")
Back to Journals » International Journal of Nanomedicine » Volume 13
A novel RGDyC/PEG co-modified PAMAM dendrimer-loaded arsenic trioxide of glioma targeting delivery system
Authors Lu Y , Han S, Zheng H, Ma R, Ping Y, Zou J , Tang HX, Zhang Y, Xu X, Li F
Received 26 May 2018
Accepted for publication 2 August 2018
Published 2 October 2018 Volume 2018:13 Pages 5937—5952
DOI https://doi.org/10.2147/IJN.S175418
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Yanping Lu,1 Shunping Han,1 Hongyue Zheng,1 Rui Ma,1 Yuting Ping,1 Jiafeng Zou,1 Hongxia Tang,1 Yongping Zhang,2 Xiuling Xu,1 Fanzhu Li1
1College of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China; 2College of Pharmaceutical Sciences, Guiyang University of Chinese Medicine, Guiyang, Guizhou, China
Background: The Traditional Chinese Medicine, arsenic trioxide (ATO, As2O3) could inhibit growth and induce apoptosis in a variety of solid tumor cells, but it is severely limited in the treatment of glioma due to its poor BBB penetration and nonspecifcity distribution in vivo.
Purpose: The objective of this study was encapsulating ATO in the modified PAMAM dendrimers to solve the problem that the poor antitumor effect of ATO to glioma, which provide a novel angle for the study of glioma treatment.
Methods: The targeting drug carrier (RGDyC-mPEG-PAMAM) was synthesized based on Arg-Gly-Asp (RGDyC) and αvβ3 integrin targeting ligand, and conjugated to PEGylated fifth generation polyamidoamine dendrimer (mPEG-PAMAM). It was characterized by nuclear magnetic resonance, fourier transform infrared spectra, Nano-particle size-zeta potential analyzer,etc. The in vitro release characteristics were studied by dialysis bag method. MTT assay was used to investigate the cytotoxicity of carriers and the antitumor effect of ATO formulation. In vitro blood-brain barrier (BBB) and C6 cell co-culture models were established to investigate the inhibitory effect of different ATO formulation after transporting across BBB. Pharmacokinetic and antitumor efficacy studies were investigated in an orthotopic murine model of C6 glioma.
Results: The prepared RGDyC-mPEG-PAMAM was characterized for spherical dendrites, comparable size (21.60±6.81 nm), and zeta potential (5.36±0.22 mV). In vitro release showed that more ATO was released from RGDyC-mPEG-PAMAM/ATO (79.5%) at pH 5.5 than that of pH 7.4, during 48 hours. The cytotoxicity of PEG-modified carriers was lower than that of the naked PAMAM on both human brain microvascular endothelial cells and C6 cells. In in vitro BBB model, modification of RGDyC heightened the cytotoxicity of ATO loaded on PAMAM, due to an increased uptake by C6 cells. The results of cell cycle and apoptosis analysis revealed that RGDyC-mPEG-PAMAM/ATO arrested the cell cycle in G2-M and exhibited threefold increase in percentage of apoptosis to that in the PEG-PAMAM/ATO group. Compared with ATO-sol group, both RGDyC-mPEG-PAMAM/ATO and mPEG-PAMAM/ATO groups prolonged the half-life time, increased area under the curve, and improved antitumor effect, significantly. While the tumor volume inhibitory of RGDyC-mPEG-PAMAM/ATO was 61.46±12.26%, it was approximately fourfold higher than the ATO-sol group, and twofold to the mPEG-PAMAM/ATO group.
Conclusion: In this report, RGDyC-mPEG-PAMAM could enhance the antitumor of ATO to glioma, it provides a desirable strategy for targeted therapy of glioma.
Keywords: arsenic trioxide, blood-brain barrier, RGDyC, PEG co-modified, glioma targeting delivery, PAMAM dendrimer
Introduction
Glioma is the cause of about 1,75,000 deaths every year in the world. It is the most common primary malignant tumor of the central nervous system,1 which mainly occurs in the brain and the glial tissue.2 A malignant glioma is considered as the most pernicious form of cancer. Glioblastoma infiltrates diffusely and uncertainly delineating the margin, so they cannot be amenable to cure by surgical resection alone.3 Currently, the strategy for glioma is surgery, combined with adjuvant chemotherapy or radiotherapy. Furthermore, chemotherapy is also primarily hindered due to poor tumor targeting, low penetration across the blood-brain barrier (BBB), and some serious side effects.4 In addition, the average median survival for patients diagnosed with malignant glioma is only 12–14 months.5 Hence, the improvement of targeting and improvement of penetration of drugs across the BBB are urgently required. Many antiglioma drugs are being researched and developed, such as doxorubicin, paclitaxel, and arsenic trioxide (ATO).6–8
Arsenic is a natural product of traditional Chinese medicine (TCM). Historically, it is famous for its effectiveness in treating hemorrhoids, carbuncle, ulcers, ringworm, cold phlegm, asthma, and malaria. However, in the past decade, ATO (As2O3), the main compound of arsenic, has become the first-line antineoplastic drug for the treatment of acute promyelocytic leukemia (APL). It has been approved by the US Food and Drug Administration (FDA) in 2000.9,10 Interestingly, some studies have shown that ATO could also inhibit growth and induce apoptosis in a variety of solid tumor cells, such as breast, liver, gastric, prostate, renal, bladder, and glioma cells.10–14 However, several limitations hinder its applications in clinical practice. The free ATO in aqueous solution exists as arsenite ions, with fast elimination in vivo, poor pharmacokinetics, and unacceptable systemic toxicity, including peripheral neuropathies and liver failure.10,15 Simultaneously, most drugs are practically ineffective because of the BBB, which restricts the delivery of most small and large molecules into the brain, and leads to a low intracranial therapeutic drug concentration.16 Besides, even the intracranial drug spreads across the whole brain, and it is difficult to localize it to the tumor site, thus resulting in side effects. Similarly, ATO is severely limited in the treatment of glioma due to its poor BBB penetration,17 nonspecificity distribution in vivo, and narrow therapeutic window.18,19 In order to overcome these shortcomings of chemotherapeutic drugs, some researches on nanovehicles were carried out. They possess advantages in improving loading capacity, long-term circulation, sustained release, and multifunctional platform. Thus, developing a multifunctional nanovehicle for ATO delivery to glioma is an urgent challenge.
There is abundant neovascularization, and a certain degree of enhanced permeability and retention (EPR) effects. Therefore, the targeted delivery system can reach the brain tumor site through the EPR effect, and then can be mediated by the receptor to the brain tumor cells.20 Therefore, the key is to find an efficient, safe, and suitable carrier, and then construct a brain glioma targeted drug delivery system, based on the identification of the specific receptor and ligand on the tumor. Increased attention has been focused on drug delivery systems with targeting or stimuli-responsive properties to prolong circulation capabilities, increase solubilization, and improve specificity to the desired sites.21–23 Also modified with high-affinity ligands, they have been used to target the tumor vessels and tumor cells. Probably, one of the most successfully identified ligand is the RGD-integrin binding peptide.24 The Arg-Gly-Asp (RGDyC) is selective to αvβ3 integrin peptides and can also combine with integrin receptors on the surface of leukocytes (neutrophils and monocytes).25,26 It has been found that both αvβ3 and αvβ5 integrins are expressed on glioma cells and vasculature.27,28 It provides an ideal strategy for targeted delivery.29 Hence, a drug delivery system enriched with αvβ3 specific ligands is potentially capable of affecting the tumors, both directly and indirectly. As seems likely, RGDyC nanoparticles could be developed for selective and preferential presentation to leukocytes.26 After that, nanoparticles could be taken up into the brain, where targeted delivery to the tumor tissues could be done. Hence, RGDyC is often used as a tumor-targeting group in a tumor-targeting drug delivery system.30
Many types of nanovehicles such as polymers, liposomes, micelles, silica nanoparticles, and dendrimers have shown their ability to deliver drugs at specific sites and target cancer cells of interest.31 Among these, polyamidoamine (PAMAM) could well turn out to be a promising carrier because of its unique characteristics.32 PAMAM has distinctive advantages, such as monodispersity, tunable size, cavity structure inside, and numerous functional groups for chemical modifications.33–35 Therefore, PAMAM can increase biodistribution of drugs, and possibly enhance the EPR effect in targeting tumors. However, the unmodified PAMAM has a positive charge under physiological conditions due to a large number of amino groups on the surface. It is easily modified while facilitating cell uptake, but it also has some hemolytic toxicity and cytotoxicity, limiting the application of therapy in vivo.36 Therefore, suitable modification of PAMAM could reduce its intrinsic toxicity and also offer a possibility that PAMAM can become a more intelligent carrier. It has been reported that PEGylation can improve biological safety and prolong biological half-life in vivo.37,38 PEGylation can effectively increase the permeation of nanoparticles in the brain, because of the prolonged blood circulation time.39 Furthermore, it is important in the treatment of brain diseases, especially gliomas. Until now, PAMAM is a potential delivery system, due to its drug delivery ability and targeting property.
Based on ATO and glioma features, we proposed a viable way to stably load ATO through a targeted drug delivery system, based on RGDyC and PEG co-modified PAMAM (RGDyC-mPEG-PAMAM/ATO) for glioma therapy.
