figure 2C). He developed oral disorder at 14 years of age and oral examination showed leukoplakia and ulcer on the tongue (Figure 2D). His mother (II5) was a carrier with normal clinical manifestations as same as unaffected person in this family and none of them had systemic disorders. Other relatives were clinically normal.
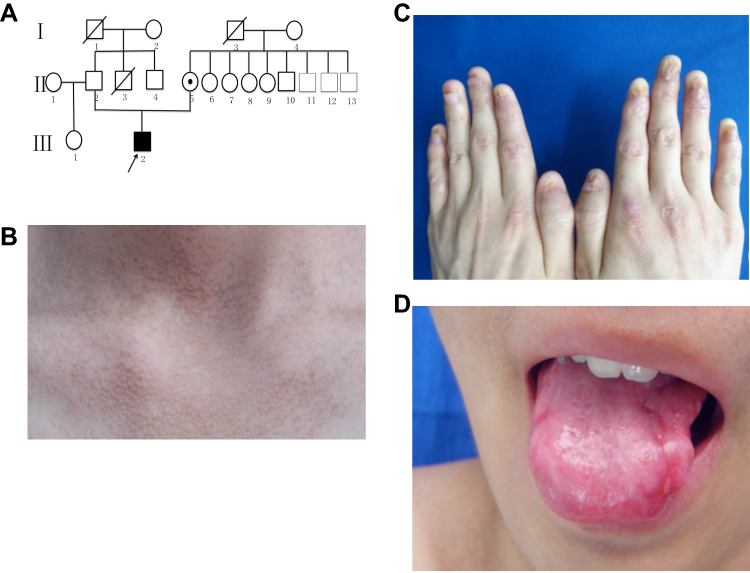 |
Figure 2 Pedigree and clinical features of the proband in the family 2. (A) The pedigree of the family. (B) The reticulate hyperpigmentation on the neck. (C) Fingernails atrophy. (D) White spots and ulcers on the tongue. |
The last case was a 22-year-old man who developed hyperpigmentation on the face, neck and upper trunk since childhood, and his parents were clinically normal (Figure 3A). At the age of 10, he was found to have abnormal features of nails and tongue. Moreover, he suffered from trachoma, pulmonary fungal infection and abnormal liver function and macrocytic anemia. His complete blood count included a red-blood-cell count (RBC) of 3.29×1012/L, hemoglobin level of 113 g/L, mean corpuscular volume of 105.3fL, and platelet count of 166×109/L. Physical examination revealed a poor health, reticulate hyperpigmented macules of the face, neck and upper trunk (Figure 3B), atrophic hypopigmented spots on the back of hands (Figure 3C), nail dystrophy (Figure 3C and D) and partial loss of the tip of the tongue (Figure 3E). Other routine laboratory examinations as well as lung function were within normal limits. Other relatives were clinically normal.
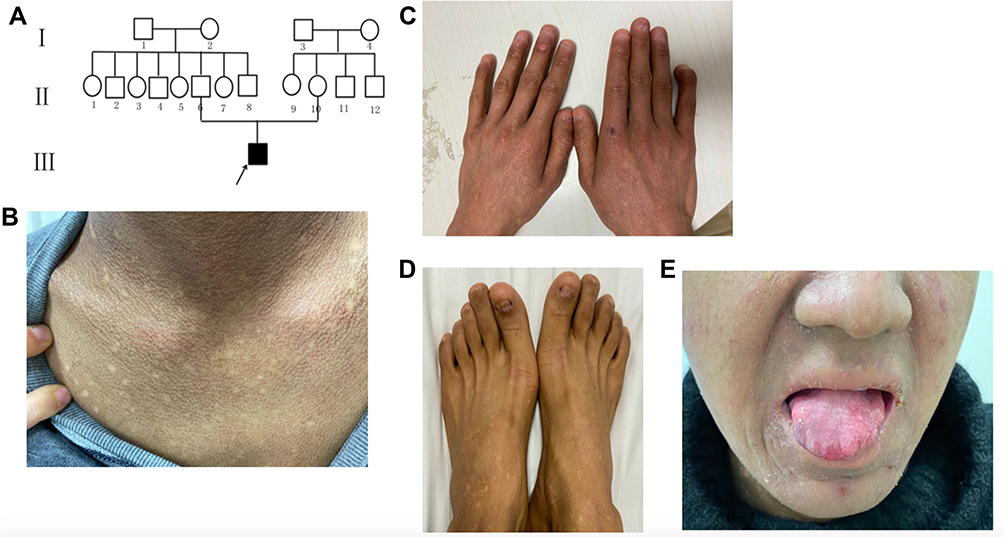 |
Figure 3 Pedigree and clinical features of the proband in family 3. (A) The pedigree of the family. (B) Reticulate scattered hyperpigmentation with hypopigmented macules on the neck. (C and D) Nails atrophy and dystrophy. (E) Oral leukoplakia and partial loss of the tongue tip. |
Whole Exome Sequencing and Data Analysis
The process of whole exome sequencing was done in Shanghai We-Health Biomedical Technology Co., Ltd. All genomic DNA samples were extracted from peripheral blood using commercial kit (TIANGEN, China). Onedrop OD1000 spectrophotometer and agarose gel electrophoresis were used to assess the quantity/quality of DNA. Exome capture was performed with xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc., Iowa, United States) and 150 base pair paired end sequencing was executed using the Illumina HiSeq4000 platform (San Diego, CA). Raw reads were aligned using the Burrows–Wheeler Aligner (BWA) and SAMtools. After removing duplicates from sorted alignment using Picard, mutations were recognized through using the Genome Analysis Toolkit (GATK v3.70). Next, the DKC1 gene mutation was confirmed by Sanger sequencing.
