Back to Journals » OncoTargets and Therapy » Volume 13
A Novel Mutation in MYH Gene Associated with Aggressive Colorectal Cancer in a Child: A Case Report and Review of Literature
Received 23 April 2020
Accepted for publication 3 August 2020
Published 25 August 2020 Volume 2020:13 Pages 8557—8565
DOI https://doi.org/10.2147/OTT.S259587
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Xin Tian, Qian Wang, Weisong Cai
Department of Clinical Oncology, Shengjing Hospital of China Medical University, Shenyang, Liaoning 110022, People’s Republic of China
Correspondence: Weisong Cai
Department of Clinical Oncology, Shengjing Hospital of China Medical University, 39 Huaxiang Road, Shenyang 110022, People’s Republic of China
Email [email protected]
Abstract: Colorectal cancer is a rare pediatric tumor. Pediatric patients with colorectal cancer present with more aggressive tumor biology and at later stages of the disease, higher proportions of signet ring and mucinous histology, and less differentiation. The effective treatment is same as that received by adults. The overall prognosis of pediatric colorectal cancer is generally poor. Genetic mutations have been identified as the cause of inherited cancer risk in some colorectal cancers. Here, we presented a case of a pediatric patient carrying a maternally derived, heterozygous MYH germline mutation (c.934– 2A>G,intron), the mutation was not reported in pediatric patients before. Also, the patient carried somatic mutations of proto-oncogene SMAD4 (R361C) and TP53 (Y234H). The patient underwent surgical resection, chemotherapy and targeted therapy, but the prognosis was not good. We also review the literature to summarize clinical features, gene mutations, management, and outcomes of pediatric colorectal cancer patient. Our results suggest that the genetic mutation of MYH together with somatic mutations of proto-oncogene SMAD4 and TP53 may lead to the early onset colorectal cancer of the patient. Although the overall prognosis of pediatric colorectal cancer is generally poor, the pathogenesis may be related to hereditary genetic mutations as was found with the MYH gene mutation in our case. Genetic screening can provide early diagnosis and improve prognosis.
Keywords: pediatric, colorectal cancer, heredity, MYH, gene mutations
Introduction
Colorectal cancer is a rare pediatric tumor, the incidence of these cases has however recently risen.1 There is extensive research on adult colorectal cancer, but pediatric colorectal cancer studies have been limited by small numbers of patients with no large institutional cohort or prospective studies to guide treatment. Genetic mutations have been identified as the cause of inherited cancer risk in some colorectal cancers.2 A better understanding of the pathogenesis, clinical features and outcomes of pediatric colorectal cancer is required. With advances in molecular biology, the utilization of next-generation sequencing technology to interrogate multiple genes simultaneously is being utilized more frequently in hereditary cancer testing.3 This can empower clinicians learn more about the disease.
In this article, we present the case of a 13 years old Chinese male with colorectal cancer carrying a maternally derived, heterozygous MYH germline mutation (c.934–2A>G,intron), also the patient carried somatic mutations of proto-oncogene SMAD4 (R361C) and TP53 (Y234H). The patient underwent surgical resection, chemotherapy and targeted therapy. We also review the literature to summarize clinical features, gene mutations, management, and outcomes of pediatric colorectal cancer patient. Our goal is to propose strategies for the evaluation and management of this rare tumor based on our clinical experience and indicators obtained from prior literature reports. Our results may uncover unique features of pediatric colorectal cancer and novel hereditary gene mutations, as well as suggest opportunities for improvements in pediatric colon cancer diagnosis and treatment.
Case Presentation
In July 2017, a 13-year-old Chinese male experienced intermittent abdominal pain lasting about 10 minutes per stint. The pain could be alleviated without treatment. No fever or diarrhea were observed. The patient subsequently developed vomiting, notably a coffee-like substance, which could not be alleviated by drugs. Gastroscopy results revealed gastric mucosal inflammation, but the patient refused to undergo further systemic examination. On the 30th of July 2017, the patient was diagnosed with an acute intestinal obstruction, with the abdominal CT scan indicating obstruction of the lower intestine and thickening of the splenic flexure (Figure 1). He underwent an emergency colostomy with a colon tumor resection. No lymph node dissection was performed because of the operation under emergency condition and considering the boy’s young age. Intraoperative findings revealed that an irregular 5cm x 3cm tumor within the intestinal wall near the splenic flexure was the cause of the obstruction. The proximal bowel tube was dilated and multiple lymph nodes were enlarged. The proximal colon was removed 5cm from the edge of the tumor as well as a small mesenteric lymph node. The patient was healthy prior to the surgery with no history of infective diseases, or allergies to food or drugs. His maternal uncle also had colorectal cancer.