Materials and methods
Materials
Generation 5 PAMAM dendrimer with an ethylenediamine core in methanol (5% in methanol, w/w; containing 128 surface primary amino groups, MW 28826), was purchased from Sigma-Aldrich Co. Ltd. (St Louis, MO, USA). Maleimide polyoxyethylene succinimidyl carboxymethyl ester (NHS-PEG-MAL, MW 3500) and methoxy polyoxyethylene succinimidyl carboxymethyl ester (mPEG-NHS, MW 3000) were purchased from JenKem Technology Co., Ltd. (Beijing, China). RGDyC (CRGDKGPDC) was synthesized by GL Biochem (Shanghai) Ltd. (Shanghai, China). ATO (As2O3, 90%) was purchased from Alfa Aesar (Shanghai, China). Arsenic standard solution (1 mg·mL−1) was purchased from Beijing Century Audiocodes Biological Technology Co., Ltd. (Beijing, China).
Laboratory Animal Center, Zhejiang Chinese Medical University (Zhejiang, China), donated the human brain microvascular endothelial cells (HBMECs) and C6 glioma cell line. They were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HBMEC and C6 glioma cells were cultured in the RPMI 1640 cell culture medium, which contained 10% FBS, penicillin (100 U·mL−1), and streptomycin (100 U·mL−1). The cells were both maintained at 37°C, with 5% CO2, and harvested after 80% confluence.
Laboratory Animal Center of Zhejiang Chinese Medical University supplied Wistar rats (weighing 200±20 g) and New Zealand white rabbits. All animal studies were performed in compliance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals by following protocols approved by the Institutional Animal Care and Use Committee at Zhejiang Chinese Medical University.
Synthesis of PEG-PAMAM
RGDyC modified PEG-PAMAM conjugate was synthesized according to the previously reported method, with slight modification.40 In short, PAMAM was dissolved in 2 mL PBS (8.0) after removal of methanol in a nitrogen stream. Then mPEG3000-NHS was added (PAMAM: PEG=1:64, molar ratio). After reacting for 48 hours at room temperature, the reaction mixture was centrifuged (4,000 rpm, 25 minutes) by an ultrafiltration tube (Millipore, Burlington, MA, USA, MWCO 30,000), to remove unreacted PEG. Thin layer chromatography (TLC) was used to test the purity of the products, until no PEG could be detected in the upper solution (methanol/chloroform=1:1, v/v, iodine vapor). White and fluffy solids (PEG-PAMAM) were retrieved by freeze-drying.
Synthesis of RGDyC-PAMAM
To synthesize RGDyC-PAMAM, 4.65 mg of RGDyC (9 μmol) was dissolved in 2 mL 0.1 M NaAc-HAc buffer (pH 6.0), and then 27.32 mg of MAL-PEG3500-NHS (9 μmol) was added and mixed by vortex for 30 seconds. The above solution was added into 2 mL of 0.05 M borate buffer (pH 9.2) with 5 mg PAMAM (0.2 μmol). The mixed solution was stirred overnight at room temperature (RGDyC: PEG: PAMAM=45:45:1, molar ratio). After the pH was adjusted to 7.0, the unreacted maleimide group was quenched by 9.2 μL of β-mercaptoethanol for another 1 hour of reaction. Then the products (RGDyC-PAMAM) were obtained by centrifugation and freeze-drying.
Synthesis of RGDyC-mPEG-PAMAM
RGDyC-mPEG-PAMAM was synthesized on the basis of RGDyC-PAMAM. The above concentrated product was redissolved in 2 mL of PBS (8.0), then mPEG3000-NHS was added (PAMAM: PEG=1:64, molar ratio) to react at room temperature for 48 hours. The unreacted PEG was removed by centrifugation using TLC to test the purity of the products, until no PEG could be detected in the upper solution and then freeze-dried.
ATO loading
PEG-PAMAM and RGDyC-mPEG-PAMAM were incubated with ATO solution (pH 7.4) and stirred for 24 hours at room temperature. The free ATO was removed by ultrafiltration centrifugation, and then freeze-dried, and stored at −80°C. The encapsulation efficiency (EE%) and drug loading efficiency (DL%) were determined by inductively coupled plasma emission spectrum (ICP, 6300, Thermo Electron Corporation, Waltham, MA, USA) as described previously.41,42
|
|
where W0 is the total dosage, W1 is the amount of free drug, and Wt is the total mass.
Preparation of fluorescein isothiocyanate (FITC)-labeled dendrimers
FITC-labeled dendrimers were prepared at a molar ratio of 12:1 (FITC: PAMAM) according to the previously reported method.43 In short, 20 μL DMSO containing 0.41 mg of FITC was added dropwise to a solution of PAMAM (0.09 μmol), or the corresponding amount of PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM in 2 mL deionized water, and stirred overnight. Then the mixture was dialyzed in the dark against deionized water for 24 hours and ultrafiltrated to remove the free FITC. The yellow fluffy solid was sequentially obtained by freeze-drying.
Characterization of conjugates
The successful synthesis of PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM were confirmed by 1H-NMR on a Mercury Plus 600 MHz spectrometer (Bruker, Karlsruhe, Germany, D2O) and Fourier-transform infrared (FT-IR) spectra (Nicolet 6700, Thermo Electron Corporation). The size distribution, polydispersity index (PDI), and zeta potentials were measured by dynamic light scattering (Malvern Nano-ZS90, Worcestershire, UK). Transmission electron microscopy (TEM) images of these nanoparticles were obtained by a transmission electron microscope (H-7650, Hitachi, Tokyo, Japan).
In vitro release
The ATO released from PEG-PAMAM and RGDyC-mPEG-PAMAM conjugates were investigated by dialysis bag method. One milliliter of ATO formulations (ATO-sol, PEG-PAMAM/ATO or RGDyC-mPEG-PAMAM/ATO, 1 mg ATO-equiv.) was sealed in a dialysis bag (MW 3500) and then submerged fully into the release medium (100 mL, pH 7.4 or 5.5), and incubated at 37°C. Samples were collected from the media at predetermined time intervals and instantly replaced with an equal volume of fresh PBS. ATO concentrations were measured by the ICP method. All experiments were performed in triplicate.
Hemolytic toxicity in vitro study
The hemolytic activity of PAMAM conjugates was evaluated as previously described in the literature.44 Briefly, 5 mL of fresh blood from a New Zealand white rabbit was collected in centrifuge tube with heparin to prevent coagulation. It was then centrifuged at 1,500 rpm for 5 minutes and washed with physiological saline (0.9%, v/v) until the supernatant became free of red color. The 2% (v/v) red blood cells (RBCs) suspension was prepared with physiological saline. Different concentrations of PAMAM, PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM were incubated with RBCs suspension for 1 hour at 37°C. In addition, distilled water and physiological saline were prepared using the same method as the positive and negative controls. After incubation, samples were centrifuged at 1,500 rpm for 10 minutes, and the supernatant absorbance was measured at 414 nm with multimode microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA). The percentage of hemolysis was calculated, and the equation that followed was
|
In vitro cytotoxicity assay
The MTT assay was used to evaluate the cytotoxicity of drug-free carriers (PAMAM, PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM) and ATO formulations (ATO-sol, PEG-PAMAM/ATO or RGDyC-mPEG-PAMAM/ATO, ATO-equiv.) on HBMEC or C6 cells. Briefly, the cells were seeded at a density of 5×103 cells/well in 96-well transparent plates in culture medium, and incubated for 24 hours at 37°C. They were then exposed to drug-free carriers or ATO formulations with different concentrations (0.001−100 μmol·L−1). After being incubated for 48 hours, the medium was removed and incubated with fresh culture media containing MTT (0.5 mg·mL−1) for 4 hours at 37°C. The medium was then replaced by 150 μL DMSO, and the absorbance at wavelength 570 nm was measured by the microplate reader (SpectraMax M2, Molecular Devices). The cell viability was calculated by dividing the OD values of samples by the OD values of blank. Each experiment, with the same concentration, was performed three times.
Cellular uptake and intracellular disposition
The intracellular localization of FITC-labeled dendrimers in C6 cells was detected by Laser scanning confocal microscope (Olympus, Tokyo, Japan). In brief, C6 cells in the logarithmic growth phase were seeded in the chambered coverslips at 4×104 cells/well and cultured at 37°C for 24 hours. They were then treated with serum-free culture media or serum-free culture media containing dendrimers (PAMAM-FITC, PEG-PAMAM-FITC, RGDyC-PAMAM-FITC, RGDyC-mPEG-PAMAM-FITC, at FITC concentrations of 10 μmol·L−1) for 2 hours. Afterward, cells were washed three times with PBS, fixed with 4% paraformaldehyde, and stained with DAPI to observe the intracellular disposition.
C6 cells in logarithmic growth phase were seeded in 12-well plates at 5×105 cells/well and incubated at 37°C for 24 hours. Subsequently, the medium was replaced by serum-free culture media or serum-free culture media containing FITC-labeled nanoparticle (PEG-PAMAM-FITC, RGDyC-PAMAM-FITC, RGDyC-mPEG-PAMAM-FITC, at FITC concentrations of 10 μmol·L−1) for 0.5, 1, 2, and 4 hours. The cells were washed three times with PBS and then harvested. The fluorescence intensity was determined by flow cytometer (Guava Easycyte, Merck Millipore, Darmstadt, Germany). Cells treated without FITC-labeled dendrimers were used as the background fluorescence. All the experiments were repeated three times.
