Back to Journals » Research Reports in Clinical Cardiology » Volume 13
A Novel Mutation in the TRPM4 Gene Associated with Congenital Long QT Syndrome: A Case Report
Authors Huang R, Luo Y, Lei Y, Li Y
Received 30 October 2021
Accepted for publication 29 December 2021
Published 13 January 2022 Volume 2022:13 Pages 1—7
DOI https://doi.org/10.2147/RRCC.S346943
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Rui Huang,1,* Yinhua Luo,2,* Yuhua Lei,1 Yuanhong Li1
1Cardiovascular Disease Center, Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi, 445000, Hubei Province, People’s Republic of China; 2Department of Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Shiyan, 442000, Hubei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuanhong Li
Cardiovascular Disease Center, Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi, 445000, Hubei Province, People’s Republic of China
Email [email protected]
Abstract: It is reported that the incidence of congenital long QT syndrome (cLQTS) is very low, with the incidence of LQTS caused by transient receptor potential melastatin 4 (TRPM4) being even lower, leading to less research on the gene TRPM4. We reported a case of an elderly male patient presenting with frequent syncopal episodes since the age of 20. Electrocardiography showed sinus bradycardia (mean heart rate 59 beats), prolonged QT interval (540ms), and torsade de pointes. His sister also had experienced an episode of syncope, but his other families had not. A novel mutation in the TRPM4 gene was discovered in this patient and his sister, according to genetic analysis. All in all, we provided a new heterozygous shift mutation (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) on TRPM4, which has never been reported in the past and thus may hopefully serve as useful feedback information for genetic pathogenesis of cLQTS caused by TRPM4 variants.
Keywords: long QT syndrome, LQTS, TRPM4, mutation
Introduction
Long QT syndrome (LQTS) is defined by a prolonged QT interval accompanied by morphological abnormalities in the T and/or U waves on the electrocardiograph (ECG).1 The primary clinical symptom of LQTS is syncope produced by ventricular arrhythmias.2–4 The clinical diagnosis of LQTS is based on a combination of the patient’s medical and family history, as well as the 12-lead ECG.5 According to the guidelines, LQTS diagnosis can be made in case the QTc is more than 460ms, and the patient presents some antecedents, most notably a family history of SCD and unexplained syncope.6
LQTS can be classified into two types based on its etiology: congenital LQTS (cLQTS) and acquired LQTS (aLQTS). While the former is a relatively rare genetic cardiovascular disease with a low incidence rate (1/2000-1/3000),7 the latter is frequently subsequent to electrolyte disorders, cardiomyopathy, cerebrovascular accidents, and autonomic dysfunction.
The pathogenesis of cLQTS is related to the mutation of genes encoding for ion channels, such as KCNH2,3,8 KCNQ1,2,9 TRPM4,10–12 and so on, and causing ion channel dysfunction with reduced repolarization ion flow and/or increased delocalization ion flow, which in turn leads to prolonged repolarization. Among ion channel genes, mutations in KCNQ1 and KCNH2, which encode voltage-gated K+ channels involved in cardiac action potential (AP) repolarization are most common,10 followed by mutations in SCN5A which encode voltage-gated Na (1.7–8%), while mutations in other genes including TRPM4 are rare (below 1% of LQTS).11 Dr. Hof and colleagues were the first to hypothesize that TRPM4 mutations cause long QT syndrome, and they detected four TRPM4 variants, including c.1321 G >A, c.1495 C >T, c.1496 G >C, and c.2531 G >A, with no changes in the key LQTS genes.11
Herein, we reported a Chinese proband with cLQTS with a new mutation (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) in the TRPM4 with the hope that this report may be helpful in future genetic studies and prospective, genetically informed research.
Case Presentation
A 75-year-old male was implanted with a permanent pacemaker 18 years ago due to a local diagnosis of bradycardia characterized by recurrent syncope since the age of 20, yet postoperative syncope continued to occur. Syncope occurred again a day before admission, and then he was taken to our hospital. Electrocardiography (ECG) at disease onset indicated sinus bradycardia, anterior wall T wave changes with visible u waves (Figure 1).