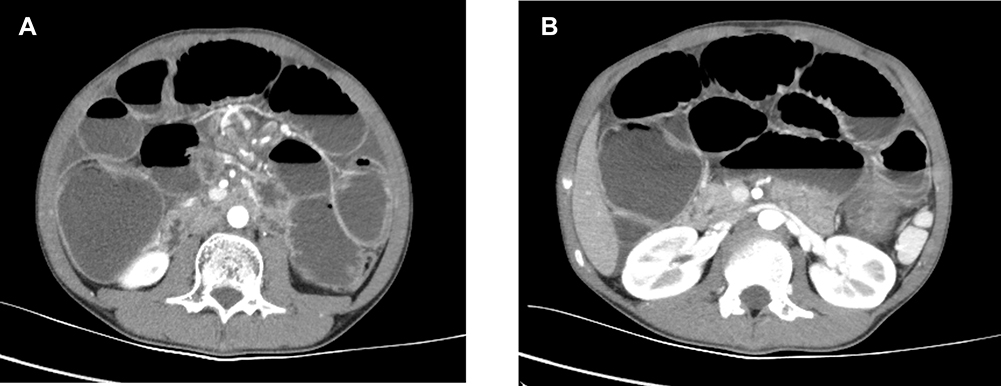 |
Figure 1 CT images at initial diagnosis. (A and B) The whole abdominal CT indicate the lower intestine obstruction and thickening of the splenic flexure. |
From August 2017 to December 2017, the patient underwent six cycles of combined mFOLFOX6 chemotherapy which consisted of oxaliplatin, leucovorin, and 5-fluorouracil. During chemotherapy, the level of CEA and CA199 was stable. An abdominal CT scan evaluation showed that the disease was stable and remained stable after the completion of the mFOLFOX6 chemotherapy regimen (Figure 2A). Oral administration of capecitabine was adopted from January 2018. The disease-free survival (DFS) was 8 months. On April 23 2018, the level of CA199 was significantly higher than before, and the abdominal CT scan suggested the presence of multiple nodules in the greater omentum and peritoneum, peritoneal effusion, and tumor progression (Figure 2B). The patient then underwent one cycle of local intraperitoneal injection of bevacizumab (avastin) + intravenous chemotherapy, and two cycles of a FOLFIRI regimen. On June 19 2018, a targeted intravenous treatment with avastin was added. On the second day of FOLFIRI chemotherapy during the third cycle, abdominal distension was obvious and abdominal pain could not be tolerated. On July 9 2018, one cycle of chemotherapy with the targeted combination of avastin and raltitrexed + oxaliplatin was started. On July 27 2018, the child presented with intestinal obstruction and disease progression, and was hospitalized for support therapy. The patient received palliative care and passed away 2 months later. The overall survival (OS) was 14 months.
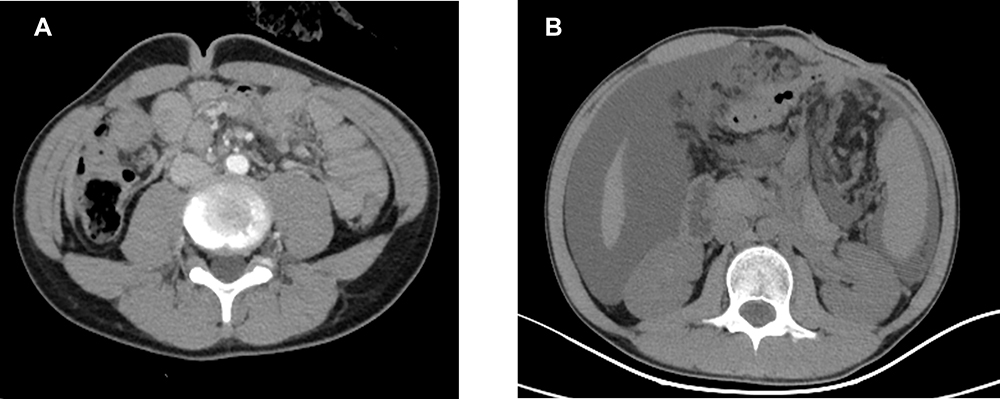 |
Figure 2 (A) CT images after 6 cyles chemotherapy. (B) CT images when the disease progressed. |
Materials and Methods
The resected specimens were fixed in 10% neutral buffered formalin and routinely processed. Paraffin embedded blocks were sectioned at a thickness of 5 μm and stained with hematoxylin and eosin. Immunohistochemical investigations were performed using paraffin embedded tissue samples.