BBB penetration in vitro
For the in vitro penetration studies, HBMEC cells were used. Briefly, HBMECs were seeded into transwell inserts (Polycarbonate Membrane Transwell Inserts of 3 μm mean pore size, Corning, NY, USA) at a density of 1×105 cells/well and incubated at 37°C for 7 days. The culture medium was changed every 2 days.45 The transepithelial electrical resistance (TEER) value was measured by transendothelial electrical resistance instrument (Millicell-ERS-2, Millipore) to test the integrity of the BBB model. Experiments were conducted only when the TEER was >250 Ω·cm2.
To evaluate the ability of the carriers crossing the BBB, FITC, PEG-PAMAM-FITC, or RGDyC-mPEG-PAMAM-FITC (at the FITC concentration of 10 μmol·L−1) were added into the corresponding insert, respectively. Two hundred microliters of medium was taken out from the acceptor compartments at 0.5, 1, 2, and 4 hours after treatment, and 200 μL of fresh medium was immediately added to the compartments. The BBB transport ratios of FITC-labeled dendrimers were evaluated by the microplate reader (Ex=495 nm, Em=525 nm).
To evaluate the antitumor ability of the ATO-loaded dendrimers after transport across the BBB, HBMEC and C6 cells co-cultural model was established. C6 cells of logarithmic growth phase were seeded into 12-well plates at 2.5×104 cells/well and incubated for 24 hours as the acceptor compartments and then combined with transwell inserts which has constructed the BBB model successfully. Afterward the ATO-sol, PEG-PAMAM/ATO, or RGDyC-mPEG-PAMAM/ATO was added into the corresponding upper compartments, at the ATO concentration of 30 μmol·L−1, respectively, and removed after 4 hours incubation. The viability of C6 cells in the acceptor compartments were measured by the MTT method after 48 hours.
Cell cycle and apoptosis analysis
C6 cells were seeded overnight at a density of 5×105 cells/well in six-well plates. Afterward, the cells were treated with different ATO formulations (1 μmol·L−1 ATO-equiv.) for 24 hours, then trypsinized, washed, and fixed with cold 75% ethanol overnight. After that, the cells were incubated with 1 mg·mL−1 RNase A and 50 μg·mL−1 propidium iodide (PI) for 30 minutes at 37°C in the dark. Ten thousand cells were acquired, and the DNA content was determined by flow cytometry (Guava Technologies, Millipore, Hayward, CA, USA). All experiments were performed in triplicate.
ATO has been reported to induce apoptosis in various human cancer cells.10 To determine whether ATO induced apoptosis in C6 cells, apoptosis was measured by flow cytometry with Annexin V-FITC/PI dual staining (BD, Oxford, UK). Cells were seeded in six-well culture plates at 5×105 cells/mL and allowed to grow overnight before being exposed to different ATO formulations. After 24 hours of drug treatment, the cells were trypsinized, washed twice with PBS, and then collected to stain with Annexin-FITC for 20 minutes in the dark. PI was added next, and the cells were incubated for 10 minutes in the dark. The cells were then analyzed within 1 hour, and the data were analyzed by means of the Modift software. All experiments were performed in triplicate.
The HBMEC and C6 cells co-cultural model was established for analyzing the cell cycle and apoptosis after the ATO-loaded dendrimers transporting across the BBB model. Different ATO formulations were added into the corresponding upper compartments and removed after being incubated for 4 hours. In addition to this, there were other steps similar to the common cycle or apoptosis steps.
Pharmacokinetic and efficacy study
To determine pharmacokinetic profiles of ATO formulations, Wistar rats (200±20 g) were randomly divided into four groups (n=6) and received intravenous injection of ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO at an ATO dose of 1 mg·kg−1. At the desired times of 0.1, 0.25, 0.50, 0.75, 1, 2, 4, 6, 8, 12, 24, 36, and 48 hours following injection, blood samples were collected and then centrifuged and stored at −80°C for further analysis. Inductively coupled plasma mass spectrometer was used to determine the serum concentration of ATO, and the working conditions were as described in our previous study.41
Therapeutic efficacy study
The antitumor efficacy of the ATO–polymer conjugates was performed in Wistar rats model. C6 cells (1×106 cells/5 μL in pH 7.4 PBS) were implanted into the right striatum (1.8 mm lateral to the bregma and 3 mm of depth) of Wistar rats by using a stereotactic fixation device with mouse adaptor, and allowed to grow for 2 weeks, when the tumor volume approximately reached 50 mm3. The glioma-bearing rats were divided into four groups (n=12): PEG-PAMAM/ATO, RGDyC-mPEG-PAMAM/ATO, ATO-sol (at a dose of 1 mg ATO-equiv./kg body weight), and saline. The tumor-bearing rats were weighed every day. The drug was given daily for the duration of 7 days. During the eighth day, six rats from each group were dissected, and tumor tissues were collected and measured. The tumor tissues were then harvested and sectioned for histopathology analyses with H&E staining. TUNEL staining was used to detect the cell apoptosis of tumor tissue, according to the manufacturer’s protocol. The remaining rats were checked for survival rates every day (n=6). The survival time was recorded and analyzed by Kaplan–Meier analysis.
|
|
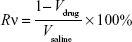
|
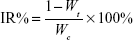
where a, b refer to the maximum and minimum diameters of the tumor, Wt and Wc refer to the mean tumor weight of treated and control groups, respectively.
Statistical analysis
All data are presented as the mean±SD. Statistical significance was determined by one-way ANOVA by SPSS software (version 20.0, IBM Inc, Armonk, NY, USA). Survival data were presented using Kaplan–Meier survival curves and analyzed by the log-rank test. P<0.05 was considered statistically significant, and extreme significance was set at P<0.01.
Results and discussion
Characterization of conjugates
The white fluffy solid products of PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM were synthesized and characterized by 1H NMR spectrum, as shown in Figure 1A. The corresponding PAMAM had multiple peaks between 2.35 and 2.77 ppm, and the appearance of characteristic peaks at 3.60–3.66 and 3.99 ppm indicated the existence of PEG in the conjugates. The appearance of characteristic peaks at 6.70 and 7.00 ppm (insert 1A) represent four hydrogen atoms on the benzene ring of RGDyC, indicating RGDyC was modified successfully. The result of 1H NMR spectra showed the successful synthesis of RGDyC-mPEG-PAMAM. In addition, according to the integration ratio of protons, there are about 32 molecules, and 7 molecules of RGDyC were grated on the periphery of PAMAM.
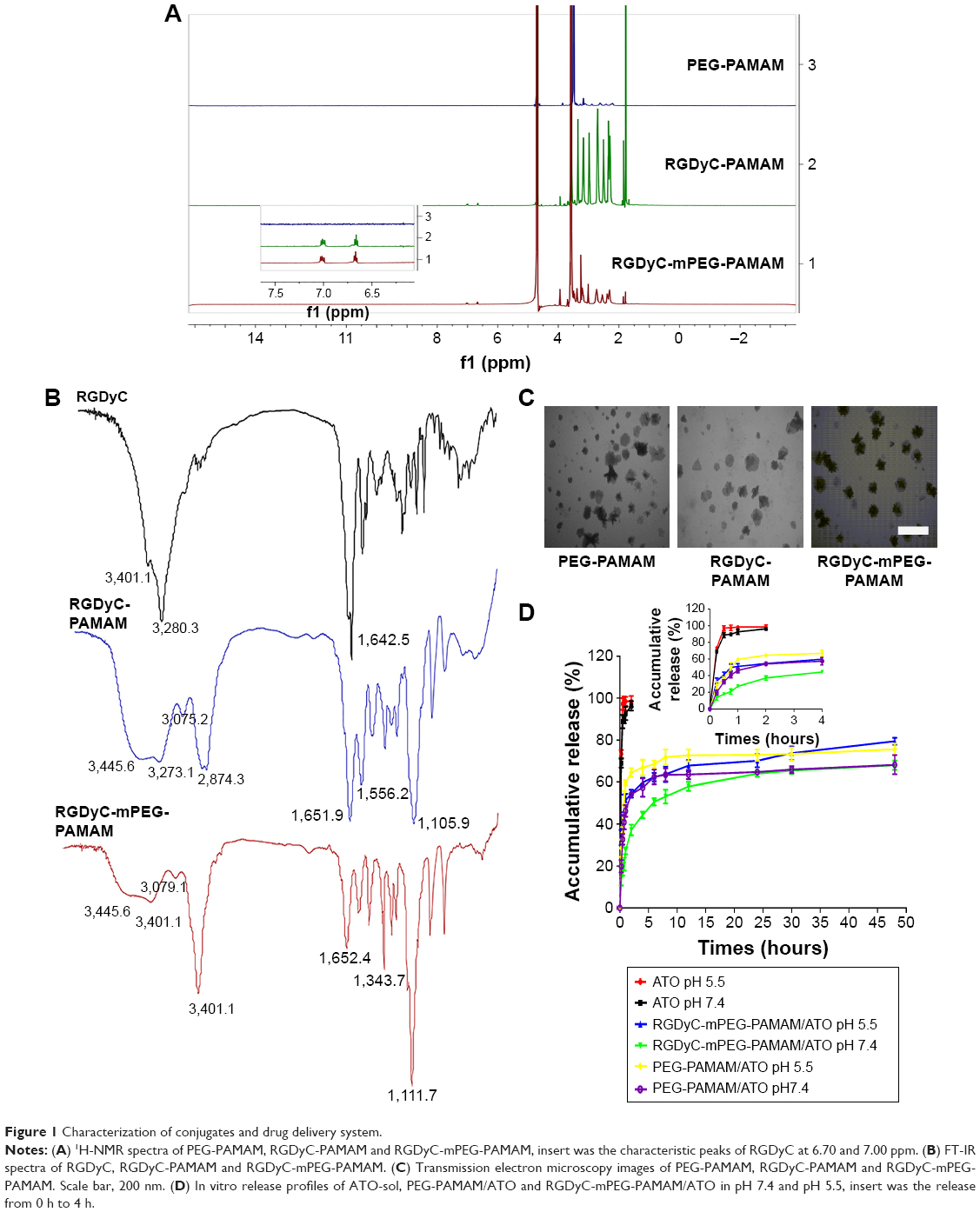 | Figure 1 Characterization of conjugates and drug delivery system. |
The FT-IR spectrum of RGDyC, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM are given in Figure 1B. Compared with the FT-IR spectrum of RGDyC-PAMAM, the peaks of PAMAM at 3,445 cm−1 and 1,651.5 cm−1 were relatively decreased, and the peaks of PEG at 2,886.6 cm−1 and 1,111.7 cm−1 were relatively enhanced. It may be caused by the mPEG modification on primary amino groups of PAMAM peripheral. In addition, peaks appeared both at 844 cm−1 and 842.7 cm−1, and presumably it was the C-H bending vibration on the binary substituted benzene ring of tyrosine, by the RGDyC structure, according to the IR and 1H NMR of RGDyC.
