")
Back to Journals » International Journal of Nanomedicine » Volume 15
A Novel Nanoparticle Preparation to Enhance the Gastric Adhesion and Bioavailability of Xanthatin
Authors Zhou Y, Zhu X, Lin S, Zhu C, Wu L, Chen R, Chen Z, Li W
Received 1 March 2020
Accepted for publication 30 June 2020
Published 14 July 2020 Volume 2020:15 Pages 5073—5082
DOI https://doi.org/10.2147/IJN.S252049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Mian Wang
Yaqian Zhou,1,* Xingyu Zhu,2,* Shangyang Lin,1 Chenqi Zhu,1 Li Wu,1 Rui Chen,1 Zhipeng Chen,1 Weidong Li1,3
1College of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, People’s Republic of China; 2Department of Pharmacy and Traditional Chinese Medicine, Jiangsu College of Nursing, Huaian 223001, People’s Republic of China; 3Engineering Center of State Ministry of Education for Standardization of Chinese Medicine Processing, Nanjing University of Chinese Medicine, Nanjing 210023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weidong Li
Engineering Center of State Ministry of Education for Standardization of Chinese Medicine Processing, Nanjing University of Chinese Medicine, Nanjing 210023, People’s Republic of China
Email [email protected]
Objective: To prepare xanthatin (XA)-loaded polydopamine (PDA) nanoparticles (PDA-XA-NPs) and to investigate their adhesion and bioavailability.
Materials and methods: PDA-XA-NPs were synthesized and characterized using transmission electron microscopy, zeta potential analysis and encapsulation efficiency analysis. Their in vitro release kinetics and inhibitory effects on gastric cancer were studied. The adhesion of PDA-XA-NPs was evaluated by in vivo imaging atlas. The pharmacokinetics of PDA-XA-NPs and XA was compared.
Results: PDA-XA-NPs had a spherical shape, a particle size of about 380 nm, an encapsulation efficiency of (82.1 ± 0.02) % and a drug loading capacity of (5.5 ± 0.1)%. The release of PDA-XA-NPs in PBS was stable and slow, without being affected by pH. The adhesion capacity of PDA-XA-NPs for mucin was significantly higher than that of bulk drug. The gastric mucosal retention of PDA-XA-NPs reached 89.1% which significantly exceeded that of XA. In vivo imaging showed that PDA-XA-NPs targeting the stomach were retained for a period of time. The pharmacokinetics study showed that PDA-XA-NPs had a longer retention time and a slower drug release than those of XA. In vitro experiments confirmed that PDA-XA-NPs exerted similar inhibitory effects on gastric cancer to those of XA, which lasted for a period of time.
Conclusion: High-adhesion NPs were constructed. Gastric cancer was targeted by orally administered PDA-XA-NPs, as a potentially feasible therapy. Eventually, the bioavailability of XA was increased.
Keywords: xanthatin, polydopamine, nanoparticles, gastric cancer, target, pharmacokinetics
Introduction
As a sesquiterpene lactone (SL) primarily isolated from Xanthium sibiricum L., xanthatin (XA) has an α-methylene-γ-butyrolactone (αMγL) moiety trans-fused to a seven-membered carbocycle and an unsaturated ketonic side chain. It has been verified as an active compound for suppressing tumor growth.1–3 SLs are a large class of botanically derived compounds with αMγL structures, as potent antimicrobial, antitumor and anti-inflammatory agents. Several SLs, such as artemisinin, thapsigargin and parthenolide, have become lead compounds in clinical cancer trials.4 Yu et al found that XA at non-toxic doses exerted potent anti-angiogenic effects by specifically targeting VEGFR2.5 Meanwhile, XA can inhibit the growth of MKN-45 cells and induce arrest in the G2 phase and apoptotic death.6 However, it is a lowly soluble drug that can be rapidly eliminated from rats after intravenous injection.7 In addition, drugs can hardly exist in the stomach for a long time due to gastric emptying.
Gastric cancer is the fifth most common cancer worldwide, but it has the third-highest mortality rate, particularly in East Asia.8 Early gastric cancers are limited to the mucosa or submucosa, while advanced ones infiltrate through the subserosa nearly to organs or metastasize.9 Currently, many therapies have been developed, including surgery, chemotherapy, radiotherapy, molecular-targeted therapy and immunotherapy,10 among which chemotherapy remains most common due to strong therapeutic efficacy on both primary and metastatic tumors.11 Besides, oral administration is usually preferred owing to high patient compliance and safety.
Gastric adhesive preparations can increase the stomach retention time of bioactive phytochemicals, for which nanoparticle (NP)-based drug delivery systems may be suitable.12 These preparations can stick on the gastric mucosa, thereby prolonging drug absorption in the stomach, allowing local targeting and improving the bioavailability. Since 2007, polydopamine (PDA) has attracted widespread attention in the fields of material chemistry, biomedicine and so on due to remarkable adhesion to almost any material surface and water solubility.13,14 In addition, PDA has been reported to promote drug adhesion.15 As a result, the gastrointestinal adhesion of XA may be augmented by PDA materials to treat gastric cancer in a targeted manner.
Thereby motivated, we herein constructed PDA-XA-NPs and investigated their gastric adhesion. The in vivo pharmacokinetics results of XA and PDA-XA-NPs were compared for the first time, and their inhibitory effects on gastric cancer in vitro were evaluated. The findings provide valuable evidence for designing targeted preparations for gastric cancer and improving the in vivo absorption of SLs represented by XA.