Whole exome sequencing (WES) and analysis were performed using both non-tumour (blood) and tumour (tissues after surgery) DNA. For blood, DNA was extracted from thawed materials using the Maxwell RSC Blood DNA Kit (AS1400, Promega, Madison, WI, USA). For tissues, DNA was extracted using the MagMAX FFPE DNA/RNA Ultra Kit (A31881, ThermoFisher, Waltham, MA, USA). The extracted DNA was sheared using a Covaris L220 sonicator, then was sequenced on an Illumina NovaSeq-6000 sequencer (Illumina, San Diego, CA, USA). Mutations from tumour sample were compared with the paired blood from the same patient to generate the list of somatic mutations. While mutations from non-tumour were determined as germline.
Results
The histopathologic diagnosis after surgery was descending colorectal mucinous adenocarcinoma (stage III), which had infiltrated the whole layer (T3), signet ring cell carcinoma can be seen microscopically, the vascular, lymphatic and nerve invasion can be found (Figure 5A and B). PTNM stage was T3NxM0. Immunostaining showed positive result for caudal-related homeobox gene 2 (CDX2) and cytokeratin 20 (CK20) (Figure 3A and B). Additionally, immunostaining showed positive result for MLH1, MSH2, MSH6 and PMS2 (Figure 3C–F), which also been found positive by whole exome sequencing. Whole exome sequencing also revealed that KRAS and NRAS genes presented as wild type, while somatic mutations of proto-oncogene SMAD4 (R361C) and TP53 (Y234H) (Figure 4B and C). Additionally, molecular analysis showed that the MYH (c.934–2A>G,intron) mutation was detected in blood samples which was determined as germline mutation. It was a heterozygous MYH mutation at the intron-exon splice site (Figure 4A). The same MYH mutation was found in his healthy mother. The patient underwent colonoscopy after surgery and no abnormalities and polyps were observed in the mucosa of the rest colon apart from the cancer (Figure 5C–F).