Sanger Sequencing
Genomic DNA was extracted from blood samples using TIANamp Blood DNA Kit (Tiangen Biotech, Beijing, China). Primers were designed with the Primer3Plus software (http://www.primer3plus.com/cgibin/dev/primer3plus.cgi). Polymerase chain reaction (PCR) amplification was performed as described previously.6 The PCR products were analyzed by 2% agarose gel electrophoresis. Purified PCR products were sequenced directly using an ABI3730xl DNA Analyzer (AppliedBiosystems, FosterCity, California, USA).
Measurement of Telomere Length
The relative telomere length was calculated by quantitative PCR (qPCR). The normalizing control gene was HBG. According to the manufacturer’s instruction, targeted DNA was added to a reaction containing the pair of primers (telomere or HBG) and SYBR ® Premix Ex Taq (MEIXUAN Biotech, Shanghai, China), in a total reaction volume of 20ul. Then, qPCR was performed on Applied Biosystems 7300/7500/7500 Fast Real-Time PCR System and StepOnePlusTMReal-Time PCR System. The standard process was 1 cycle of 95°C for 5 min followed by 40 cycles of 95°C for 15 sec, 58°C for 30 sec and 72°C for 45 sec. Data collection and analysis used 7500 Fast Real-Time PCR software v2.0. Data were collected from repeated reactions for each sample. The standard deviation of Ct value was lower than 0.5, then the repeated values were received. The relative telomere length of normal population was regarded as 1. The final results were shown as 2−ΔΔCt, which represented the relative telomere length of DC patients in comparison with the normal population of certain age.
Primer sequences for telomere and HBG were as follows:
TEL - forward: 5’-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3’.
TEL - reverse: 5’-GGCTTGCCTACCCTTACCCTTACCCTTACCCTTACCCT-3’.
HBG - forward: 5’-GCTTCTGACACAACTGTGTTCACTAGC-3’.
HBG - reverse: 5’-CACCAACTTCATCCACGTTCACC-3’.
DKC1 Protein Structure Visualization and Function Prediction
Three-dimensional structures of DKC1 protein encoded by the wild-type and mutated DKC1 gene (NM_001363.5) were predicted using I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER-MR/). Furthermore, we visualized the architectures using PyMOL (PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC). Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) were applied to predict the possible impact of an amino acid substitution on the structure and function of the DKC1 protein.
Results
Clinical Features
All affected individuals were males and had classical mucocutaneous features (Figures 1–3). Skin pigmentation and nails changes developed in about the first decade of life.