Materials and Methods
Materials
XA (purity ≥98%) and psoralen (purity ≥98%) were obtained from Nanjing World Biological Technology Co., Ltd. (China). Dopamine hydrochloride and cis-platinum (DDP) were purchased from Sigma Aldrich (St. Louis, MO, USA). Cy7-N-hydroxysuccinimide ester (Cy7) was purchased from Lumiprobe (USA). Cell counting kit-8 (CCK-8) was purchased from Shanghai Yisheng Biotechnology Co., Ltd. (China). BCA protein assay kit (enhancement mode) was purchased from Beyotime Biotechnology Co., Ltd. (China). LC-mass spectrometry (MS) grade acetonitrile was obtained from Merck (Darmstadt, Germany). Ultrapure water (18.25 Ω) was daily prepared with a Milli-Q water purification system (Millipore Corporation, Bedford, MA, USA). Aqueous ammonia solution (28.0–30.0% NH3 by weight), DMSO and other reagents were analytically pure and commercially available.
Gastric cancer cell line MKN-45 was purchased from KeyGEN Biotechnology Co., Ltd. (China).
Apparatus
Hydrodynamic diameters and zeta potentials were measured by Zetasizer Nano ZS90 dynamic light scattering (DLS) analyzer (Malvern Instruments, UK). DF-101S magnetic stirrer, SL16R high-speed centrifuge (Thermo Scientific, USA) and 0.00001 g electronic balance (Sartorius BT25S, Germany) were used. E2695 Separations Module was equipped with 2489 UV/visible detector, and the data were processed by EmpowerTM Software Build (Waters, USA).
All separations were performed on a UPLC system (Shimadzu, Kyoto, Japan) equipped with an LC-30AD binary pump, an auto sampler (Model SIL-30SD) and an online degasser (DGU-20A5R). MS was conducted by using 5500 Triple Quad tandem mass spectrometer with an electrospray ionization source (AB Sciex, Concord, Ontario, Canada).
Preparation of PDA-XA-NPs
Synthesis of PDA: The detailed preparation process is shown in Figure 1. Briefly, 0.25 mL of aqueous ammonia (28–30%) was added into a mixture of 4 mL ethanol and 9 mL distilled water in a 50 mL round-bottomed flask under mild magnetic stirring. Then, 50 mg of dopamine hydrochloride dissolved in 1 mL of distilled water was injected into the above solution. The solution color gradually changed from pale yellow to dark brown. The reaction was allowed to proceed for 24 h. Finally, PDA was obtained by centrifugation at 13,000 r·min−1 for 15 min and then washed 3 times with distilled water to remove unpolymerized dopamine.
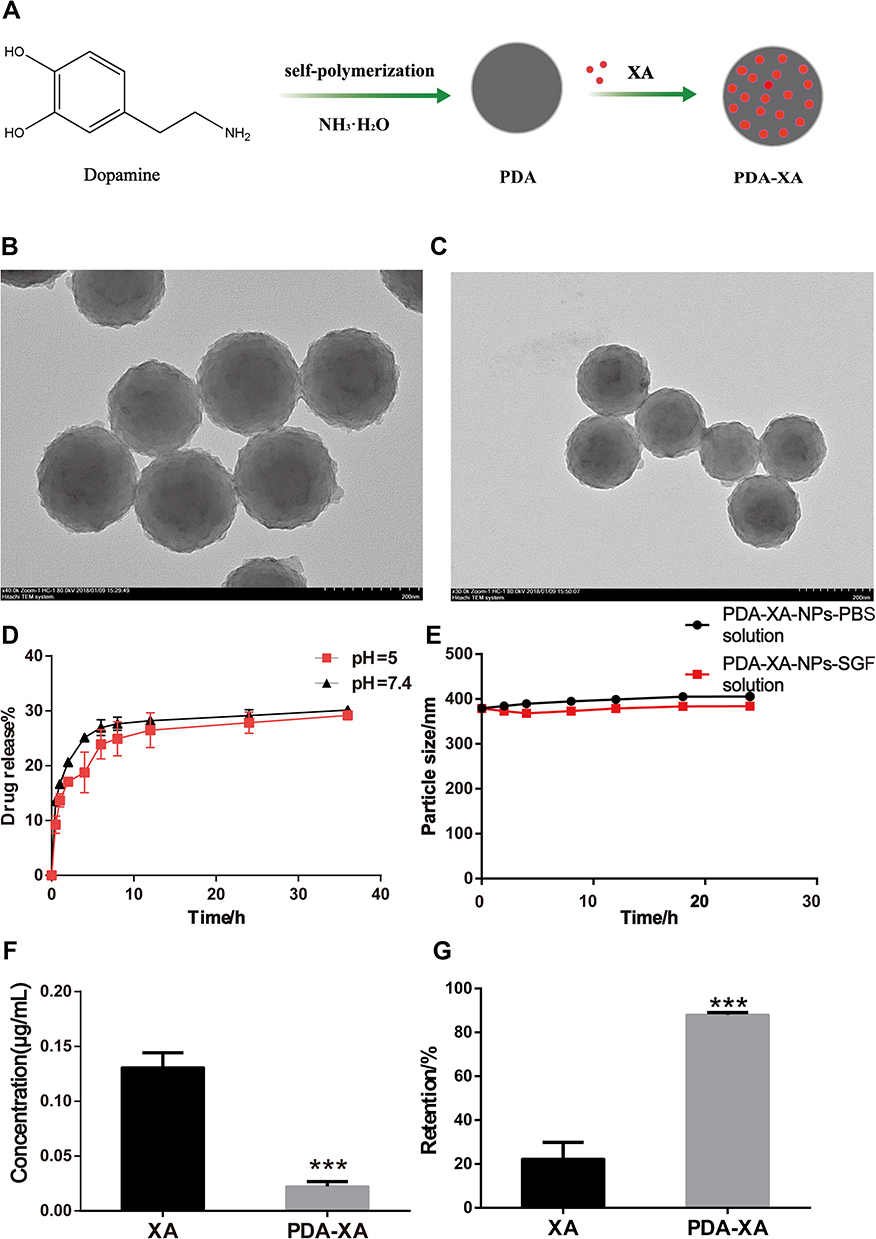 |
Figure 1 (A) Schematic diagram of synthetic process of PDA-XA-NPs. (B) TEM image of PDA-XA-NPs. (C) TEM image of PDA. (D) In vitro release of PDA-XA-NPs in PBS at different pH values. (E) Stability of PDA-XA-NPs in PBS and SGF. (F) Protein contents in supernatant of XA and PDA-XA-NPs (***P < 0.001 vs XA). (G) Gastric mucosal retentions of XA and PDA-XA (***P < 0.001 vs XA). |
Drug loading: PDA-XA-NPs were prepared by mixing 5 mg distilled water dispersion of PDA with XA in DMSO under magnetic stirring at room temperature for 12 h. The product was purified by centrifugation at 13,000 r·min−1 for 15 min and washed with distilled water three times to remove unloaded XA.
Characterizations of PDA and PDA-XA-NPs
Measurement of Particle Size and Zeta Potential
The particle sizes and zeta potentials of PDA and PDA-XA-NPs water dispersions were measured by Malvern Zetasizer ZS90 system.