These results indicated that PEG chains and RGDyC were successfully conjugated to the periphery of PAMAM dendrimers. The grafting ratio of PEG and RGDyC attached to PAMAM were estimated using the proton integration method; about 31 molecules of PEG were grafted on PAMAM, and 10 molecules of FITC were labeled at each PAMAM dendrimer on average.
The size, zeta potential, and PDI of RGDyC-mPEG-PAMAM were 21.60±6.81 nm, 5.36±0.22 mV, and 0.56±0.02, respectively. The result showed that the size increased and the zeta potential decreased after the nanoparticles modification, but it still remained positive (Table 1). Nanoparticles with a particle size of 10–100 nm and a certain amount of positive charge, are more favorable for tumor therapy. When the particle size is <10 nm, it is easily removed by the kidney, and when it is >100 nm, it cannot be recognized by the tumor vasculature.46 From the perspective of nanoparticle size, RGDyC-PAMAM and RGDyC-mPEG-PAMAM in this study are promising drug carriers.
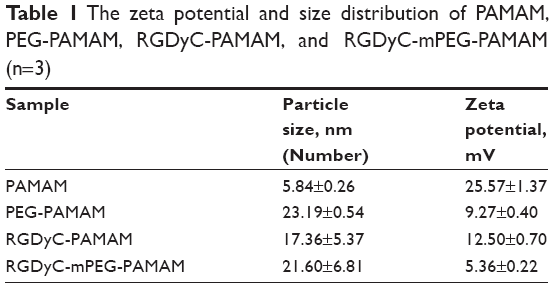 | Table 1 The zeta potential and size distribution of PAMAM, PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM (n=3) |
TEM images (Figure 1C) further displayed that RGDyC-mPEG-PAMAM had a good dispersion. However, aggregation may appear due to decreased intermolecular repulsive force caused by the reduced zeta potential. The shape of RGDyC-mPEG-PAMAM was spherical and dendritic.
Drug loading and in vitro release
The EE% of PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM as measured by ICP was 32.97%, 31.68%, and 34.82%, and DL% was 2.67%, 2.58%, and 2.82%, respectively.
The in vitro release profiles of ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO were carried out in a simulated physiological (pH 7.4) and tumor environment (pH 5.5; Figure 1D). Compared with the burst release of ATO-sol (~95% in 2 hours), ATO-loaded dendrimers displayed an obvious sustained and pH-dependent release. ATO released from PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO in pH 5.5 was about 72.9%, 70.2%, and in pH 7.4, it was about 64.8%, 64.0%, respectively, after 24 hours. Additionally, the release was steady after 24 hours, while the release rate of PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO at pH 5.5 was 75.6% and 79.5%, and at pH 7.4, it was 68.1% and 68.2%, respectively, at 48 hours. The sustained release property of ATO suggested that the drug molecules were well embedded in the cavities of PAMAM, and the release was accelerated at pH 5.5 due to the partial protonation of the PAMAM dendrimers.
In vitro cytotoxicity
The results of hemolytic toxicity of the carriers showed that the hemolysis rate of unmodified PAMAM reached 99.0% at a concentration of 2 mg·mL−1, after incubating for 1 hour (Figure 2A). However, the toxicity of PEG-PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM was significantly reduced to 3.5%, 10.6%, and 2.8%, respectively, after PEG modified. There was a significant difference between the hemolytic toxicity of PAMAM, RGDyC-PAMAM, and RGDyC-mPEG-PAMAM (P<0.05), but no difference in that of PEG-PAMAM and RGDyC-mPEG-PAMAM (P>0.05). It indicated that the PEG modification could significantly attenuate the hemolytic toxicity of PAMAM, particularly at high concentrations, the results of hemolytic toxicity showed that the hemolytic rate of unmodifed PAMAM was 99.0% after incubated with RBCs for 1 hour, and the hemolytic rate of unmodified PAMAM was 99.0%. Even at a high concentration (2 mg·mL−1) of PAMAM, the hemolysis rate of PEG modification group was <5%, which could be regarded safe for intravenous administration.47 We speculated that it might relate to the neutralization of positive potential on the PAMAM surface. However, RGDyC-PAMAM was about 10.6% at a concentration of 2 mg·mL−1. Thus, the RGDyC-PAMAM was not suitable for later experiments. In conclusion, the result showed the necessity of detoxification modification of PAMAM and modified with PEG displayed good blood compatibility and safety for further application.
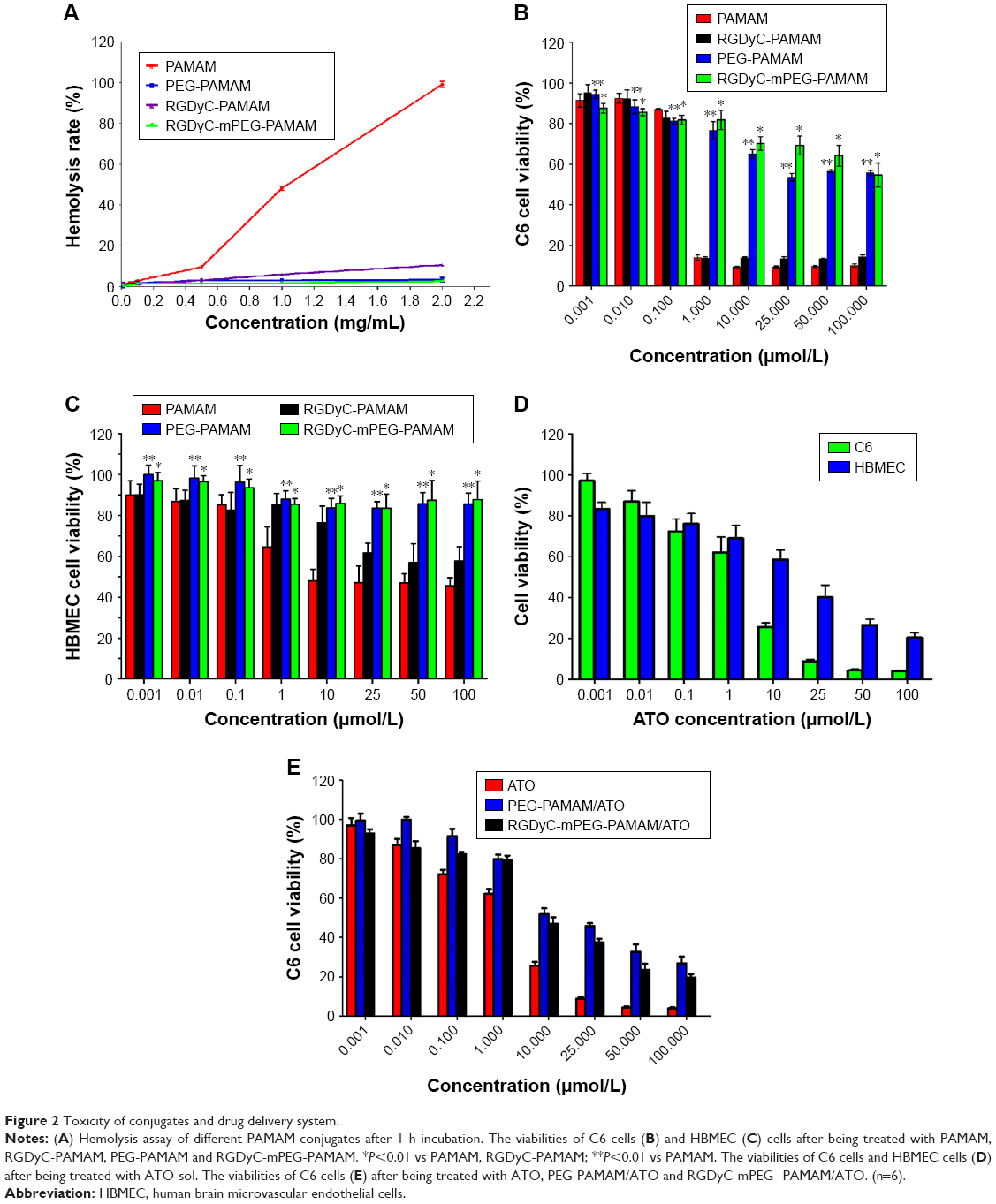 | Figure 2 Toxicity of conjugates and drug delivery system. |
To evaluate the in vitro cytotoxicity of drug-free carriers, the viability of C6 cells and HBMEC treated with PAMAM conjugates is shown in Figure 2B and C. There was no significant cytotoxicity observed in either group at low concentration (<0.1 μmol·L−1). However, at high concentration (100 μmol·L−1), the cell viability in RGDyC-mPEG-PAMAM group was significantly higher than that in PAMAM group, indicating that it was a suitable characteristic for carriers. C6 cells were more susceptible to carrier toxicity. In addition, the viability of cells treated with PEG-PAMAM and RGDyC-mPEG-PAMAM was both higher than that of cells treated with unmodified PAMAM and RGDyC-PAMAM. This further indicated that PEG modification could decrease the cytotoxicity at concentrations >1 μmol·L−1. The toxicity of PAMAM is related to the positive charge of the surface amino group, which can lead to cell membrane damage, thinning, leakage, and destruction of acidified endosomes through proton sponge effect into the cytoplasm.48 Therefore, modification is necessary to reduce the toxicity of PAMAM.