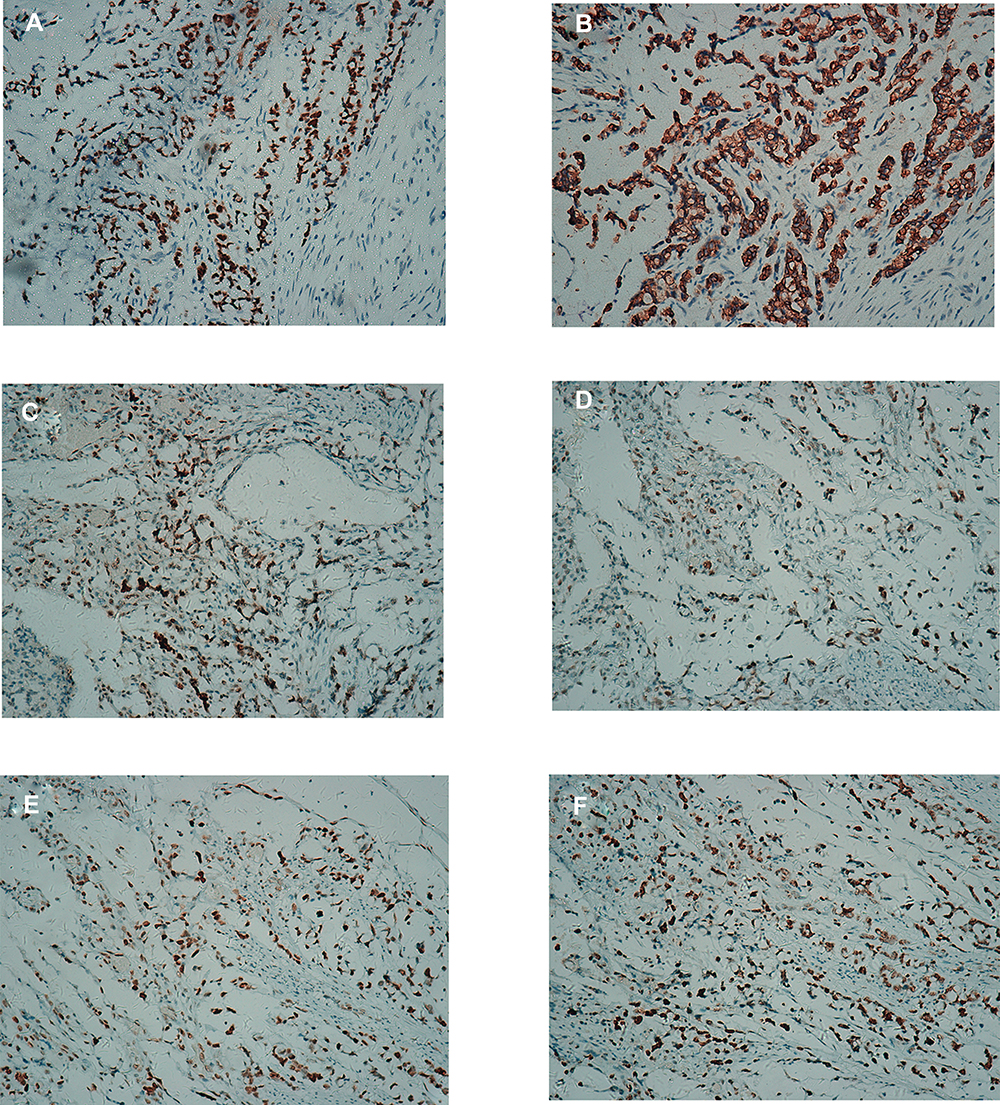 |
Figure 3 Immunohistochemical results. (A) CDX2 (+). (B) CK20(+). (C) MLH1(+). (D) MSH2(+). (E) MSH6(+). (F) PMS2(+). |
 |
Figure 4 The sequencing results of the patient. (A) Heterozygous monoallelic MYH mutation (c.934–2A>G,intron). (B) SMAD4 mutation (R361C). (C) TP53 mutation (Y234H). |
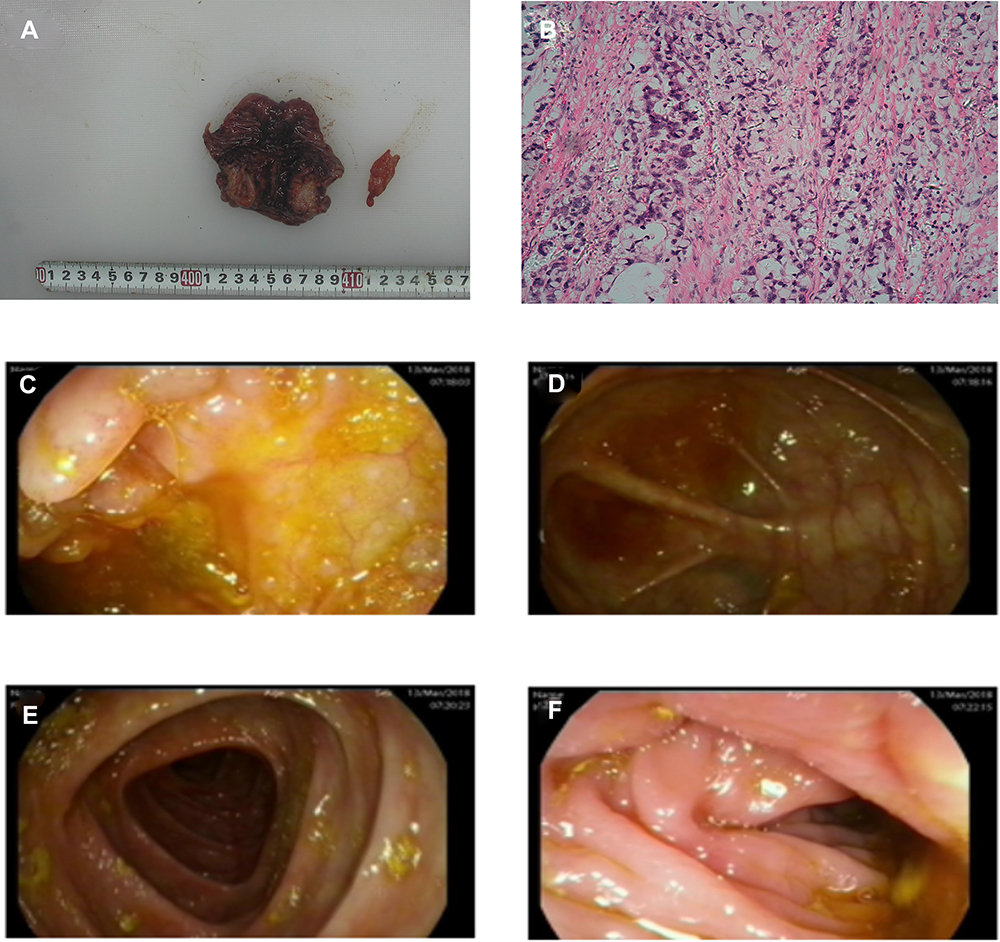 |
Figure 5 Histopathologic diagnosis and colonoscopy after surgy. (A and B) Histopathologic diagnosis, descending colorectal mucinous adenocarcinoma (stage III), which had infiltrated the whole layer (T3), signet ring cell carcinoma can be seen microscopically, the vascular, lymphatic and nerve invasion can be found. (C–F) Colonoscopy after surgy, no abnormalities were observed in the mucosa of the large intestine. |
Discussion
Colorectal carcinoma in children and adolescents is uncommon, the mean age of diagnosis was 15.6 years.4 A higher percentage of males was found in cases involving children/adolescents.5 Lesions were most commonly found in the right hemicolon, followed by rectum, transverse colon, and left hemicolon.6 The most common chief complaints were abdominal pain, hematochezia, abdominal distension, and diarrhea.7 Many patients underwent emergency surgery due to acute intestinal obstruction.8 They were more likely to present with stage III or IV disease, angiolymphatic invasion, perineural invasion, and distal tumors.9 With respect to histology, pediatric patients were diagnosed with mucinous adenocarcinoma and signet ring cell carcinoma more frequently than adults.10 Pediatric patients also had significantly worse histological grade with a higher proportion of poorly differentiated tumors.11 For pediatric patients, genetic diseases such as familial polyposis of the colon, Gardner’s syndrome, Turcot’s syndrome, Peutz–Jegher’s syndrome, ulcerative colitis, familial occurrence of