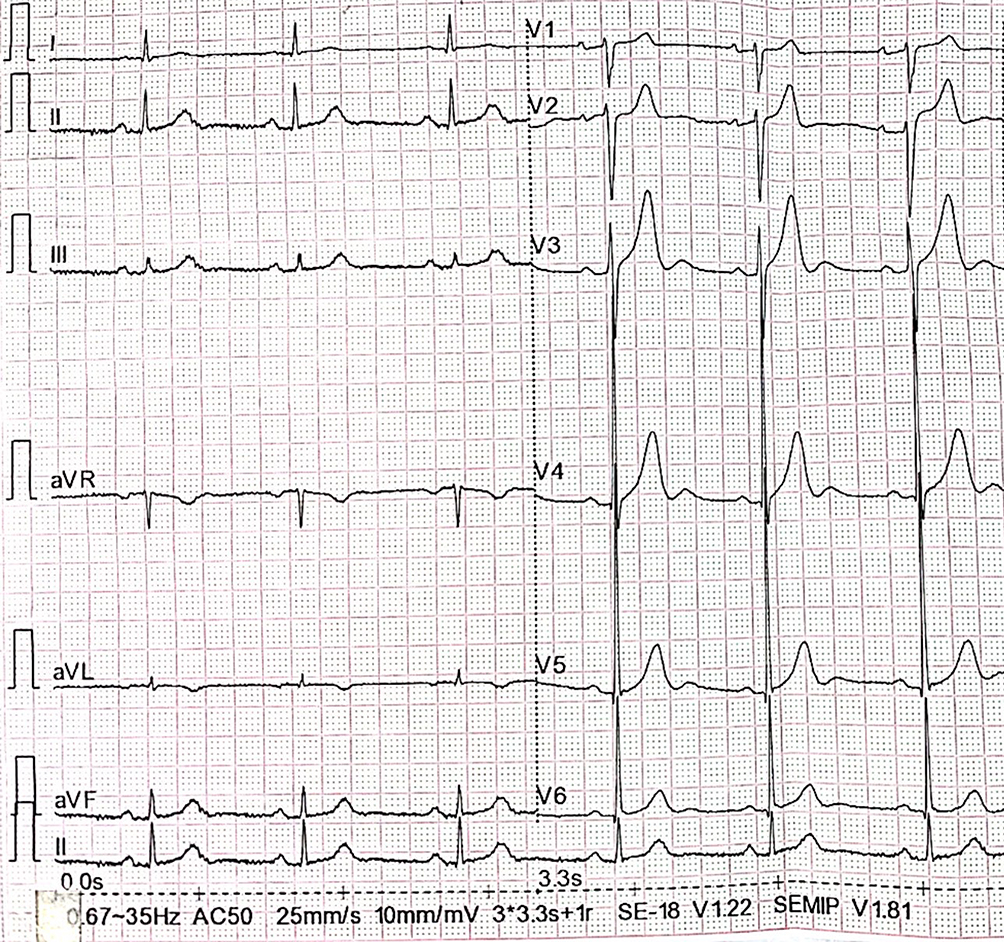 |
Figure 1 The admission ECG showed sinus bradycardia with QTc interval 432ms and U wave. |
On admission, the following vital signs were recorded: blood pressure of 135/88mmHg, pulse rate of 59 beats per minute, the body temperature of 36.4°C, and breathing rate of 18 beats per minute. Physical examination revealed no evidence of heart failure or pathological nervous system features.