The Mutations of DKC1 Gene
A novel missense variant c.92A>C (p. Q31P) in exon 3 of the DKC1 gene (NM_001363.5) was identified in affected members (III1, 2) in family 1 (Figure 4A), thus confirming the diagnosis of DC in the proband and X-linked recessive inherited pattern. Other relatives were clinically normal and unable to go to hospital for sequencing. In the second pedigree, the proband (III2) was identified to have a transition mutation of C to T in the coding region at nucleotide position 1058 (Figure 4B) which caused a replacement of the alanine at amino acid residue 353 by valine in dyskerin. His mother (II5) carried a heterozygous mutation of 1058C to T (Figure 4C). No such mutation was found in his father, and other family members had no genetic examination. Similarly, the diagnosis of DC was definite and X-linked recessive inheritance was clear. As the only one affected individual of family 3, the proband (III1) showed the same variant c.1058C>T in exon 11 of the DKC1 gene (Figure 4D) resulting in an amino acid change of A353V in dyskerin, however, his mother did not have sequencing because of death. His father was genetically normal.
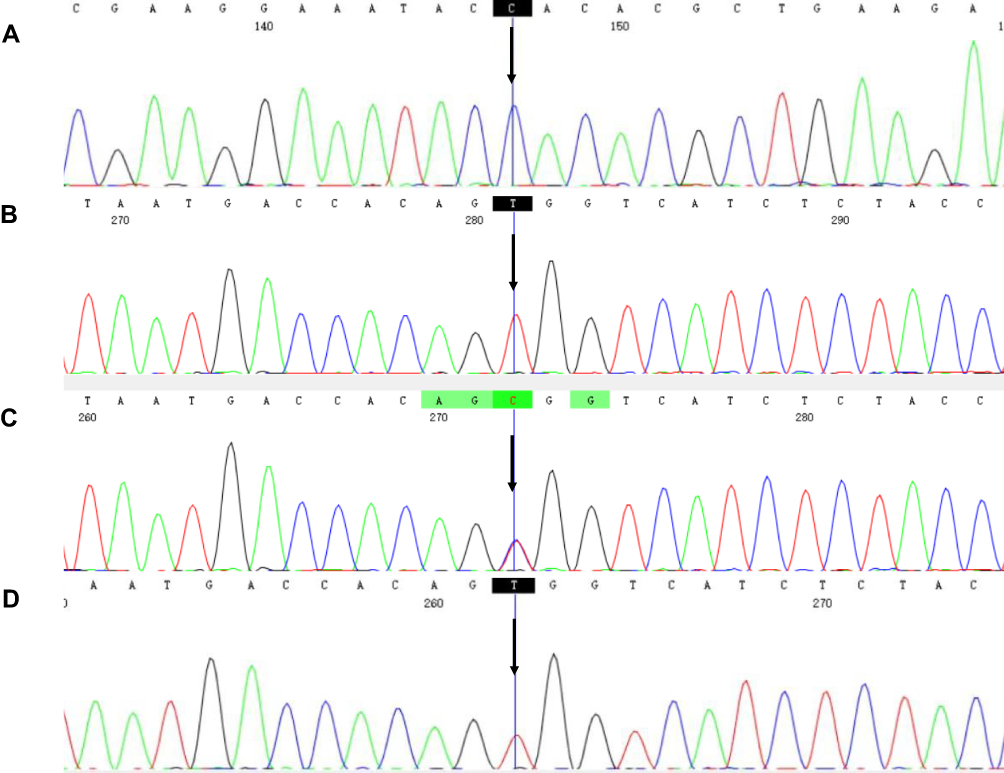 |
Figure 4 DKC1 gene mutations in three DC families. (A) A hemizygous mutation c.92A>C was identified in III1 and III2 of family 1. (B) Mutation c.1058C>T was identified in the proband (III2) in family 2. (C) A heterozygous mutation c.1058C>T was detected in II5 in family 2. (D) A hemizygous mutation c.1058C>T was identified in the proband in family 3. The black arrow indicated mutated bases. |
The Telomere Length
Four DC patients in three families conducted the telomere length testing. The values of 2−ΔΔCt were 0.831, 0.803, 0.887 and 0.815, respectively, which indicated that the telomere lengths of our DC patients were shorten.