colorectal cancer, and Bloom’s syndrome have been reported as predisposing pathologies that increase the risk of colorectal carcinoma.12,13 The most common genetic factors include classic FAP (familial adenomatous polyposis), Lynch syndrome and MYH-associated polyposis.14 The proportion of patients with a diagnosis of FAP and microsatellite instability (MSI) was higher in young patients.15
Recently, the base excision repair gene MYH has been revealed as being the causative gene of multiple colorectal adenomas, which is an autosomal recessive disorder with a high risk of colorectal cancer.16 The MYH gene product is part of the base-excision repair (BER) pathway, which is involved in defending against oxidative DNA damage. Functionally, MYH helps prevent G:C to T:A transversions caused by oxidative stress to highly mutagenic DNA bases.17 The MYH-associated polyposis (MAP) accounted for less than 1% of the total number of colorectal cancer cases.18 Patients with a double allele MYH mutation have an almost 80% risk of developing colorectal cancer and an equally high risk of developing adenomatous polyps, while patients with a single allele MYH mutation have an increased risk of colorectal cancer or polyps.19 Biallelic (homozygous or compound heterozygous) and monoallelic MYH germline mutations have been detected in Caucasian patients with multiple colorectal adenomas having 3–100 polyps.20 The frequency of biallelic germline mutations of the MYH gene is reported as between 7% and 42% in patients with multiple colorectal adenomas.21 Patients with multiple adenomatous polyps, or those under 50 years of age who have not been diagnosed with MSI stable colorectal cancer, should be tested for the MYH gene. Patients with multiple adenomatous polyps should undergo colonoscopy once a year.22 Partial or complete colectomy may be selected according to the size or characteristics of polyps.