After admission, repeated electrocardiograms suggested prolonged QT intervals, sinus bradycardia, and T wave changes (Figure 2). Ambulatory ECG also showed sinus bradycardia (mean heart rate 59 beats), prolonged QT interval (540ms), and torsade de pointes (Figure 3). What’s more, the electrodes on the patient’s pacemaker were discovered to be depleted for nearly five years. Laboratory data showed a slightly elevated level of troponin, as well as N-terminal-pro-brain natriuretic peptide, while other laboratory indexes including hepatic and renal function, electrolytes, coagulation, and inflammatory indexes were normal. We also performed a cranial MRI on this patient, and no neurological lesion was found that could cause syncope. Echocardiography indicated no abnormalities in cardiac structure, and the left ventricular ejection fraction was 61%. Moreover, selective coronary angiography was performed, indicating that the patient has no apparent pathological stenosis in the coronary arteries.
 |
Figure 2 (A–C) During the hospitalization, the ECG showed the dynamic changes of T wave; the longest QTc interval was 540ms. |
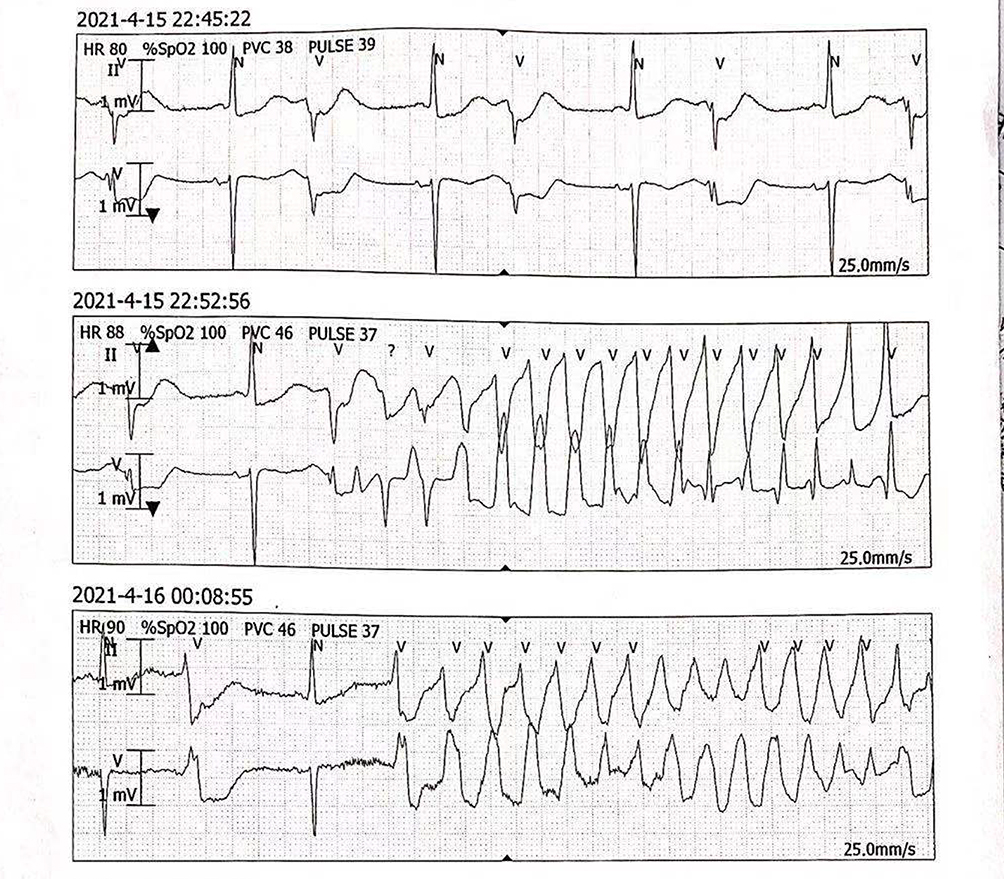 |
Figure 3 Electrocardiogram monitoring shows torsion de pointes tachycardia. |
According to the above results and the diagnostic criteria of LQTS, a highly suspected diagnosis of LQTS was finally made (Rating 6.5 out of 5).1,13,14
Then the etiology of LQTS was further explored. For no acquired LQTS associated risk factors such as electrolyte disorders, cardiomyopathy, cerebrovascular accidents, and autonomic dysfunction were found in the patient’s previous medical history and related examinations after admission, we are suspicious of the patient’s Genetics of LQTS.