Morphological Observation of NPs
The morphologies of NPs were observed by a transmission electron microscope (TEM).
Determination of Encapsulation Efficiency and Loading Capacity
The encapsulation efficiency and loading capacity of PDA were determined by an HPLC UV-vis spectrometer, and the supernatant was collected. The dose of added drug subtracted by that in the supernatant was the dose of XA in PDA-XA-NPs.
Analysis method:
Column: HIBAR ods-c18 (250 mm × 4.6 mm, 5 μm); mobile phase: water (a), methanol (b), 40:60, isoelution; flow rate: 1.0 mL·min−1; column temperature: 25°C; detection wavelength: 278 nm; injection volume: 10 μL.
The encapsulation efficiency and loading capacity were calculated according to the following formulas:
EE (%) = (M - M1)/M × 100%
LC (%) = (M - M1)/W × 100%
M1: the amount of free drug; M: total amount of added drug; W: mass of NPs after removal of free drug.
Stability of NPs in Different Media
PDA-XA-NPs were mixed with the same volumes of PBS and simulated gastric fluid (SGF). The particle size was measured at 0, 2, 4, 8, 12, 18 and 24 h, respectively. The stability of PDA-XA-NPs in different media was evaluated by observing the turbidity or precipitation.
Adhesion of NPs16,17
Interaction Between NPs and Mucin
1 mg·mL−1 suspension liquid of mucin was prepared with pH 3.0 hydrochloric acid solution and centrifuged for 10 min at 4000 r·min−1. The supernatant was taken and divided into two groups. For the PDA-XA-NPs group, 1 mL of mucin solution was mixed with 10 mg of PDA-XA-NPs. For the XA group, 1 mL of mucin solution was mixed with an equivalent amount of XA (0.56 mg). The centrifuge tube was placed in a 37°C water bath for 1 h, followed by centrifugation for 10 min at 13,000 r·min−1. The supernatant was taken, and the protein content therein was determined according to the instructions of BCA kit.
NP Tissue Retention Experiment
After 24 h of fasting, rats were anesthetized by intraperitoneally injecting 10% chloral hydrate. The whole stomach was taken out and cut along the greater curvature. The inner wall of the stomach was carefully rinsed with 0.1 mol·L−1 HCl solution. Subsequently, 3.6 mg of XA-loaded NPs and an equivalent amount of XA (0.20 mg) prepared with SGF were uniformly coated on the gastric mucosa and dried at 40°C. Then, the gastric mucosa was placed in a dryer containing saturated potassium nitrate solution and fully hydrated for 30 min. The slides were placed on a petri dish at an oblique angle of 45°C, and the gastric mucosa was rinsed with 0.1 mol·L−1 HCl solution at the flow rate of 100 r·min−1 with a constant flow pump. After collection of the eluent, the XA amount was detected by HPLC, and NPs were counted by an automatic cell counter. The number of NPs in the gastric mucosa was calculated by subtracting that of the leach.
In vivo Imaging
Preparation of PDA-XA-Cy7-NPs
Firstly, 9 mL of water and 1 mL of PDA-XA-NPs water dispersion were added to a 50 mL flask and mixed evenly. Then, 0.01 mg of Cy7 was added and mixed evenly. The solution gradually turned bluish green. After the reaction under magnetic stirring for 24 h, the solution was centrifuged at 13,000 r·min−1 for 15 min. The lower layer was washed with water 3 times and dispersed in 1 mL of water to obtain PDA-XA-Cy7-NPs.
