")
Back to Journals » The Application of Clinical Genetics » Volume 7
A novel nonsense mutation of the KAL1 gene (p.Trp204*) in Kallmann syndrome
Authors El Husny AS, Raiol-Moraes M, Fernandes-Caldato M, Ribeiro-dos-Santos Â
Received 18 March 2014
Accepted for publication 7 May 2014
Published 30 September 2014 Volume 2014:7 Pages 177—182
DOI https://doi.org/10.2147/TACG.S64280
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Antonette Souto El Husny,1 Milene Raiol-Moraes,1 Milena Coelho Fernandes-Caldato,2,3 Ândrea Ribeiro-dos-Santos1
1Laboratory of Human and Medical Genetics, Federal University of Pará, 2João de Barros Barreto University Hospital, Federal University of Pará, 3University Center of Pará, CESUPA, Belém, Pará, Brazil
Objective: To describe a novel KAL1 mutation in patients affected by Kallmann syndrome.
Setting: Endocrinology Clinic of the João de Barros Barreto University Hospital – Federal University of Pará, Brazil.
Methods: Clinical examination, hormone assays and sequencing of exons 5, 6 and 9 of the KAL1 gene in four Brazilian brothers with Kallmann syndrome.
Results: Detected a novel KAL1 mutation, c.612G>A/p.Trp204*, in four hemizygous brothers with Kallmann syndrome, and five heterozygous female family members.
Conclusion: The novel p.Trp204* mutation of the KAL1 gene results in the production of a truncated anosmin-1 enzyme in patients with Kallmann syndrome. This finding broadens the spectrum of pathogenic mutations for this disease.
Keywords: Kallmann syndrome, KAL1 gene, anosmin-1, mutation
Introduction
Deficiencies in gonadotropins and sex hormones are detrimental for various stages of sexual maturation and lead to hypogonadotropic hypogonadism (HH).1 The isolated form of HH is rare; however, its association with certain olfactory deficits (anosmia/hyposmia) that are part of Kallmann syndrome (KS) – OMIM 300836 – makes it more common.
KS can occur sporadically or be hereditary, and its incidence is 1:8,000 in males and 1:40,000 in females.2,3 Hereditary cases have been well documented but sporadic cases are more common, which suggests that de novo mutations are an important cause.4–6
There are at least six well-known genes involved in the etiology of KS: the KAL1 gene, which encodes the anosmin-1 glycoprotein; the FGFR1 (KAL2) gene, which encodes fibroblast growth factor receptor 1; the PROKR2 (KAL3) and PROK2 (KAL4) genes, which encode the prokineticin-2 receptor and prokineticin-2, respectively; and the CDH7 (KAL5) and FGF8 (KAL6) genes. Other genes have been reported to be associated with KS: NELF, WDR11, HS6ST1, SEMA3A, HESX1.7,8
X-linked KS (X-KS) typically involves mutations in the KAL1 gene, which is found on chromosome Xq22.3; such mutations can alter the form and function of the anosmin-1 protein, which is involved in the migration of olfactory neurons and neurons that produce gonadotropin-releasing hormone. Without anosmin-1, migration is impaired, which affects the patient’s olfaction and gonadotropic hormone production.6,9,10
In combination with the profile of HH and anosmia/hyposmia, a patient may present with various other alterations; the following are some common findings: unilateral renal agenesis; cavus foot; cleft lip and palate; synkinesis; and auditory, visual or other sensory disturbances.7 In any case, great phenotypic heterogeneity has been demonstrated in various studies, especially those of Quinton et al,11 Oliveira et al12 and Hardelin and Dodé,13 suggesting that the activity of epigenetic factors or modifying genes COULD determine phenotypic variability.
Detailed clinical assessment of patients who are affected by HH and have anosmia/hyposmia has led to the discovery of other significant changes in this group, and adding genetic analysis permits appropriate genetic counseling and helps molecular understanding of KS.
Materials and methods
Ethical issues
Ethical consent was obtained according to the Declaration of Helsinki. Ethical approval was obtained from the Brazilian National Committee on Research Ethics (CEP/HUJBB – Protocol Number 142/2008). All of the patients assessed agreed to participate in the study by signing an informed consent form.