After obtaining the informed consent, we conducted whole-exome sequencing (WES) on the patient and his family to confirm our diagnosis. Gene testing revealed that the patient carried a TRPM4 heterozygous shift mutation (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133). Moreover, WES analysis of his family members revealed that his sister carried the same TRPM4 mutation as the patient (Figure 4), but his two brothers and son did not. Regrettably, the proband’s parents have all died, and hence their genes have not been obtained.
 |
Figure 4 The results of genetic testing showed the proband and his sister carried a TRPM4 heterozygous shift mutation (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) (A), while his two brothers and son did not (B). |
Because of the high risk of sudden cardiac death, we recommend implanting a cardioverter defibrillator (ICD) for the patient. Due to economic reasons, the patient refused. Due to the patient’s strong preference for cautious treatment, we administered Shengsong Yangxin Capsule as a placebo.
Discussion
cLQTS is a rare cardiac disorder inherited in an autosomal trait, with an estimated incidence of 1:2000–1:3000.7 It is accepted that cLQTS is a rare ion channelopathy, and a host of genes were described to be responsible for LQTS. So far, 15 genes with more than 7000 mutations have been considered to be associated with cLQTS.15 Among the six genes encode for a pore-forming ion channel, while others encode for regulatory subunits or proteins. Mutations in KCNQ1 (30–35%) and KCNH2 (25–30%) encoding voltage-gated K+ channels involved in cardiac action potential (AP) repolarization are the most common among ion channel genes,10,14 followed by mutations in SCN5A, which encode voltage-gated Na+ (1.7–8%).11,14 In comparison, mutations in other genes, including TRPM4 are rare (below 1% of LQTS).11,12,14
As far as the pathology of LQTS, it is generally known that QT duration depends on both ventricular AP duration and AP propagation within the ventricle and ion channel dysfunction with reduced repolarization ion flow and/or increased delocalization ion flow leads to prolonged repolarization. According to a sack of animal experiments on TRPM4, TRPM4 affects cardiac electrophysiological activity through nonselective cation permeability, which leads to cLQTS.11 Unfortunately, additional research is required to decipher the biological mechanism underlying TRPM4-induced loss of function of nonselective cation permeability.
Above all, gene test counts for cLQTS. The importance of gene detection for cLQTS lies in exploring its pathogenic mechanism and its treatment, for the drugs targeted specific ion channels can be used with exerting maximal effects.
In our case, a new mutation site on TRPM4 (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) was discovered through whole-exon detection, which can provide a brand-new direction for gene screening of patients with cLQTS and further complements its diagnostic criteria. As for the detail of gene tests, we used PolyPhen2 to predict whether a new mutation is damaging to the resultant protein function. And then, according to the relevant literature, we did consider that TRPM4 is as same amino acid change as a previously established pathogenic variant regardless of nucleotide change after searching the OMIM database. But the absence of the literature for molecular pathology makes us failure to achieve the information of damaged protein. At last, combined clinical history, ECG, and the results of gene test, it was suspected that TRPM4 mutation (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) was the pathogenic variant.
In the treatment of cLQTS, beta-blockers effectively prevent cardiovascular disease and ventricular arrhythmia, and ICD implantation is regarded as the ultimate therapy.16 Because of the high risk of sudden cardiac death, we recommend implanting a cardioverter defibrillator (ICD) for the patient. Due to economic reasons, the patient refused, and we administered a placebo.
The incidence of cLQTS is very low, with the incidence of LQTS caused by TRPM4 being even lower, leading to less research on the gene TRPM4. Therefore, we reported a new mutation in TRPM4 (NM_017636: exon4: c.434delC, p. Ala145ValfsTer133) to provide more evidence for gene screening, to improve the detection rate of healthy gene carriers or patients in the early incubation stage, thereby providing further complements to the clinical data of the study about TRPM4. Notwithstanding its limitation such as lack of this patient’s past clinical data that can help to compare the symptom before and after the permanent pacemaker implantation, detailed information of the pedigree of this patient’s parents and so on, this report does hopefully serve as useful feedback information for genetic pathogenesis of cLQTS caused by TRPM4 variants.