The Prediction of Potential Effects of Mutations
The variant c.92A>C (p. Q31P) was predicted to be “possibly damaging” and “deleterious” with Polyphen-2 and SIFT, respectively. The variant c.1058C>T (p. A353V) was predicted to be “benign” and “natural” with Polyphen-2 and SIFT, respectively.
The mutation c.92A>C caused the replacement of a polarly amino acid glutamine by a non-polarly proline. In a three-dimensional structure, mutated PRO-31 could form a hydrogen bond with VAL-27 at the distance of 3.1 Å (Figure 5A). Wild-type GLN-31 could form a hydrogen bond with VAL-27 and the distance was 3.3Å, GLN-31 also could form a hydrogen bond with SER-280 at the distance of 3.5 Å (Figure 5B). Variant c.1058C>T (p. A353V) could not change the polarity of amino acids. Mutated VAL-353 could form a hydrogen bond with THR-351, THR-357, and SER-356 at the distance of 2.9 Å, 3.3 Å and 2.4 Å, 2.6 Å and 2.4 Å, respectively (Figure 5C). Wild-type ALA-353 could form a hydrogen bond with THR-352, THR-357 and SER-356; the distance was 2.7Å, 3.4 Å and 2.4 Å, 2.0 Å and 2.5 Å, respectively (Figure 5D). Both mutations locally affected the protein structure.
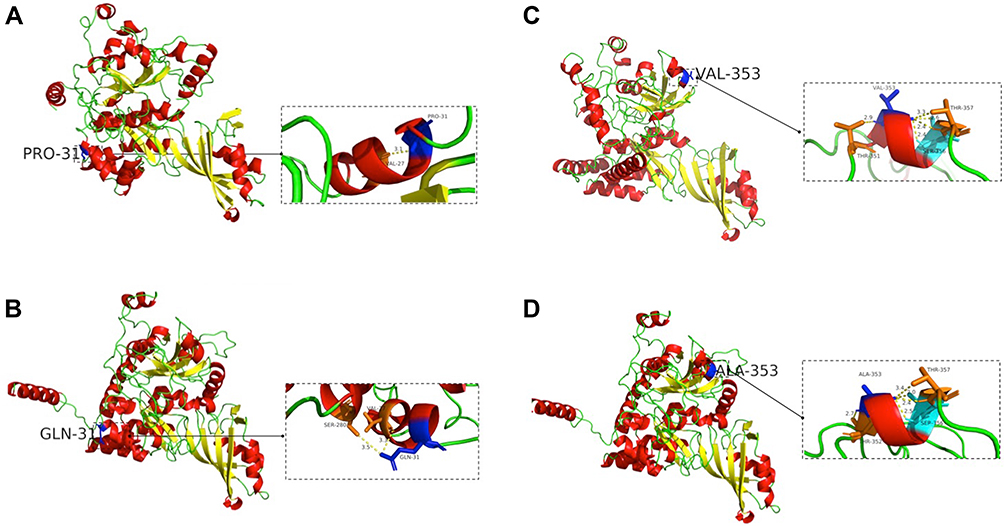 |
Figure 5 Three-dimensional structure of dyskerin. (A) PRO replaced GLN at position 31, and the hydrogen bond between amino acids changed. (B) Three-dimensional structure of wild-type with GLN 31. (C) ALA was replaced by VAL at position 353, and the hydrogen bond between amino acids changed. (D) Three-dimensional structure of wild-type with ALA 353. The left picture showed overall map of protein three-dimensional structure, and the right small picture showed partial map of protein three-dimensional structure. |
Discussion
According to diagnostic criteria,1 our four patients in three families with mucocutaneous triad (reticulate pigmentation, nail dystrophy, and oral leukoplakia) were considered to have classic DC. A research about DC patients in Chinese mainland shows that 87% of the patients have mucocutaneous symptoms and 13% of the patients with two or three symptoms of the classical triad.3 Abnormal skin pigmentation usually appears within 10 years of age.2 Reticulate hyperpigmentation is a typical feature, but hypopigmentation is also been reported.7 Lesions tend to occur on sun-exposed areas such as the neck, trunk and limbs.7–9 Commonly reported nail changes are atrophy and longitudinal ridges, and involvement of the fingernails is more severe than toenails.2,10 As the most common dental finding, oral leukoplakia is associated with an increased risk of head and neck squamous cell carcinoma (HNSCC) (primarily tongue) and approximately 30% of oral leukoplakia can be transformed to carcinoma.11,12 It is investigated that erosive oral leukoplakia and carcinoma develop about 20–30 years of age.12 In general, DC-affected individuals have 100-fold higher risk for developing HNSCC and anogenital SCC compared with general population.13 Significant cancers in DC are HNSCC, acute myeloid leukemia, non-Hodgkin lymphoma and anogenital SCC.13