In our case, the pediatric patient carried a heterozygous monoallelic MYH mutation (c.934–2A>G, intron), also he carried somatic mutations of proto-oncogene SMAD4 (R361C) and TP53 (Y234H). As we mentioned above, MYH was genetic related gene which causes polyposis and colorectal cancer. MYH-associated polyposis (MAP), an autosomal recessive condition caused by biallelic pathogenic variants in MYH, is characterized by multiple colorectal adenomas arising in adulthood and increased colorectal cancer risk. Lifetime risk for colorectal cancer among patients with biallelic pathogenic variants in MYH is estimated to be at least 43 to almost 100% in the absence of timely surveillance. Whether monoallelic pathogenic variants in MYH also increase cancer risk is unclear, although there is some evidence suggesting a slight increase, especially when there is a family history of colon cancer. We also gave his mother genetic test, and his mother was found same MYH mutation as him. The MYH mutation which was found in the patient and his mother was not reported in other pediatric cases. Also the patient carried somatic mutations of proto-oncogene SMAD4 (R361C) and TP53 (Y234H). SMAD4 and TP53 were confirmed as colorectal cancer pathogenic genes. The MYH mutation in our case was a heterozygous mutation at the intron-exon splice site, it is located near the transcript region, intron-exon splice site sequences is related to the stability of DNA, during transcription, when the mutation located at the splice site, the pre-mRNA may altered, leading to protein changes and disease. This may increase cancer risk, we believe the genetic mutation of MYH (c.934–2A>G,intron) together with somatic mutations of proto-oncogene SMAD4 and TP53 may lead to the early onset colorectal cancer of the patient.
Treatment of colorectal cancer in pediatric patients is not different from that seen in adults.
Initial treatment of pediatric colorectal cancer usually involved surgery, with most patients undergoing radical or palliative surgery.23 According to TNM staging, adjuvant chemotherapy and radiotherapy are required. Most patients received postoperative adjuvant chemotherapy consisting of FOLFOX (oxaliplatin, 5‑fluorouracil [5‑FU] plus leucovorin), XELOX (oxaliplatin plus Xeloda) and irinotecan. The effective regimens were also same as those prescribed to adults.24 The use of radiotherapy and targeted therapy in adolescents remains to be controversial, in the absence of pediatric prospective clinical trials, treatment guidelines must be extrapolated from adult trials.10 The overall prognosis of pediatric colon cancer is generally poor. Recurrences presented 3–17 months from the time of original diagnosis, while the 3‑ and 5‑ year survival rates were 34.9% and 23.2%, respectively, with a median survival time of 26 months from initial diagnosis.25 The most important predictor of mortality was disease stage. Therefore, early detection of colorectal cancer in young patients through extensive evaluation will contribute to more cases being reported. This, in turn, will affect disease prognosis.
Our work has several limitations. One of the main limitations of the study is that we did not get the genetic testing results of the patient’s father. The correlation between the genetic mutations of the child and the father is not clear. Another limitation is we do not have enough follow-up data for the patient’s mother. She should undergo a colonoscopy every 3–5 years.
Conclusion
Overall, the study summarizes the clinical symptoms, stage, histopathology, pathology, gene mutations and heredity, treatment, and prognosis of pediatric patients with colorectal cancer. Since colorectal cancer rarely occurs in pediatric patients, early diagnosis and management is difficult. Poorer outcomes in these patients appear to be owing to worse tumor biology and stage. Hereditary gene mutations, such as MYH gene variants, are known to exist and were detected in our case study. The necessity of early screening for the detection of colorectal cancer in subjects with a family history of early onset colorectal cancer should be emphasized. Lastly, the results of this study show that genetic screening may provide early diagnosis, which can consequently improve prognosis.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The committees of Shengjing Hospital of China Medical University approved to publish the case details.
Consent for Publication
The patient’s parents provided written informed consent for the case details to be published.