Abbreviation
cLQTS, congenital long QT syndrome; LQTS, long QT syndrome; ECG, electrocardiograph; AP, action potential; ICD, implanting cardioverter defibrillator; WES, whole-exome sequencing; TRPM4, transient receptor potential melastatin 4; aLQTS, acquired LQTS.
Data Sharing Statement
All relevant data supporting the conclusions of this article are included within the article.
Ethical Approval
The need for institutional ethics approval for this case report was waived. Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Consent for Publication
The patient has provided informed consent for the publication of the case. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgments
Dr. Rui Huang and Dr. Yinhua Luo are co-first authors for this study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported in part by the National Natural Science Foundation of China (82160072) and the Science and Technology Support Project of Enshi Science and Technology Bureau (D20210024).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Vohra J. The long QT syndrome. Heart Lung Circ. 2007;16(Suppl 3):S5–S12. doi:10.1016/j.hlc.2007.05.008
2. Beiyin G, Tingliang L, Lei Y, et al. Head-up tilt test induces T-wave alternans in long QT syndrome with KCNQ1 gene mutation: case report CARE-compliant article. Medicine. 2020;99(20):e19818.
3. Henk-Jan B, Lucia B. Orgasm induced torsades de pointes in a patient with a novel mutation with long-QT syndrome type 2: a case report. Eur Heart J Case Rep. 2018;2(2):yty062.
4. Joel G, Kinsley H, Amanda W, et al. Recurrent torsades with refractory QT prolongation in a 54-year-old man. Am J Case Rep. 2018;19:1515.
5. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963. doi:10.1016/j.hrthm.2013.05.014
6. Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. [2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death]. Kardiol Pol. 2015;73(10):795–900. Croatian. doi:10.5603/KP.2015.0190
7. Zumhagen S, Stallmeyer B, Friedrich C, et al. Inherited long QT syndrome: clinical manifestation, genetic diagnostics, and therapy. Herzschrittmacherther Elektrophysiol. 2012;23(3):211–219. doi:10.1007/s00399-012-0232-8
8. Du F, Wang G, Wang D, et al. Targeted next generation sequencing revealed a novel deletion-frameshift mutation of KCNH2 gene in a Chinese Han family with long QT syndrome: a case report and review of Chinese cases. Medicine. 2020;99(16):e19749. doi:10.1097/MD.0000000000019749
9. Motoi N, Marehiko U, Ryota E, et al. A novel KCNQ1 nonsense variant in the isoform-specific first exon causes both jervell and Lange-Nielsen syndrome 1 and long QT syndrome 1: a case report. BMC Med Genet. 2017;18(1):1–6.
10. Amin AS, Pinto YM, Wilde AA. Long QT syndrome: beyond the causal mutation. J Physiol. 2013;591(17):4125–4139. doi:10.1113/jphysiol.2013.254920
11. Hof T, Liu H, Sallé L, et al. TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med Genet. 2017;18(1):31. doi:10.1186/s12881-017-0397-4
12. Guinamard R, Bouvagnet P, Hof T, et al. TRPM4 in cardiac electrical activity. Cardiovasc Res. 2015;108(1):21–30. doi:10.1093/cvr/cvv213
13. Hayashi K, Konno T, Fujino N, et al. Impact of updated diagnostic criteria for long QT syndrome on clinical detection of diseased patients: results from a study of patients carrying gene mutations. JACC Clin Electrophysiol. 2016;2(3):279–287. doi:10.1016/j.jacep.2016.01.003
14. Neira V, Enriquez A, Simpson C, et al. Update on long QT syndrome. J Cardiovasc Electrophysiol. 2019;30(12):3068–3078. doi:10.1111/jce.14227
15. Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 2014;10(1):29–33. doi:10.14797/mdcj-10-1-29
16. Betge S, Schulze-Bahr E, Fitzek C, et al. [Long QT syndrome causing grand mal epilepsy: case report, pedigree, therapeutic options, and review of the literature]. Nervenarzt. 2006;77(10):1210–1217. German. doi:10.1007/s00115-006-2118-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.