Other mucocutaneous features include hair loss, early graying and palmoplantar hyperkeratosis and so on. A study shows that the severity of mucocutaneous phenotypes has a positive relation with higher-risk genotypes and poorer prognosis.14 Therefore, careful examination of mucocutaneous features is important for diagnosis and prediction of outcomes. Moreover, close monitoring should be suggested once patients are diagnosed with DC for preventing malignancies. In patients with classic mucocutaneous features, the diagnosis of DC is apparent. However, some patients present with aplastic anemia lacking mucocutaneous features and the diagnosis is challenging.
The deficiency of telomere biology resulting in short telomeres and limitation of cellular replication can affect multiple organs, especially those undergoing rapid replication.4 Our four DC patients were confirmed to have shorter telomere lengths than normal people of the same age. Moreover, the proband of family 3 had infections, liver complication and anemia. The clinical complications include bone marrow failure, pulmonary fibrosis, liver disease and immunologic abnormalities and other medical disorders.4 Bone marrow failure is the principal cause of premature mortality. It is estimated that 80% of the patients with classic DC develop at least one cell lineage cytopenia by 30 years of age.15 Then it progresses into pancytopenia and eventually evolves into myelodysplastic syndromes (MDS).13 Because of low numbers of B-lymphocytes and dysfunction of T-lymphocytes, DC-affected individuals suffer from recurrent infections including virus, fungi and bacteria.16 Pulmonary complications are reported in more than 20% of the DC patients, and pulmonary fibrosis is the most frequent pulmonary manifestation.17 The mutation of DKC1, TERC and PARN genes is related to pulmonary fibrosis.17,18 The clinical management of DC is complex, and allogeneic hematopoietic stem cell transplant (allo-HSCT) is the only curative therapy for bone marrow failure.1
In our study, all of the patients were males and their parents and sisters presented with normal phenotype, suggesting the X-linked recessive inheritance in these three pedigrees. The substitution of nucleotide 92A with C in exon 3 of the DKC1 gene leading to a Q31P mutation in family 1 is not reported before, and a mutation of 1058C to T in exon 11 of the DKC1 gene leading to an A353V mutation in the last two families has been reported several times.