Description of the samples
A clinical and molecular study was conducted with four KS patients and 12 of their family members, for a total of 16 individuals. The clinical study of those affected assessed the following parameters: 1) pattern of family inheritance; 2) stage of sexual maturation; 3) anthropometric data; 4) hormone levels (follicle-stimulating hormone [FSH], luteinizing hormone [LH], testosterone, estradiol, thyroid-stimulating hormone [TSH], free T4); 5) other associated conditions including cryptorchidism, renal malformation, synkinesis, and others; and 6) molecular analysis of the KAL1 gene (NM_000216.2).
The family members underwent an assessment focused on the investigation of anosmia, identification of associated malformations and genetic study.
The olfactory assessment was conducted using the semiological test of the first cranial nerve based on the presentation of test tubes containing ten different common everyday odors to be identified by the patients and their family members. Chloroform was used to test for false positives because the olfactory nerve is not required to detect it.
Molecular analysis
DNA was extracted from peripheral blood samples taken from each individual by the phenol-chloroform method.14 Exons 5, 6 and 9 of the KAL1 gene (NM_000216.2) were selected for amplification by polymerase chain reaction (PCR) because they contained the greatest number of mutations described at the time of the present study.15 The primers used for amplification reaction are shown in Table S1. The final reaction volume was 25 μL and included 100 ng of template DNA, 0.1 μM dNTPs, 0.1 μM of each primer, 1.5 μM MgCl2, 1× PCR buffer (10 mM Tris-HCl, pH 8.4, and 50 mM KCl) and 1 U Platinum® Taq DNA Polymerase (Invitrogen). After initial denaturing for 10 min at 95°C, the samples underwent 30 cycles under the following parameters: denaturing at 94°C for 1 minute, annealing at 54°C for 1 minute for exon 5, and at 56°C for 1 minute for exons 6 and 9, and extension at 72°C for 1 minute. The samples were checked by electrophoresis on a 1.5% agarose gel.
The sequencing reaction was performed using 1 μL of purified PCR product, 0.5 μL reverse primer, 2.0 μL of Big Dye Terminator V3.1 Cycle Sequencing Kit (Applied Biosystems®) and sterile deionized water to a final volume of 20 μL. The reaction was performed on a thermocycler with an initial denaturing at 96°C for 2 minutes, followed by 36 cycles of 96°C for 30 seconds, 54/56°C for 20 seconds (for exon 5, and exons 6 and 9, respectively), and 60°C for 4 minutes. The samples were treated and sent to an ABI 3110 sequencer for electrophoresis on 4.5% acrylamide gel (10 mL of 18.5 acrylamide:1 bis-acrylamide, 36 g of urea and 60 mL of 1× TBE). The sequencing results were interpreted using the ABI Analysis Software®.
Results
Four male patients in the third decade of life presented with the clinical and laboratory profile of HH (Table 1) combined with anosmia confirmed by the semiological test. Four other features were observed that are common in KS (Table 2): unilateral renal agenesis, cryptorchidism, synkinesis and dental agenesis. Nystagmus, ptosis, auricular dysgenesis and cleft lip/palate were not found in any patient.