Cytotoxicity against HBMEC and C6 cells was compared with ATO-sol (Figure 2D). It showed that C6 cells were more sensitive to ATO toxicity, and the IC50 values of ATO on C6 cells and HBMEC cells were 6.77 and 12.91 μmol·L−1, respectively. When concentration was >1 μmol·L−1, the cytotoxicity of ATO was significantly different on the HBMEC and C6 cells (P<0.01). Moreover, compared with PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO groups’ C6 cell survival rate (Figure 2E), ATO-sol group had better inhibition when directly treated on C6 in vitro, which may be related to the release of ATO from the drug delivery system. RGDyC-modified PAMAM showed greater inhibition on C6 cells than the unmodified group (P<0.05) at concentrations >10 μmol·L−1, indicating the targeting ability of RGDyC on glioma C6 cells.
Intracellular disposition and cellular uptake
To evaluate the targeting effect of RGDyC systems on C6 cells, confocal laser scanning microscopy and flow cytometry were used to conduct intracellular disposition and cellular uptake experiments. The intracellular disposition in Figure 3A showed the cellular fluorescence distribution of FITC-labeled dendrimers after 2 hours incubation with C6 cells. FITC-labeled dendrimers showed green fluorescence and the nuclei stained with DAPI showed blue fluorescence. C6 cell uptake of both PAMAM and the modified carrier was observed, but the PAMAM group had the strongest fluorescence intensity, whereas the PEG-PAMAM had the weakest fluorescence intensity. RGDyC modification could increase the carrier uptake by C6 cells, which is consistent with the results of cell uptake. With the masking of positive charge through a small modification in the flexible long-chain PEG, the in vitro toxicity of PAMAM was significantly reduced and the cell uptake also inevitably decreased.
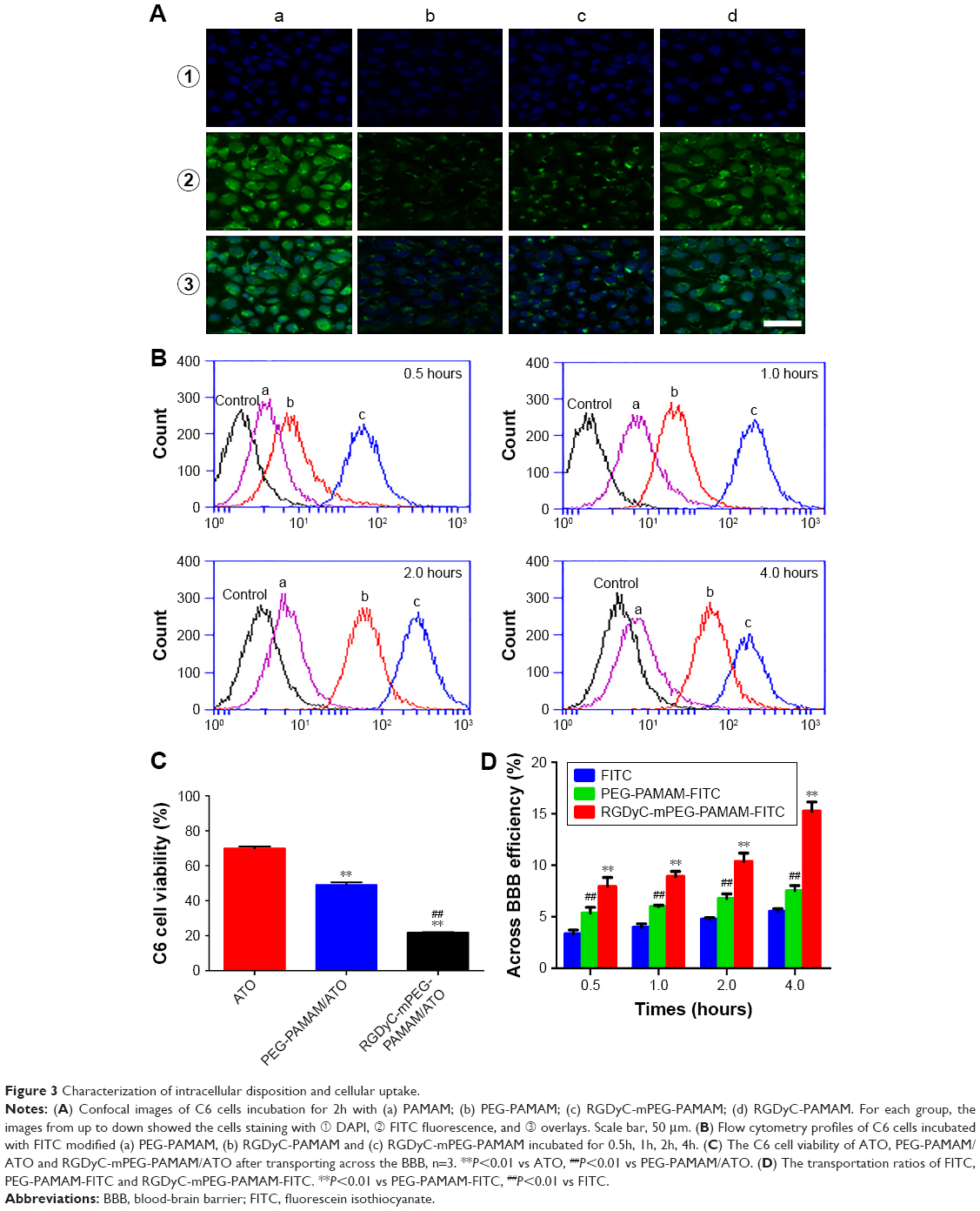 | Figure 3 Characterization of intracellular disposition and cellular uptake. |
As shown in Figure 3B, C6 cells could uptake each carrier group, after incubating for 0.5, 1, 2, and 4 hours. The amount of uptake was the maximum in unmodified RGDyC-PAMAM group. The RGDyC-PEG-PAMAM group was second, but the uptake was significantly decreased in the PEG-PAMAM group; this was mainly due to modification of the long-chain PEG, which could mask the positive charge of the PAMAM surface, and further hinder interaction with the cells. The modification of RGDyC, an integrin receptor, could increase the cellular uptake of the carrier to some extent. It further proved that RGDyC could be used as a tumor-targeting group. After incubating for 4 hours, fluorescence intensity of each group was as follows: RGDyC-PAMAM>RGDyC-mPEG-PAMAM>PEG-PAMAM, and there was a significant difference between each group (P<0.01), which indicated that modification of the tumor targeting functional group can increase the affinity between carriers and tumor cells, and the uptake of carriers by the tumor cell, to some extent. However, in our studies, the RGDyC modification effect on cell uptake was less than the PEG shielding effect on the positive charge of PAMAM. Compared with RGDyC-mPEG-PAMAM, RGDyC-PAMAM showed higher hemolytic toxicity in vitro. Considering the safety of a drug delivery system, we select RGDyC-mPEG-PAMAM as the main research object in the following experiment.