The DKC1 gene mutation can be found in XLR-DC and Hoyeraal-Hreidarsson syndrome (HHS).4 HHS is considered as a severe variant form of DC, and can be diagnosed by cerebellar hypoplasia combined with DC-related features or four of the six common HHS features: intrauterine growth retardation, developmental delay, microcephaly, cerebellar hypoplasia, immunodeficiency, or aplastic anemia.4,15 As reported, the mutation of c.1156G>A in the DKC1 gene is found both in a DC patient and an HHS patient in one family.19 Since A353V mutation is also found in an HHS case, showing the same mutation has widely various phenotypes.20 DC-affected males owing to DKC1 mutations often present with classic mucocutaneous triad and early onset bone marrow failure.20 Female carriers with DKC1 pathogenic mutations occasionally show subtle DC-related phenotypes because of skewed X-chromosome inactivation and epigenetics and other mechanisms.21
To our knowledge, DKC1 gene contains 15 exons that are translated into a ~58kDa L-shaped protein, dyskerin, which primarily presents with five domains: pseudouridine synthase domain (TRUB), pseudouridine synthase and archaeosine transglycosylase domain (PUA), dyskerin-like domain (DKCLD), nuclear and nucleolar localization signals (NLS/NoLS), amino-terminal extension (NTE), and carboxy-terminal extension (CTE).22,23 As previously noted, mutations found in DKC1 mainly cause amino acid substitutions. Mutation sites are primarily distributed in exons 2–6 and 9–12, and c.1058C>T (p. A353V) in exon 11 is described as the most frequent site which is known to occur de novo in many cases.6,24,25 The cause of this hotspot site may be that CpG-to-TpG mutation is relatively common in human DNA.26 A353V mutation localizes to the PUA domain, the RNA-binding motif in dyskerin, and the transformation of amino acid affects the interaction between dyskerin and H/ACA RNAs.27 A study reports that A353V mutation has the most severe effect on the telomere maintenance. The mechanism of DC may be related to WNT signaling.28 The mutation of c.92A>C in exon 3 causes replacement of a polarly amino acid glutamine by a non-polarly proline. Exons 3–5 encode the DKCLD domain, in which mutations may affect the interaction within domains.23
As known using PolyPhen-2 and SIFT softwares, Q31P mutation may have a damaging impact on the function of dyskerin. However, the specific mechanism is unclear. We suspect this mutation leads to malfunction of dyskerin resulting in phenotypes of DC. Further studies are necessary to understand the role of mutation in the pathogenesis of DC.
Conclusion
In summary, a novel mutation c.92A>C (p. Q31P) in the DKC1 gene was detected in family 1, and a missense mutation c.1058C>T (p. A353V) in the DKC1 gene was detected in the last two families. The substitution of amino acid Q31P was predicted to locally change the structure of dyskerin and to be deleterious. Similarly, the substitution of amino acid A353V was predicted to locally change the structure of dyskerin and to be natural. These results improve our understanding of DC and broaden the mutation spectrum of the DKC1 gene.
Abbreviations
DC, dyskeratosis congenita; HNSCC, head and neck squamous cell carcinoma; MDS, myelodysplastic syndromes; allo-HSCT, allogeneic hematopoietic stem cell transplant; HHS, Hoyeraal–Hreidarsson syndrome; PUA, pseudouridine synthase and archaeosine transglycosylase domain; DKCLD, dyskerin-like domain.
Ethics Statement
Ethics Committee at the Suzhou Dushu Lake Hospital approved the study. This study was complied with the Declaration of Helsinki. The participants provided their written informed consent to participate in this study and allowed to publish images and clinical details.
Acknowledgments
We want to thank all patients and their families who participated in this study.
Funding
This project was supported by the Suzhou Science and Technology Program (SZM2021007) and Hainan Province Clinical Medical Center.
Disclosure
Chunyu Yuan and Dongmei Deng are co-first authors. Mrs Yujiang Li and Mrs Min Li report grants from Suzhou Science and Technology Program (SZM2021007), grants from Hainan Province Clinical Medical Center, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita - update 2015. Eur J Hum Genet. 2015;23(4):558. doi:10.1038/ejhg.2014.170
2. AlSabbagh MM. Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges. 2020;18(9):943–967. doi:10.1111/ddg.14268