In vivo Imaging and Detection of Mice
ICR mice were fasted for 24 h. PDA-XA-Cy7-NPs were prepared with 1 mg·mL−1 saline, and the mice were administered by gavage at 10 μL·g−1 body weight. After administration, the mice were anesthetized at 2, 8 and 24 h, respectively. An in vivo imaging system of small animals was used to observe the mice and to record the fluorescence signals in their abdomens at different time points.
Pharmacokinetics of PDA-XA-NPs in Rats
Preparation of Stock Solutions and Quality Control Samples
Stock solutions were prepared by dissolving XA (800 μg·mL−1) and psoralen (100 μg·mL−1) in methanol. XA working solution (8000 ng·mL−1) and internal standard working solution (100 ng·mL−1) were obtained by diluting the stock solutions with methanol.
Calibration samples with the final XA concentrations of 1.0, 7.8, 15.6, 62.5, 250, 500 and 800 ng·mL−1 respectively were prepared by spiking 90 μL of blank plasma with 10 μL of XA working solutions and 10 μL of internal standard working solutions into.
Quality control samples with the final XA concentrations of 3.0, 62.5 and 500 ng·mL−1 respectively were prepared. All the solutions prepared above were stored at 4°C before analysis.
Sample Preparation
Internal standard working solution (10 μL) was spiked into 100 μL of plasma samples. The mixture was added 1 mL of ethyl acetate, extracted through vortex for 3 min and centrifuged at 12,000 r·min−1 for 5 min, and the supernatant was transferred into another centrifuge tube and dried by nitrogen flow. The residue was re-dissolved with 100 μL of methanol and then centrifuged at 12,000 r·min−1 for 5 min. The supernatant was collected and stored at 4°C before analysis.
Pharmacokinetics Study
Fifteen male Sprague-Dawley rats were obtained from Shanghai Jiesjie Experimental Animals Co., Ltd. (China) and housed in the laboratory with controlled temperature and humidity for a week, with free access to food and water. Afterwards, they were randomly divided into three groups and fasted overnight before drug administration. Group 1 and group 2: XA was administered intravenously (2.0 mg·kg−1; dissolved in a mixture comprising 85% physiological saline, 10% Pluronic F-68 and 5% DMSO) and orally (100 mg·kg−1; dissolved in 0.5% carboxymethyl cellulose sodium solution), respectively. Group 3: PDA-XA-NPs were administered orally (XA: 65 mg·kg−1; dissolved in physiological saline). Blood samples (approximately 0.15 mL) were collected from the ocular choroidal vein into heparinized tubes 0, 0.08, 0.17, 0.25, 0.50, 1.00, 2.00, 4.00, 6.00, 8.00, 10.00, 12.00 and 24.00 h after drug administration, and centrifuged at 4500 r·min−1 for 5 min at 4°C to separate the plasma that was stored at −80°C before analysis. The study was approved by the Animal Ethics Committee of Nanjing University of Chinese Medicine, and all experiments were performed following Guidelines for the Management and Use of Experimental Animals.
In vitro Efficacy Evaluation
MKN-45 cells in the logarithmic growth phase were inoculated in a 96-well plate at the density of 1×104. The blank wells contained no cells, and the cells in control wells were untreated. The edges were filled with PBS (100 μL per well), and the cells were incubated at 37°C for 24 h until they adhered to the wall. XA and PDA-XA-NPs water dispersions at the concentrations of 0 μM (DMSO control group), 5 μM, 10 μM and 20 μM, respectively, were prepared with 1640 medium, 100 μL of which was added into each well for 4 h, 8 h and 24 h of co-incubation. After incubation, 100 μL of 10% CCK-8 solution was added to each well, and the 96-well plate was incubated at 37°C for 2 h. The absorbance at 490 nm was determined with a standard enzyme solution.
Statistical Analysis
All data were statistically analyzed, and the quantitative data were expressed as mean ± standard deviation (SD). Pharmacokinetics parameters were obtained by the non-compartmental method using DAS 2.0 software. Figures were plotted by GraphPad software. P < 0.05 was considered statistically significant.
Results
Size and Zeta Potential of NPs
As shown in Figure 2, the hydrodynamic size of PDA is (330 ± 16.3) nm, and that of PDA-XA-NPs is slightly larger ((379.3 ± 15.3) nm). As exhibited in Figure 3, the zeta potential barely changes after drug loading. The polydispersity index (PDI) values of PDA and PDA-XA-NPs were lower than 0.2, suggesting that the prepared NPs were uniform and well dispersed in water (Table 1).