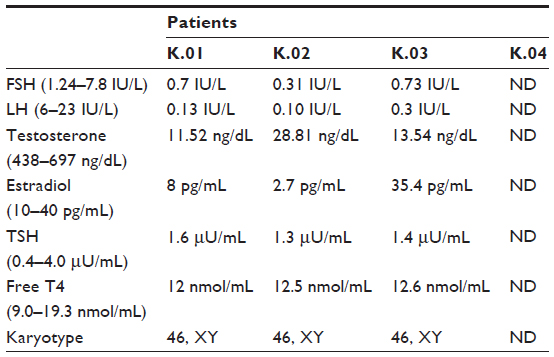 | Table 1 Summary of laboratorial tests in the first assessment |
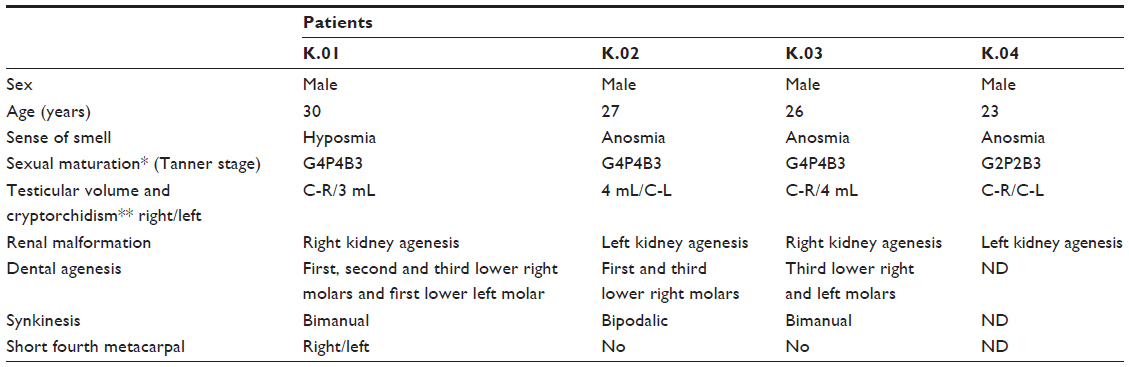 | Table 2 Summary of clinical findings in Kallmann syndrome patients |
The molecular analysis detected the transversion of G to A at position 612 on exon 5 of the KAL1 gene. This mutation was hemizygous in the four affected brothers. Five females heterozygous for this mutation were identified in the family, including the patients’ mother (Figure 1). The same mutation was not found in the remaining seven family members. This novel mutation generates a stop codon instead of a tryptophan amino acid at position 204 of the anosmin-1 protein.
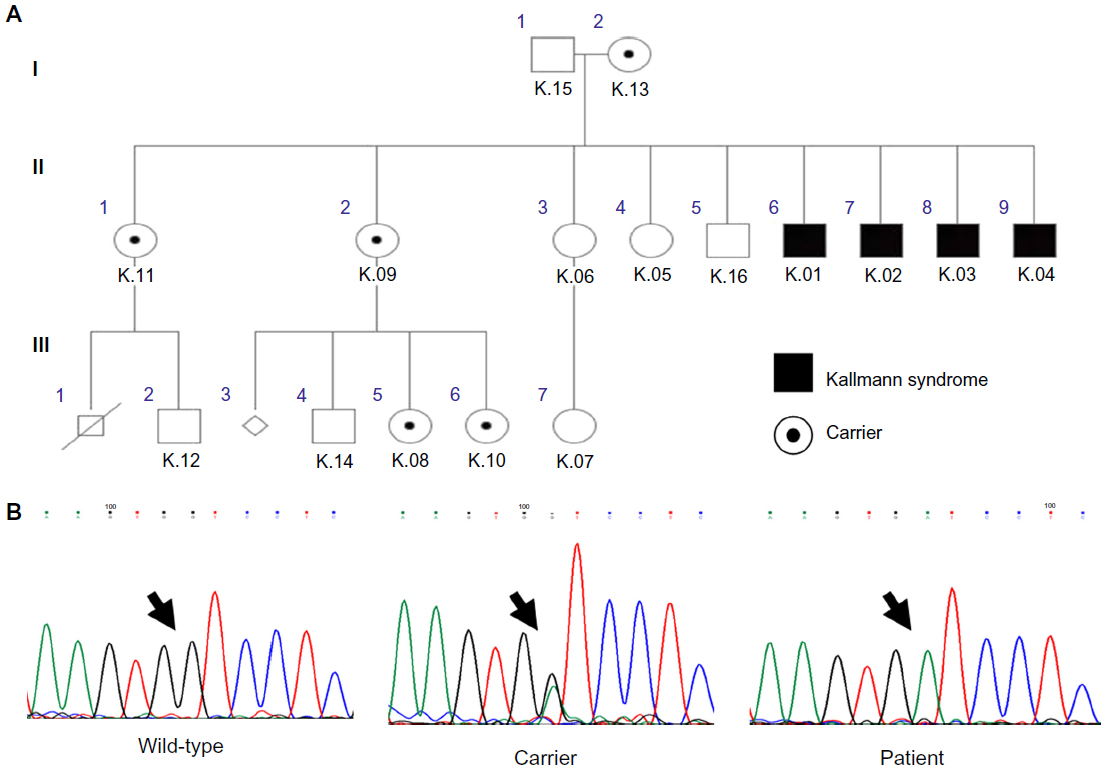 | Figure 1 The inheritance pattern for the family studied shows the hemizygous affected individuals with Kallmann syndrome, and heterozygous carriers who were detected by genetic analysis (A). The chromatogram patterns found in the study are shown above with mutations indicated by an arrow (B). |
Discussion
KS is one of the most common causes of HH.4 In male patients (who have a greater incidence) who present with a clear picture including delayed puberty and, therefore, likely hypogonadism, it is appropriate to question or investigate their olfactory function with possible investigation oriented toward KS. In the cases studied, the complaint of difficulty perceiving odors aided the clinical study.