BBB penetration in vitro
In vitro BBB model was established to evaluate the BBB penetration ability of ATO formulations. Compared with ATO-sol, PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO (Figure 3C) had better inhibition activity of C6 cell after BBB penetration (P<0.01). In addition, the BBB penetration ability of RGDyC modified group was significantly enhanced (P<0.01). Combined with cytotoxicity and BBB penetration in vitro assay, although ATO-sol showed the best inhibition on C6 cells in vitro, it had poor inhibition in the BBB model. It is consistent with the previous report that it is difficult for ATO to cross the BBB.41 Compared with the unmodified group, RGDyC modification showed a higher inhibition rate, and it further illustrated the rationality of using RGDyC as a tumor targeting ligand.
As shown in Figure 3D, after incubating for 240 minutes, the transportation ratios of FITC, PEG-PAMAM-FITC, or RGDyC-mPEG-PAMAM-FITC were less 4.53%, 7.53%, and 15.26%, respectively. The BBB penetration transportation ratios of RGDyC-mPEG-PAMAM-FITC were significantly enhanced with the modified RGDyC, compared with PEG-PAMAM-FITC (P<0.01), while a time-dependent transport profile was observed, especially in the groups of RGDyC-mPEG-PAMAM-FITC.
However, studies have shown that the interaction between receptors and ligands could occur only in close proximity.49 So the target molecules cannot carry nanoparticles forward to the target site. The targeting nanoparticles are also required to rely on EPR effect to reach the target site. The target molecules interact with the cell surface receptor and then promote the nanoparticles into the cell. We believed that, modification with targeting ligands could improve the amount and rate of the nanoparticle intake into tumor cells, so that the target nanoparticles in the blood circulation are mainly distributed to the tumor sites.
Cell cycle and apoptosis analysis
To determine the possible effect of cell cycle phases, C6 cells were treated with ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO for 24 hours. As shown in Figure 4A and B, the cell population was arrested in G2-M with 34.08%, 26.25%, and 29.96% of ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO, respectively, whereas the control group in that phase was only 10.97%. The results suggested that the ATO formulations could inhibit the proliferation of C6 at G2-M phase. However, after transporting across the BBB model, C6 cell cycle perturbations induced by ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO at G2-M phase were 13.91%, 11.4%, and 23.89%, respectively, compared with the control group. The alteration of G2-M phase ratios further proved that RGDyC-mPEG-PAMAM/ATO had the best inhibitory effect on C6 cells after they crossed the BBB in vitro.
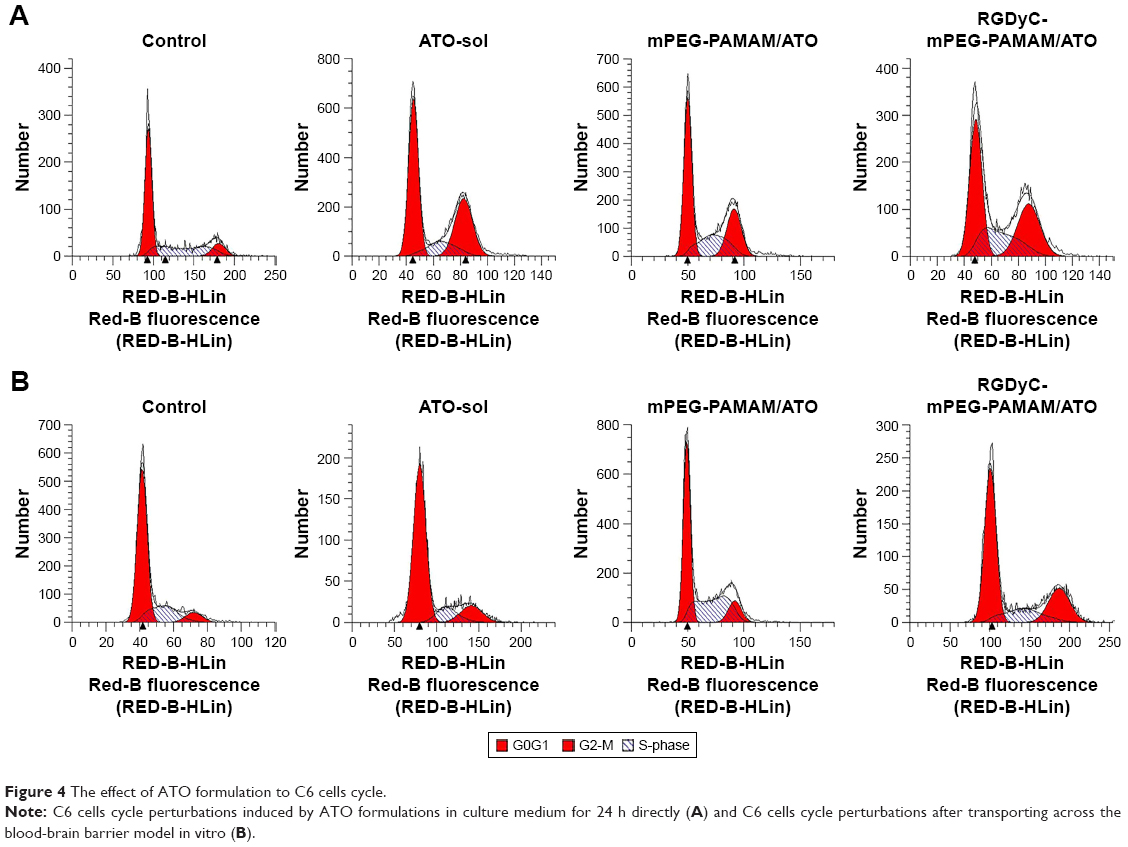 | Figure 4 The effect of ATO formulation to C6 cells cycle. |
The quantitative analysis of the apoptosis activity in control, and treated with different ATO formulations, was measured by flow cytometry. FITC dye and PI were used to label and differentiate between live, apoptotic and dead cells. FITC is apoptotic cell permeant, and PI is apoptotic cell impermeant. As shown in Table 2, ATO-sol induced the most cell apoptosis; however, RGD-mPEG-PAMAM/ATO had been confirmed to have the greatest effect, when compared with other groups, on inducing cancer cell apoptosis after cell incubation for 24 hours in the HBMEC and C6 cells co-cultural model. It could be explained that ATO-sol directly contacted the tumor cells and affected them, whereas the ATO in nanoparticles must be released before it could play a role. At the same time, the amount of its release is also limited, according to the release assay in vitro. But in the BBB model, the ATO-sol was limited across the BBB, and RGD-mPEG-PAMAM/ATO had better tumor targeting for improving endocytosis, thereby enhancing antiproliferation and cell apoptosis. Although there was only a 12% difference between PEG-PAMAM/ATO and RGD-mPEG-PAMAM/ATO groups, the RGDyC-mPEG-PAMAM/ATO group showed close to a threefold increase in apoptosis cells percentage than the PEG-PAMAM/ATO group.
 | Table 2 The apoptosis assay of C6 cells induced by ATO formulations in culture medium for 24 hours with or without across BBB model in vitro (n=3) |
Pharmacokinetic and efficacy study
The pharmacokinetic behaviors of ATO formulations were investigated, after intravenous administration of the four formulations of saline, ATO-sol, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO. The corresponding pharmacokinetic parameters are depicted in Figure 5A and Table 3. The results showed that ATO-sol had a short retention time in the body and was eliminated fast. Compared with the rapidly removed ATO-sol in plasma, PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO had a longer blood circulation time. After the PAMAM carrier encapsulated ATO, the elimination half-life (T1/2β) of PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO was 3.21 and 4.05 times higher than that of ATO-sol. The area under the curve (AUC0→t) was 3.52 and 2.93 times higher than that of ATO-sol, and the mean residence time was 3.45 and 4.30 times higher than that of ATO-sol, respectively. But the clearance rate was significantly reduced compared with that of ATO-sol (P<0.01). All these results manifested that the clearance of the drug in the plasma was significantly slower and the circulation profile was longer, after ATO was encapsulated by PAMAM. The RGDyC-mPEG-PAMAM/ATO potentially improved the tumor targeting and therapeutic index in vivo.