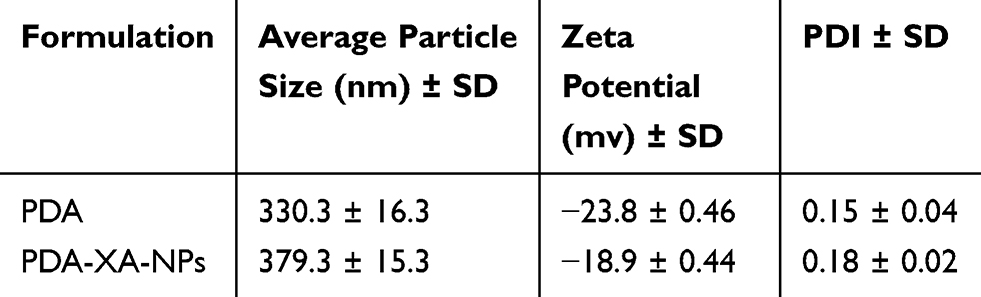 |
Table 1 Particle Sizes and Zeta Potentials of PDA and PDA-XA-NPs |
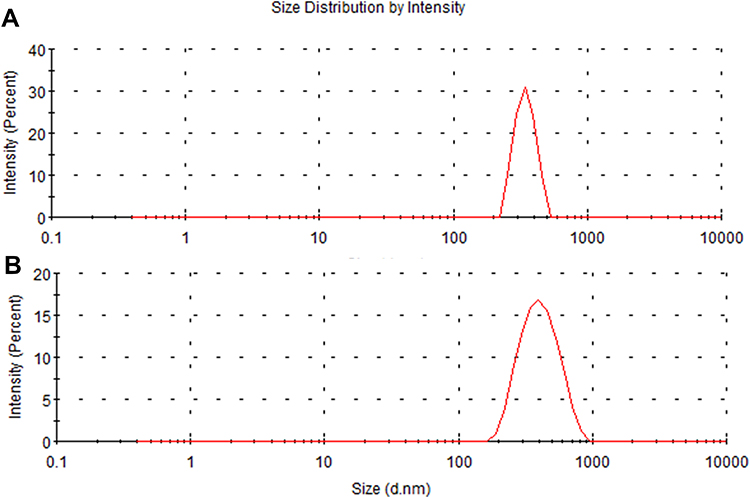 |
Figure 2 Particle size distributions of PDA (A) and PDA-XA-NPs (B). |
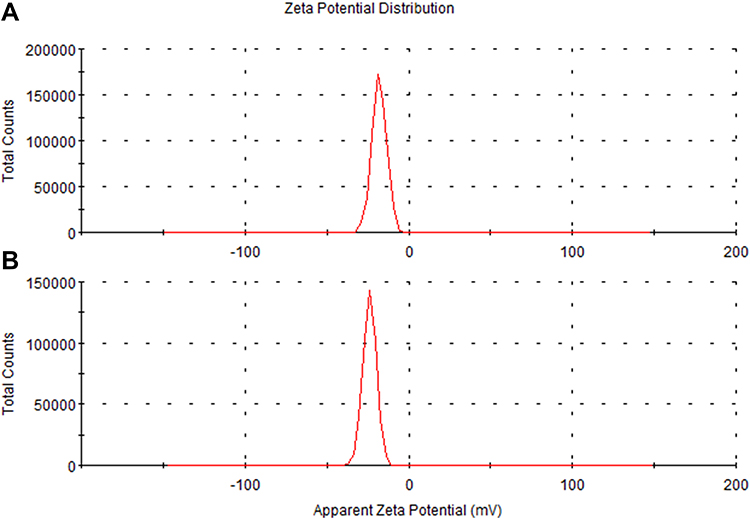 |
Figure 3 Zeta potentials of PDA-XA-NPs (A) and PDA (B). |
TEM Images
A homogeneous system with small and uniform particles was observed. They had a spherical shape, without obvious fracture or adherence among each other. The particle size is consistent with that measured by DLS. After XA loading, the appearance or morphology of PDA NPs did not change significantly, and the particle size increased slightly (Figure 1B and C).
Encapsulation Efficiency and Loading Capacity
HPLC Analysis
HPLC was conducted under the following conditions. Reversed phase column: Thermo-C18 Endcapped (250 mm × 4.6 mm, 5 μm); mobile phase: water-methanol (V/V) ratio of 40/60; flow rate: 1.0 mL·min−1; detection wavelength: 278 nm; column temperature: 25°C; injection volume: 10 μL; standard calibration curve for XA determination: Y = 58,324.4x - 22,545.4, r2 = 0.9999; linear range: 0.7–45 μg·mL−1.
The encapsulation efficiency (%EE) of PDA-XA-NPs was (82.1 ± 0.02) %, and the loading capacity (%LC) was (5.5 ± 0.1)%.
In vitro Release of PDA-XA-NPs at Different pH Values
As shown in Figure 1D, the release behaviors of PDA-XA-NPs at different pH values (pH = 5, 7.4) were consistent, indicating that the release of prepared NPs in PBS was stable and not affected by pH. In the beginning, the NPs were suddenly released, which may be attributed to the easy shedding of XA adsorbed on the NP surface. The final release rate was about 30%.
Stability of NPs in Different Media
After 24 h of culture in PBS and SGF, the particle size of PDA-XA-NPs did not change significantly. As presented in Figure 1E, there is no turbidity or accumulation, and the stability is still high. Hence, PDA-XA-NPs were suitable for intragastric administration.
Adhesion of NPs
Interaction Between NPs and Mucin
According to Figure 1F, the protein content of the PDA-XA-NPs group is significantly lower than that of the XA group (P < 0.001). Thus, PDA-XA-NPs were strongly adsorbed onto the gastric mucosa.
NP Tissue Retention
PDA-XA-NPs adhered to the gastric mucosa of rats significantly better than XA did (P < 0.001) (Figure 1G).
In vivo Imaging Atlas
PDA-XA-NPs remained in the stomach for 2 h and 8 h, so they targeted the stomach and were slowly released. At 24 h, almost no drug was left in the mice, except for a small amount of residue in the stomach (Figure 4E), which demonstrated the gastric targeting effect of PDA-XA-NPs.