Acknowledgments
The authors would like to thank the pathologist Xiaofei Li from Shengjing Hospital of China Medical University as well as the patient and his family.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Sultan I, Rodriguez-Galindo C, El-Taani H, et al. Distinct features of colorectal cancer in children and adolescents: a population-based study of 159 cases. Cancer. 2010;116(3):758–765. doi:10.1002/cncr.24777
2. Attard TM, Lawson CE. Comparison of the demographic characteristics of pediatric and adult colorectal cancer patients: a national inpatient sample based analysis. World J Pediatr. 2019;15(1):37–41. doi:10.1007/s12519-018-0187-x
3. Pritchard CC, Smith C, Salipante SJ, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14(4):357–366. doi:10.1016/j.jmoldx.2012.03.002
4. Poles GC, Clark DE, Mayo SW, et al. Colorectal carcinoma in pediatric patients: a comparison with adult tumors, treatment and outcomes from the National Cancer Database. J Pediatr Surg. 2016;51(7):1061–1066. doi:10.1016/j.jpedsurg.2015.11.005
5. Tricoli JV, Seibel NL, Blair DG, et al. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628–635. doi:10.1093/jnci/djr094
6. You YN, Xing Y, Feig BW, et al. Young-onset colorectal cancer: is it time to pay attention? Arch Intern Med. 2012;172(3):287–289. doi:10.1001/archinternmed
7. Kravarusic D, Feigin E, Dlugy E, et al. Colorectal carcinoma in childhood: a retrospective multicenter study. J Pediatr Gastroenterol Nutr. 2007;44(2):209–211. doi:10.1097/01.mpg.0000252195.84084.52
8. Goldberg J, Furman WL. Management of colorectal carcinoma in children and young adults. J Pediatr Hematol Oncol. 2012;34(2):S76–79. doi:10.1097/MPH.0b013e31824e38c1
9. Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc. 2014;89(2):216–224. doi:10.1016/j.mayocp.2013.09.006
10. Al-Tonbary Y, Darwish A, El-Hussein A, et al. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29–33. doi:10.1016/j.hemonc
11. Singer G, Hoellwarth ME. Colorectal carcinomas in children: an institutional experience. Pediatr Surg Int. 2012;28(6):591–595. doi:10.1007/s00383-012-3090-x
12. Datta RV, LaQuaglia MP, Paty PB. Genetic and phenotypic correlates of colorectal cancer in young patients. N Engl J Med. 2000;342(2):137–138. doi:10.1056/NEJM200001133420216
13. Power DG, Gloglowski E, Lipkin SM. Clinical genetics of hereditary colorectal cancer. Hematol Oncol Clin North Am. 2010;24(5):837–859. doi:10.1016/j.hoc.2010.06.006
14. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–932. doi:10.1056/NEJMra012242
15. Terdiman JP, Gum JR, Conrad PG, et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor micro satellite instability before germline genetic testing. Gastroenterology. 2001;120(1):21–30. doi:10.1053/gast.200.20874
16. Poley JW, Wagner A, Hoogmans MM, et al. Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer. 2007;109(11):2349–2356. doi:10.1002/cncr.22697
17. Win AK, Dowty JG, Cleary SP, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology. 2014;146(5):1208–1211. doi:10.1053/j.gastro.2014.01.022
18. Win AK, Cleary SP, Dowty JG, et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. 2011;129(9):2256–2262. doi:10.1002/ijc.25870
19. Win AK, Reece JC, Dowty JG, et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int J Cancer. 2016;139(7):1557–1563. doi:10.1002/ijc.30197
20. Pervaiz M, Eppolito A, Schmidt K. Papillary thyroid cancer in a patient with MUTYH-associated polyposis (MAP). Fam Cancer. 2010;9(4):595–597. doi:10.1007/s10689-010-9366-1
21. Win AK, Hopper JL, Jenkins MA. Association between monoallelic MUTYH mutation and colorectal cancer risk: a meta-regression analysis. Fam Cancer. 2011;10(1):1–9. doi:10.1007/s10689-010-9399-5
22. Lubbe SJ, Di Bernardo MC, Chandler IP, et al. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27(24):3975–3980. doi:10.1200/JCO.2008.21.6853
23. Blumer SL, Anupindi SA, Adamson PC, et al. Sporadic adenocarcinoma of the Colon in children: case series and review of the literature. J Pediatr Hematol Oncol. 2012;34(4):e137–141. doi:10.1097/MPH.0b013e3182467f3e
24. Hill DA, Furman WL, Billups CA, et al. Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J Clin Oncol. 2007;25(36):5808–5814. doi:10.1200/JCO.2007.12.6102
25. Saab R, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177–192. doi:10.2165/00148581-200810030-00006
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.