3. Li F, Li W, Qiao X, Xie X. Clinical features of dyskeratosis congenita in mainland China: case reports and literature review. Int J Hematol. 2019;109(3):328–335. doi:10.1007/s12185-018-02582-x
4. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2019;12(12):1037–1052. doi:10.1080/17474086.2019.1662720
5. Savage SA, Dokal I, Armanios M, et al. Dyskeratosis congenita: the first NIH clinical research workshop. Pediatr Blood Cancer. 2009;53(3):520–523. doi:10.1002/pbc.22061
6. Ding YG, Zhu TS, Jiang W, et al. Identification of a novel mutation and a de novo mutation in DKC1 in two Chinese pedigrees with dyskeratosis congenita. J Invest Dermatol. 2004;123(3):470–473. doi:10.1111/j.0022-202X.2004.23228.x
7. Kelmenson DA, Hanley M. Dyskeratosis congenita. N Engl J Med. 2017;376(15):1460. doi:10.1056/NEJMicm1613081
8. Stoopler ET, Shanti RM. Dyskeratosis congenita. Mayo Clin Proc. 2019;94(9):1668–1669. doi:10.1016/j.mayocp.2019.04.032
9. Otoshi R, Baba T, Shintani R, et al. Diverse pathological findings of interstitial lung disease in a patient with dyskeratosis congenita. Intern Med. 2021;60(8):1257–1263. doi:10.2169/internalmedicine.5143-20
10. Fernández García MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014;5:157–167. doi:10.2147/JBM.S47437
11. Bongiorno M, Rivard S, Hammer D, Kentosh J. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;124(4):e239–e242. doi:10.1016/j.oooo.2017.08.001
12. Liu AQ, Deane EC, Prisman E, Durham JS. Dyskeratosis congenita and squamous cell cancer of the head and neck: a case report and systematic review [published online ahead of print, 2021 Oct 15]. Ann Otol Rhinol Laryngol. 2021;34894211047470. doi:10.1177/00034894211047470
13. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018;103(1):30–39. doi:10.3324/haematol.2017.178111
14. Ward SC, Savage SA, Giri N, et al. Beyond the triad: inheritance, mucocutaneous phenotype, and mortality in a cohort of patients with dyskeratosis congenita. J Am Acad Dermatol. 2018;78(4):804–806. doi:10.1016/j.jaad.2017.10.017
15. Okal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi:10.1182/asheducation-2011.1.480
16. Allenspach EJ, Bellodi C, Jeong D, et al. Common variable immunodeficiency as the initial presentation of dyskeratosis congenita. J Allergy Clin Immunol. 2013;132(1):223–226. doi:10.1016/j.jaci.2012.11.052
17. Gardos G, Cole JO. Maintenance antipsychotic therapy: is the cure worse than the disease? Am J Psychiatry. 1976;133(1):32–36. doi:10.1176/ajp.133.1.32
18. Dvorak LA, Vassallo R, Kirmani S, et al. Pulmonary fibrosis in dyskeratosis congenita: report of 2 cases. Hum Pathol. 2015;46(1):147–152. doi:10.1016/j.humpath.2014.10.003
19. Olivieri C, Mondino A, Chinello M, et al. Clinical heterogeneity in a family with DKC1 mutation, dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome in first cousins. Pediatr Rep. 2017;9(3):7301. doi:10.4081/pr.2017.7301
20. Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–359. doi:10.3324/haematol.2011.055269
21. Xu J, Khincha PP, Giri N, Alter BP, Savage SA, Wong JM. Investigation of chromosome X inactivation and clinical phenotypes in female carriers of DKC1 mutations. Am J Hematol. 2016;91(12):1215–1220. doi:10.1002/ajh.24545
22. Garus A, Autexier C. Dyskerin: an essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA. 2021;27(12):1441–1458. doi:10.1261/rna.078953.121
23. Cerrudo CS, Mengual Gómez DL, Gómez DE, Ghiringhelli PD. Novel insights into the evolution and structural characterization of dyskerin using comprehensive bioinformatics analysis. J Proteome Res. 2015;14(2):874–887. doi:10.1021/pr500956k
24. Zhao XY, Zhong WL, Zhang J, Ma G, Hu H, Yu B. Dyskeratosis congenita with DKC1 mutation: a case report. Indian J Dermatol. 2020;65(5):426–427. doi:10.4103/ijd.IJD_716_18
25. Ratnasamy V, Navaneethakrishnan S, Sirisena ND, et al. Dyskeratosis congenita with a novel genetic variant in the DKC1 gene: a case report. BMC Med Genet. 2018;19(1):85. doi:10.1186/s12881-018-0584-y
26. Marrone A, Mason PJ. Dyskeratosis congenita. Cell Mol Life Sci. 2003;60(3):507–517. doi:10.1007/s000180300042
27. Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107(7):2680–2685. doi:10.1182/blood-2005-07-2622
28. Gu BW, Apicella M, Mills J, et al. Impaired telomere maintenance and decreased canonical WNT signaling but normal ribosome biogenesis in induced pluripotent stem cells from X-linked dyskeratosis congenita patients. PLoS One. 2015;10(5):e0127414. doi:10.1371/journal.pone.0127414
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.