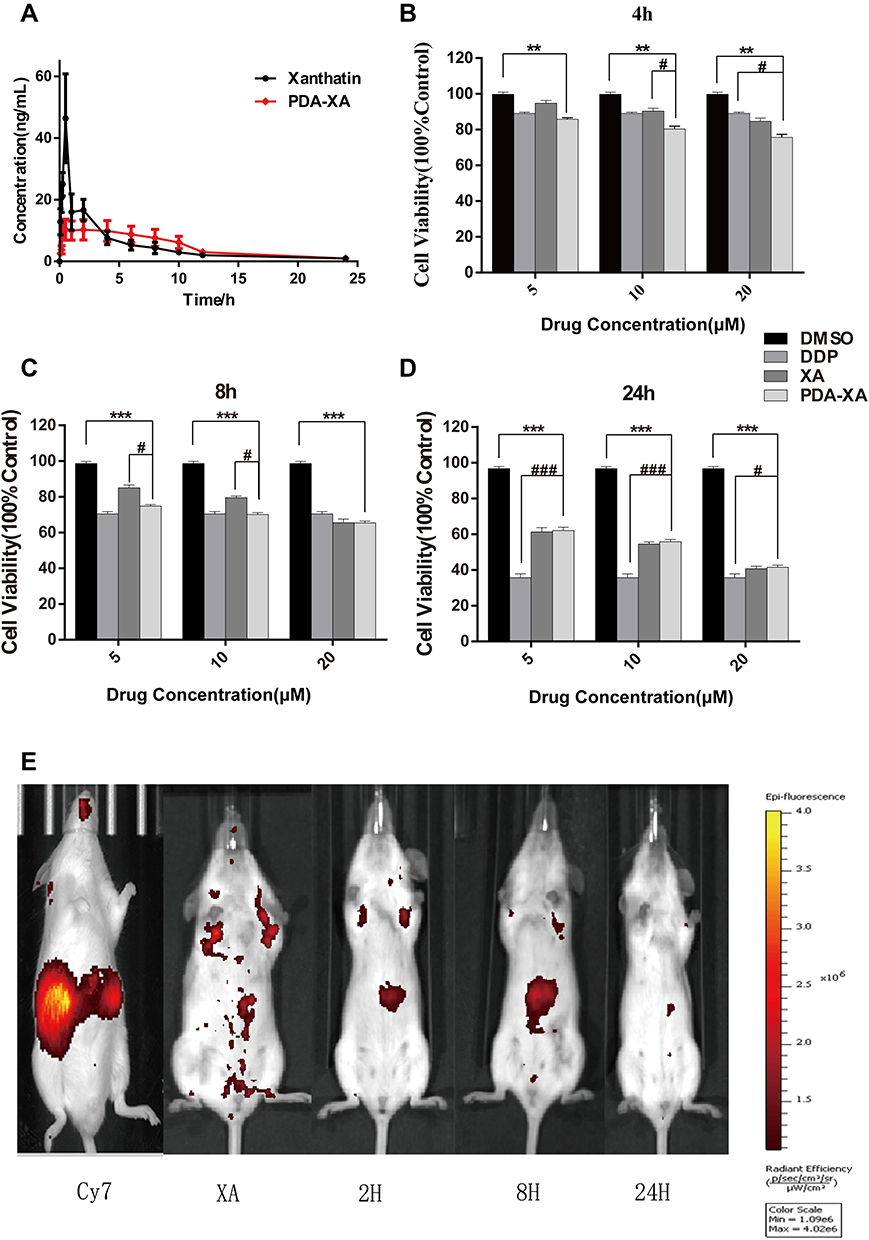 |
Figure 4 Plasma concentration of XA vs time curves after intragastric administration with XA and PDA-XA-NPs. (B–D) Viability of MKN-45 cells after treatment with XA, PDA-XA-NPs, DDP and DMSO. Incubation for 4 h (B); 8 h (C); 24 h (D). Data are represented as mean ± SD (n=6; vs DMSO **P < 0.01, ***P < 0.001, vs DDP #P < 0.05, ###P < 0.001). (E) Imaging atlas of mice in vivo. |
Pharmacokinetics in Rats
The plasma concentration-time profiles of XA after different treatments based on the validated method are shown in Figure 4A, and the main pharmacokinetics parameters calculated by DAS 2.0 software are listed in Table 2. The bioavailability of XA was calculated by F = AUC0-t (test) × D standard/AUC0-t (standard) × D test. The absolute bioavailability of XA was 3.2%, and that of PDA-XA-NPs was 4.1%. Additionally, the relative bioavailability of XA was 128.1%.