X-linked inheritance is the best clinically characterized form of the disease.11 Some associated conditions are more common in this type of inheritance, such as dental agenesis, synkinesis and renal agenesis.16 All of these features were observed in the probands, reinforcing the suspicion for X-linked inheritance.
The cryptorchidism found in the four patients should be of high importance because it is often the most evident clinical feature, especially in pediatric patients when it is not possible to apply olfactory tests and hormone levels are of little value. In such cases, it is important to look for other signs such as the presence of micropenis. Additionally, a family history of the characteristic signs of KS should be investigated.17
Renal agenesis is present in 35%–40% of X-linked KS cases.18 Other malformations of the urinary tract can also be found in such patients, including horseshoe kidney, kidney rotation, hydronephrosis, duplicated ureter, etc.19 The importance of imaging exams, especially abdominal ultrasound, is clear because they can show renal abnormalities that are not clinically evident. Therefore, such exams should be considered imperative for KS patients.
Georgopoulos et al20 and Sato et al16 reported a greater incidence of right renal agenesis with a tendency for KS patients to present with this side affected, especially those with a KAL1 mutation. Left renal agenesis is more commonly found in individuals without KS. The mechanisms that explain these findings are still unknown.
Synkinesis, which is considered the marker of X-linked inheritance and was present in the probands in this study, can be explained by an absence of inhibitory fibers that connect the two motor cortices, resulting in fewer fibers in the corpus callosum or even agenesis.11 These alterations were not found in the studied patients with magnetic resonance imaging.
An analysis of the type of mutation that generates a protein truncated at codon 204 revealed interrupted synthesis at the first fibronectin type III-like domain. This finding agrees with the analysis by Georgopoulos et al,20 who found many mutations in this domain related to renal alterations. A more recent study could not specifically attribute this renal phenotype to any of the genes known to cause the syndrome.7
Most cases require lifelong sex steroid hormone replacement to maintain sexual features and bone density, although there have been cases in which the profile of hypogonadism reversed after a period of hormone replacement21 in patients who had not been genetically screened and in patients with detected mutations in the KAL122 and KAL2/FGFR1 genes.23
Reversal of the disease is achieved when the individual maintains normal serum levels of sex hormones after a long period without hormone replacement, allowing the possibility of endogenous maintenance of complete virilization and fertility.22 Although the estimated number of reversible cases is low, Quinton et al21 have suggested that they are underestimated due to interruption of treatment by the patient, now eugonadal, without any effect on their clinical progression. Additionally, there may be a lack of follow-up by specialized professionals. Therefore, there may be reversible cases that remain unidentified.
Advances in the understanding of the genetic bases of KS have allowed increasingly complex molecular studies that can provide additional information on the pathogenesis of the disease and the genotype–phenotype associations.7 Although it has been widely studied, novel recessive mutations of the KAL1 gene are still being described in the literature, as in the present study and in a recent analysis of Chinese probands.24
In conclusion, the clinical and molecular studies conducted herein broadened the spectrum of the known pathogenic recessive mutations related to KS. The results will serve as the basis for appropriate genetic counseling to inform patients and their families about the disease and its repercussions, especially reproductive issues. Additionally, counselors can warn them about the risk of recurrence in the family and suggest early diagnosis of future patients so that treatment can be initiated at the ideal time.
Acknowledgments
The authors thank all of the patients and the Endocrinology team from João de Barros Barreto University Hospital – UFPA and the Genetics team from the Laboratory of Human and Medical Genetics – UFPA. Financial support was received from UFPA(PROPESP)/FADESP and CAPES.