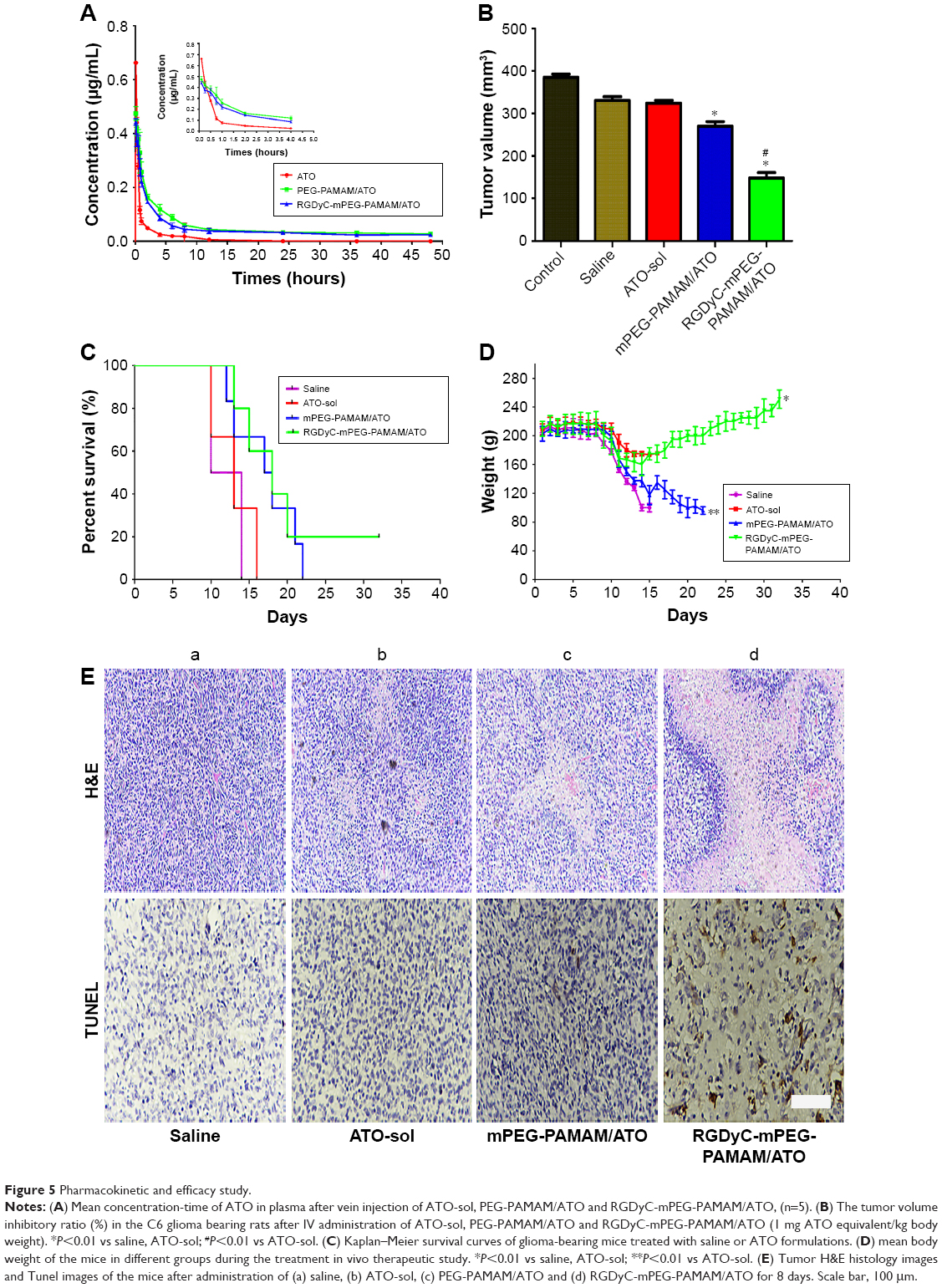 | Figure 5 Pharmacokinetic and efficacy study. |
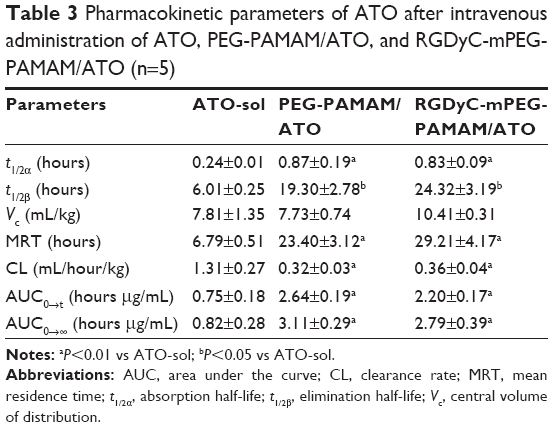 | Table 3 Pharmacokinetic parameters of ATO after intravenous administration of ATO, PEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO (n=5) |
ATO has been approved by the FDA for the treatment of refractory APL, and a variety of explanations have been proposed for its treatment mechanism, which include active oxygen-mediated apoptosis, promoting cell differentiation, inhibiting neovascularization, and cell migration and invasion.50–53 However, the clinical application of ATO is limited due to its rapid renal clearance and dose-limiting toxicity. In our study, ATO-sol released quickly in vitro at pH 5.5 and 7.4, and the cumulative release rate after 2 hours had reached >95%. After being encapsulated into RGDyC-mPEG-PAMAM and PEG-PAMAM, it had significantly sustained release and pH-sensitive properties. There was no significant difference between PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO groups in pharmacokinetic experiments. Therefore, we speculated that the sustained release and the long circulation features of the drug delivery system were mainly due to the modification of PEG on the PAMAM surface. This was consistent with the previously reported literature.37,38 The steric hindrance and a hydrophilic protective layer were formed on the surface of nanocomposites when the PEG hydrophilic chains were introduced. This made them escape from the reticular endothelial system and then extend the circulation time in vivo.
Therapeutic efficacy study
To evaluate the therapeutic effect of ATO-polymer conjugates, Wistar rats bearing glioma were used in this study. The tumor volume shown in Figure 5B was 324.21±7.02, 2,700.41±10.44, and 148.46±12.26 with ATO-sol, mPEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO, respectively. Compared with other groups, RGDyC-mPEG-PAMAM/ATO showed a significant reduction in tumor volume (P<0.01).
Survival curve of C6 glioma-bearing rats after treatment with different formulas is shown in Figure 5C; the median survival time of rats was 18.5±2.3, 21±3.7, 23±5.6, and 30±7.4 days, after treatment with saline, ATO-sol, mPEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO, respectively (P<0.05). The body weight was taken on a daily basis (Figure 5D); the saline group and ATO-sol group showed obvious weight loss, whereas the other groups did not. We speculated that the tumors grow rapidly in the saline group, and the free ATO caused toxicity in the ATO-sol group, which caused weight loss. However, PEGylated PAMAM-encapsulated ATO could reduce the toxicity of free ATO in blood circulation, and the RGDyC modification had the ability of targeting integrin αvβ3 overexpressed in tumor cells; thus, RGDyC-mPEG-PAMAM/ATO had the best antitumor efficacy. Overall, RGDyC-mPEG-PAMAM/ATO, with targeting and controlled release, showed the best inhibitory effect on tumor growth. Meanwhile, the phenomenon of hair loss was serious in ATO-sol group, and other groups had no hair loss. It is indicated that the free ATO may have skin irritation, but it is weakened after being loaded by the delivery system.
To further assess the antitumor efficacy, we conducted H&E staining of tumor tissues. The apoptosis of different groups (Figure 5E) showed that the tumor tissue displayed a necrotic response after treatment with ATO-sol, mPEG-PAMAM/ATO, and RGDyC-mPEG-PAMAM/ATO. The RGDyC-mPEG-PAMAM/ATO group exhibited the largest area of cell necrosis compared with that in ATO-sol and mPEG-PAMAM/ATO groups.
The cell apoptosis in tumor tissue was also evidently identified by TUNEL assay, in which the dead cells were stained brown. As shown in Figure 5E, the tumor tissue treated with normal saline group revealed malignancy. Nucleus of the glioma cells was large and oval shaped with more chromatin and visible binucleolates. In contrast, tumor sections treated with RGDyC-mPEG-PAMAM/ATO clearly exhibited severe apoptosis and a significant tissue loss across a large tumor area. The rats injected with RGDyC-mPEG-PAMAM/ATO had much more remarkable cell apoptosis induced than when treated with saline and mPEG-PAMAM/ATO. These results reinforced the conclusion that RGDyC-mPEG-PAMAM/ATO exhibited the best antitumor efficacy among all the groups, which was consistent with the H&E result. Considering the above results, RGDyC-mPEG-PAMAM/ATO significantly inhibited glioma tumor growth in Wistar rats by suppression of cell proliferation and induction of apoptosis. We speculated the enhanced efficacy of RGDyC-mPEG-PAMAM/ATO may be caused by an ideal particle size, which was suitable for crossing the BBB. The tumor targeting ability of peptide RGDyC could be specifically recognized by integrin αvβ3, so RGDyC-mPEG-PAMAM/ATO had a better antitumor effect when compared with mPEG-PAMAM/ATO.
Conclusions
A series of ATO delivery systems, with PEG modification at a molar ratio of 64:1, and different drug conjugations (PEG-PAMAM/ATO and RGDyC-mPEG-PAMAM/ATO), were evaluated in vitro and in vivo glioma models. Results indicated that the drug delivery systems were tumor targeting and had long circulation, after modification with RGDyC and PEG. Additionally, the antitumor efficacy in vitro and in vivo was improved by RGDyC-mPEG-PAMAM/ATO remarkably, compared with unencapsulated ATO. In summary, RGDyC-mPEG-PAMAM/ATO might be a promising strategy for glioma targeting therapy.