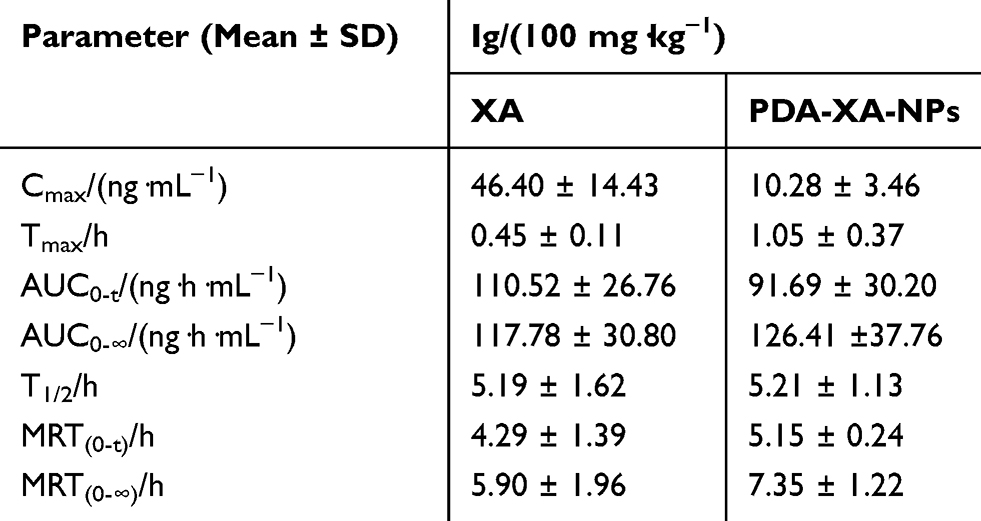 |
Table 2 Pharmacokinetics Parameters of XA vs Time Curves After Intragastric Administration with XA and PDA-XA-NPs |
MRT(0-t)/h of XA was extended after modification by PDA, but without a significant difference, inferring that PDA NPs prolonged the retention time of XA in rats after oral administration. In addition, the PDA group had longer Tmax (h) and higher oral bioavailability of XA than those of the XA group. Hence, PDA modification may elevate the oral bioavailability.
In vitro Pharmacokinetics
As shown in Figure 4B–D, the effect of PDA-XA-NPs on cell viability is enhanced with elapsed incubation time. At 4 h and 8 h, the influence of PDA-XA-NPs on cell viability surpassed that of XA (Figure 4B and C). However, the difference became insignificant after incubation for 24 h (Figure 4D). It is well documented that DDP is one of the most commonly used positive drugs to conquer cancer cells.18 In this study, the outcomes of PDA-XA-NPs and DDP groups were similar at 4 h and 8 h (Figure 4B and C). Besides, the PDA-XA-NPs group showed extremely significant differences from the DMSO group throughout this experiment. Therefore, PDA-XA-NPs were cytotoxic to MKN-45 cells in vitro and can potentially combat gastric cancer.
Discussion
Gastrointestinal adhesive preparation plays a local or systemic role and has high bioavailability by prolonging the adhesion time in the gastrointestinal mucosa and the absorption time. Adhesion can be evaluated in vitro by detecting adhesive force, rheological properties, surface energy and fluorescent signals.19 The in vivo detection methods mainly include gamma-scintillation scanning tracing, isotope tracing and fluorescent sensing. In this study, the adhesion of nanopreparation was tested based on the interaction with mucin and the retention in the gastric mucosa. The adsorbability of nanopreparation onto mucin was significantly higher than that of bulk drug. Moreover, the retention of this preparation in the gastric mucosa reached 89.1% which significantly exceeded that of bulk drug, verifying its excellent adhesion.
Like most SLs, XA has an extremely low oral bioavailability (only 3.2%). Zhang et al20 administered rats with the extract of Radix Aucklandiae equivalent to 15.7 mg·kg−1 costunolide and 71.9 mg·kg−1 dehydrocostuslactone through gavage, and found that the corresponding Cmax values were 19.84 ng·mL−1 and 493.00 ng·mL−1, respectively. Therefore, clarifying the oral bioavailability of SLs is of great significance to their application.
Given that MRT(0-t)/h of PDA-XA-NPs was significantly longer than that of XA, the NPs prolonged the retention time of XA in rats by adhering to the gastrointestinal mucosa. Since Tmax was extended 2.33-fold, the absorption time of XA in vivo was longer, which may be ascribed to the slow absorption of XA owing to the adhesion of NPs to the gastrointestinal mucosa. After PDA modification, the oral bioavailability of XA was raised. The currently available studies concerning dopamine-loaded NPs for drug delivery have focused on the synthesis process as well as in vivo and in vitro pharmacokinetics.21 Although the modification with dopamine-loaded NPs can boost the therapeutic effects of anticancer drugs and allow phototherapy and thermal therapy,22 the targeting to gastric tumors or the enhancement of oral bioavailability has seldom been referred. Herein, the low and slow absorption of XA into the blood results from the strong adhesion of NPs to the gastric mucosa. In short, the fabricated NPs indeed targeted the stomach.
The adhesion of PDA-XA-NPs can be evaluated in vivo. For example, the movement process of the preparation in the gastrointestinal tract over time can be directly observed by using γ-scintillation scanning tracing. To further elucidate the gastric retention of PDA-XA-NPs, their distribution in various tissues were further explored. Nevertheless, the NPs in the experimental design were only able to target the stomach, so further modification is in need to realize tumor cell targeting.
Conclusion
In summary, we have successfully prepared PDA-XA-NPs and investigated their physicochemical characteristics. The oral bioavailability of XA was improved, as evidenced by the pharmacokinetics in vivo, which may be related to the strong adhesion of PDA. For the first time, this study fabricated a PDA NP preparation for pharmacokinetics research, paving the way for developing gastric adhesive preparations in the future.