Disclosure
The authors have no conflicts of interest in this work.
References
Tusset C, Trarbach EB, Silveira LFG, Beneduzzi D, Montenegro LR, Latronico AC. Clinical and molecular aspects of congenital isolated hypogonadotropic hypogonadism. Arq Bras Endocrinol Metabol. 2011;55:501–511. | |
Dodé C, Hardelin JP. Clinical genetics of Kallmann syndrome. Annales d’Endocrinologie. 2010;71:149–157. | |
Ribeiro RS, Abucham J. Kallmann syndrome: a historical, clinical and molecular review. Arq Bras Endocinol Metabol. 2008;52:8–17. | |
Jansen C, Hendricks-Stegeman BI, Jansen M. A novel nonsense mutation of the KAL gene in two brothers with Kallmann syndrome. Horm Res. 2000;53:207–212. | |
Hardelin JP. Kallmann syndrome: towards molecular pathogenesis. zMol Cell Endocrinol. 2001;179:75–81. | |
Cadman SM, Kim SH, Hu Y, González-Martínez D, Bouloux PM. Molecular pathogenesis of Kallmann syndrome. Horm Res. 2007;67:231–242. | |
Costa-Barbosa FA, Balasubramanian R, Keefe KW, et al. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. 2013;98(5):E943–E953. | |
Newbern K, Natrajan N, Kim HG, et al. Identification of HESX1 mutations in Kallmann syndrome. Fertil Steril. 2013;99(7):1821–1837. | |
Cariboni A, Pimpinelli F, Colamarino S, et al. The product of X-linked Kallmann’s syndrome gene (KAL-1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet. 2004;13:2781–2791. | |
Fechner A, Fong S, Mcgovern P. A review of Kallmann syndrome: genetics, pathophysiology and clinical management. Obstet Gynecol Surv. 2008;63:189–193. | |
Quinton R, Duke VM, De Zoysa PA, Platts AD, et al. The Neuroradiology of Kallmann’s syndrome: a genotypic and phenotypic analysis. J Clin Endocrinol Metab. 1996;81(8):3010–3017. | |
Oliveira LMB, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86(4):1532–1538. | |
Hardelin JP, Dodé C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex Dev. 2008;2:181–193. | |
Sambrook J, Russel DW. Molecular Cloning: a Laboratory Manual. 3rd ed. New York: Cold Spring Harbor; 2001. | |
Trarbach EB, Baptista MTM, Garmes HM, Hackel C. Molecular analysis of KAL1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients. J Endocrinol. 2005;187:361–368. | |
Sato N, Katsumata N, Kagami M, et al. Clinical assessment and mutation analisys of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FRFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079–1088. | |
Zenaty D, Bretones P, Lambe C, et al. Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1). Mol Cell Endocrinol. 2006;254–255:78–83. | |
Trarbach EB, Monlleo IL, Porciuncula CGG, Fontes MIB, Baptista MTM, Hackel C. Similar intersticial deletions of the KAL-1 gene in two Brazilian families with X-linked Kallmann syndrome. Genet Mol Biol. 2004;27:337–341. | |
Zenteno JC, Méndez JP, Maya-Núñez G, Ulloa-Aguirre A, Kofman-Alfaro S. Renal abnormalities in patients with Kallmann syndrome. BJU International. 1999;83:383–386. | |
Georgopoulos NA, Koika V, Galli-Tsinopoulou A, et al. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril. 2007;88:1311–1317. | |
Quinton R, Cheow HK, Tymms DJ, Bouloux PMG, Wu FCW, Jacobs HS. Kallmann’s syndrome: is it always for life? Clin Endocrinol. 1999;50:481–485. | |
Ribeiro RS, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol. 2007;156:285–290. | |
Pitteloud N, Acireno JS, Meysing AU, et al. Reversible Kallmann syndrome, delayed puberty and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005;90(3):1317–1322. | |
Zhang S, Xu H, Wang T, Liu G, Liu J. The KAL1 pVal610Ile mutation is a recessive mutation causing Kallmann syndrome. Fertil Steril. 2013;99(6):1520–1523. |
Supplementary material
 | Table S1 Polymerase chain reaction primers used for amplification of KAL1 exons 5, 6 and 9, their annealing temperature and product size |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.