Acknowledgments
National Natural Science Foundation of China (Nos 81473361, 81673607, and 81603303) and New Products of TCM Senile Diseases Co-Innovation Center of Hubei (No 15111903) financially supported this work. We thank Feiye Zhu (Department of Analysis and Testing, Zhejiang Chinese Medical University, China) for the TEM measurements. We thank Dr Huajun Zhao (Department of Pharmacology, Zhejiang Chinese Medical University, China) for providing us with technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14(Suppl 5):v1–v49. | ||
Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. | ||
Liu D, Auguste DT. Cancer targeted therapeutics: from molecules to drug delivery vehicles. J Control Release. 2015;219:632–643. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Stan AC, Casares S, Radu D, Walter GF, Brumeanu TD. Doxorubicin-induced cell death in highly invasive human gliomas. Anticancer Res. 1999;19(2A):941–950. | ||
Prados MD, Schold SC, Spence AM, et al. Phase II study of paclitaxel in patients with recurrent malignant glioma. J Clin Oncol. 1996;14(8):2316–2321. | ||
Kumthekar P, Grimm S, Chandler J, et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J Neurooncol. 2017;133(3):589–594. | ||
Peng Y, Zhao Z, Liu T, et al. Smart human-serum-albumin-As2O3 nanodrug with self-amplified folate receptor-targeting ability for chronic myeloid leukemia treatment. Angew Chem Int Ed Engl. 2017;56(36):10845–10849. | ||
Zhang Z, Liu H, Zhou H, et al. A facile route to core-shell nanoparticulate formation of arsenic trioxide for effective solid tumor treatment. Nanoscale. 2016;8(7):4373–4380. | ||
Cheung WM, Chu PW, Kwong YL. Effects of arsenic trioxide on the cellular proliferation, apoptosis and differentiation of human neuroblastoma cells. Cancer Lett. 2007;246(1–2):122–128. | ||
Akhtar A, Xiaoyan Wang S, Ghali L, Bell C, Wen X. Recent advances in arsenic trioxide encapsulated nanoparticles as drug delivery agents to solid cancers. J Biomed Res. 2017;31(3):177–188. | ||
Sun Y, Wang C, Wang L, Dai Z, Yang K. Arsenic trioxide induces apoptosis and the formation of reactive oxygen species in rat glioma cells. Cell Mol Biol Lett. 2018;23:13. | ||
Cheng Y, Li Y, Ma C, et al. Arsenic trioxide inhibits glioma cell growth through induction of telomerase displacement and telomere dysfunction. Oncotarget. 2016;7(11):12682–12692. | ||
Chen H, Macdonald RC, Li S, Krett NL, Rosen ST, O’Halloran TV. Lipid encapsulation of arsenic trioxide attenuates cytotoxicity and allows for controlled anticancer drug release. J Am Chem Soc. 2006;128(41):13348–13349. | ||
Song XL, Liu S, Jiang Y, et al. Targeting vincristine plus tetrandrine liposomes modified with DSPE-PEG2000-transferrin in treatment of brain glioma. Eur J Pharm Sci. 2017;96:129–140. | ||
Zhou J, Meng R, Sui X, Li W, Yang B. Various tolerances to arsenic trioxide between human cortical neurons and leukemic cells. Sci China C Life Sci. 2006;49(6):567–572. | ||
Emadi A, Gore SD. Arsenic trioxide – an old drug rediscovered. Blood Rev. 2010;24(4–5):191–199. | ||
Au WY, Kwong YL. Arsenic trioxide: safety issues and their management. Acta Pharmacol Sin. 2008;29(3):296–304. | ||
Chung EJ, Cheng Y, Morshed R, et al. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials. 2014;35(4):1249–1256. | ||
Wang H, Huang Q, Chang H, Xiao J, Cheng Y. Stimuli-responsive dendrimers in drug delivery. Biomater Sci. 2016;4(3):375–390. | ||
Hsu HJ, Bugno J, Lee SR, Hong S. Dendrimer-based nanocarriers: a versatile platform for drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(1):e1409. | ||
Li Z, Fan Z, Xu Y, et al. pH-Sensitive and thermosensitive hydrogels as stem-cell carriers for cardiac therapy. ACS Appl Mater Interfaces. 2016;8(17):10752–10760. | ||
Zhang X, Yao S, Liu C, Jiang Y. Tumor tropic delivery of doxorubicin-polymer conjugates using mesenchymal stem cells for glioma therapy. Biomaterials. 2015;39:269–281. | ||
Saraswathy M, Knight GT, Pilla S, Ashton RS, Gong S. Multifunctional drug nanocarriers formed by cRGD-conjugated βCD-PAMAM-PEG for targeted cancer therapy. Colloids Surf B Biointerfaces. 2015;126:590–597. | ||
Qin J, Chen D, Hu H, Cui Q, Qiao M, Chen B. Surface modification of RGD-liposomes for selective drug delivery to monocytes/neutrophils in brain. Chem Pharm Bull. 2007;55(8):1192–1197. | ||
Zhan C, Meng Q, Li Q, Feng L, Zhu J, Lu W. Cyclic RGD-polyethylene glycol-polyethylenimine for intracranial glioblastoma-targeted gene delivery. Chem Asian J. 2012;7(1):91–96. | ||
Kim YM, Park SC, Jang MK. Targeted gene delivery of polyethyleneimine-grafted chitosan with RGD dendrimer peptide in αvβ3 integrin-overexpressing tumor cells. Carbohydr Polym. 2017;174:1059–1068. | ||
Zhu S, Qian L, Hong M, Zhang L, Pei Y, Jiang Y. RGD-modified PEG-PAMAM-DOX conjugate: in vitro and in vivo targeting to both tumor neovascular endothelial cells and tumor cells. Adv Mater. 2011;23(12):H84–H89. | ||
Li Q, Xu W. Novel anticancer targets and drug discovery in post genomic age. Curr Med Chem Anticancer Agents. 2005;5(1):53–63. | ||
Kesharwani P, Iyer AK. Recent advances in dendrimer-based nanovectors for tumor-targeted drug and gene delivery. Drug Discov Today. 2015;20(5):536–547. | ||
Luong D, Kesharwani P, Deshmukh R, et al. PEGylated PAMAM dendrimers: enhancing efficacy and mitigating toxicity for effective anticancer drug and gene delivery. Acta Biomater. 2016;43:14–29. | ||
Liu R, Sun M, Liu X. Interplay of stimuli-responsiveness, drug loading and release for a surface-engineered dendrimer delivery system. Int J Pharm. 2014;462(1–2):103–107. | ||
Ma P, Sun Y, Chen J, et al. Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate. Drug Deliv. 2018;25(1):153–165. | ||
Thanh VM, Nguyen TH, Tran TV, et al. Low systemic toxicity nanocarriers fabricated from heparin-mPEG and PAMAM dendrimers for controlled drug release. Mater Sci Eng C Mater Biol Appl. 2018;82:291–298. | ||
Taghavi Pourianazar N, Mutlu P, Gunduz U. Bioapplications of poly(amidoamine) (PAMAM) dendrimers in nanomedicine. J Nanopart Res. 2014;16(4):2342. | ||
Bai S, Ahsan F. Synthesis and evaluation of pegylated dendrimeric nanocarrier for pulmonary delivery of low molecular weight heparin. Pharm Res. 2009;26(3):539–548. | ||
Bai S, Thomas C, Rawat A, Ahsan F. Recent progress in dendrimer-based nanocarriers. Crit Rev Ther Drug Carrier Syst. 2006;23(6):437–495. | ||
Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34–47. | ||
Zhang L, Zhu S, Qian L, Pei Y, Qiu Y, Jiang Y. RGD-modified PEG-PAMAM-DOX conjugates: in vitro and in vivo studies for glioma. Eur J Pharm Biopharm. 2011;79(2):232–240. | ||
Xiao X, Liu Y, Guo M, et al. pH-Triggered sustained release of arsenic trioxide by polyacrylic acid capped mesoporous silica nanoparticles for solid tumor treatment in vitro and in vivo. J Biomater Appl. 2016;31(1):23–35. | ||
Fei W, Zhang Y, Han S, et al. RGD conjugated liposome-hollow silica hybrid nanovehicles for targeted and controlled delivery of arsenic trioxide against hepatic carcinoma. Int J Pharm. 2017;519(1–2):250–262. | ||
Oddone N, Zambrana AI, Tassano M, Porcal W, Cabral P, Benech JC. Cell uptake mechanisms of PAMAM G4-FITC dendrimer in human myometrial cells. J Nanopart Res. 2013;15(7):1776. | ||
Russ V, Günther M, Halama A, Ogris M, Wagner E. Oligoethylenimine-grafted polypropylenimine dendrimers as degradable and biocompatible synthetic vectors for gene delivery. J Control Release. 2008;132(2):131–140. | ||
He H, Li Y, Jia XR, et al. PEGylated Poly(amidoamine) dendrimer-based dual-targeting carrier for treating brain tumors. Biomaterials. 2011;32(2):478–487. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Zhang J, Han J, Zhang X, et al. Polymeric nanoparticles based on chitooligosaccharide as drug carriers for co-delivery of all-trans-retinoic acid and paclitaxel. Carbohydr Polym. 2015;129:25–34. | ||
Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Berenson JR, Yeh HS. Arsenic compounds in the treatment of multiple myeloma: a new role for a historical remedy. Clin Lymphoma Myeloma. 2006;7(3):192–198. | ||
Baj G, Arnulfo A, Deaglio S, et al. Arsenic trioxide and breast cancer: analysis of the apoptotic, differentiative and immunomodulatory effects. Breast Cancer Res Treat. 2002;73(1):61–73. | ||
Miller WH, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62(14):3893–3903. | ||
Park MJ, Lee JY, Kwak HJ, et al. Arsenic trioxide (As2O3) inhibits invasion of HT1080 human fibrosarcoma cells: role of nuclear factor-kappaB and reactive oxygen species. J Cell Biochem. 2005;95(5):955–969. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.