Acknowledgment
The authors thank the College of Pharmacy, Nanjing University of Chinese Medicine for their support and acknowledge the award of Engineering Center of State Ministry of Education for Standardization of Chinese Medicine Processing, Nanjing University of Chinese Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Takeda S, Nishimura H, Koyachi K, et al. (^|^#8211;)-Xanthatin induces the prolonged expression of c-Fos through an N-acetyl-L-cysteine (NAC)-sensitive mechanism in human breast cancer MDA-MB-231 cells. J Toxicol Sci. 2013;38(4):547–557. doi:10.2131/jts.38.547
2. Takeda S, Matsuo K, Yaji K, et al. (-)-Xanthatin Selectively Induces GADD45γ and Stimulates Caspase-Independent Cell Death in Human Breast Cancer MDA-MB-231 Cells. Chem Res Toxicol. 2011;24(6):855–865. doi:10.1021/tx200046s
3. Takeda S, Noguchi M, Matsuo K, et al. (-)-Xanthatin up-regulation of the GADD45γ tumor suppressor gene in MDA-MB-231 breast cancer cells: role of topoisomerase IIα inhibition and reactive oxygen species. Toxicology. 2013;305:1–9. doi:10.1016/j.tox.2012.12.019
4. Ghantous A, Gali-Muhtasib H, Vuorela H, et al. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15(15–16):668–678. doi:10.1016/j.drudis.2010.06.002
5. Yu Y, Yu J, Pei CG, et al. Xanthatin, a novel potent inhibitor of VEGFR2 signaling, inhibits angiogenesis and tumor growth in breast cancer cells. Int J Clin Exp Pathol. 2015;8(9):10355–10364.
6. Zhang L, Tao L, Ruan J, et al. Xanthatin induces G2/M cell cycle arrest and apoptosis in human gastric carcinoma MKN-45 cells. Planta Med. 2012;78(9):890–895. doi:10.1055/s-0031-1298481
7. Yan C, Li H, Wu Y, et al. Determination of xanthatin by ultra high performance liquid chromatography coupled with triple quadrupole mass spectrometry: application to pharmacokinetic study of xanthatin in rat plasma. J Chromatogr B. 2014;947–948:57–61. doi:10.1016/j.jchromb.2013.12.006
8. Mihmanli M, Ilhan E, Idiz UO, et al. Recent developments and innovations in gastric cancer. World J Gastroenterol. 2016;22(17):4307. doi:10.3748/wjg.v22.i17.4307
9. Song Z, Wu Y, Yang J, et al. Progress in the treatment of advanced gastric cancer. Tumor Biol. 2017;39(7):1010428317714626. doi:10.1177/1010428317714626
10. Coccolini F, Montori G, Ceresoli M, et al. Advanced gastric cancer: what we know and what we still have to learn. World J Gastroenterol. 2016;22(3):1139. doi:10.3748/wjg.v22.i3.1139
11. Cao Y, Depinho RA, Ernst M, et al. Cancer research: past, present and future. Nat Rev Cancer. 2011;11(10):749–754. doi:10.1038/nrc3138
12. Wang Z, Duan Y, Duan Y. Application of polydopamine in tumor targeted drug delivery system and its drug release behavior. J Control Release. 2018;290:56–74. doi:10.1016/j.jconrel.2018.10.009
13. Batul R, Tamanna T, Khaliq A, et al. Recent progress in the biomedical applications of polydopamine nanostructures. Biomater Sci. 2017;5(7):1204–1229. doi:10.1039/C7BM00187H
14. Mrowczynski R. Polydopamine-Based Multifunctional (Nano)materials for Cancer Therapy. ACS Appl Mater Interfaces. 2018;10(9):7541–7561. doi:10.1021/acsami.7b08392
15. Wu F, Li J, Zhang K. Multi-Functional Coating Based on Hyaluronic Acid and Dopamine Conjugate for Potential Application on Surface Modification of Cardiovascular Implanted Devices. ACS Appl Mater Interfaces. 2016;8(1):109–121. doi:10.1021/acsami.5b07427
16. Tao Y. Study on Acyclovir-Loaded Bioadhesive Microspheres. Fudan University; 2009.
17. Song Y. The Preparation of Bioadhesive Sustained-Release Capsule of Captopril and Its Evaluation in vivo and in vitro. Ocean University of China; 2005.
18. Evaluation of the betulinic Acid-Cisplatin conjugate APC and its precursor DE9B for the treatment of human malignant glioma. Chem Biol Interact. 2019;undefined(undefined):108841
19. Yiqun S. Preparation and Study of Radix Puerariae Flavonoids Adhesive and Floating Preparation for Gastrointestinal Tract. Beijing: Peking Union Medical College; 2014.
20. Zhang J, Hu X, Gao W, et al. Pharmacokinetic study on costunolide and dehydrocostuslactone after oral administration of traditional medicine Aucklandia lappa Decne. by LC/MS/MS. J Ethnopharmacol. 2014;151(1):191–197. doi:10.1016/j.jep.2013.10.024
21. Nie J, Cheng W, Peng Y, et al. Co-delivery of docetaxel and bortezomib based on a targeting nanoplatform for enhancing cancer chemotherapy effects. Drug Deliv. 2017;24(1):1124–1138. doi:10.1080/10717544.2017.1362677
22. Zhuang H, Su H, Bi X, et al. Polydopamine nanocapsule: a theranostic agent for photoacoustic imaging and chemo-photothermal synergistic therapy. ACS Biomater Sci Eng. 2017;3(8):1799–1808. doi:10.1021/acsbiomaterials.7b00260
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.