Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
A novel missense KIT mutation causing piebaldism in one Chinese family associated with café-au-lait macules and intertriginous freckling
Authors Jia W, Xiao X, Wu J, Ma Y, Ge Y, Li Q, Mao Q, Li C
Received 8 October 2014
Accepted for publication 21 November 2014
Published 21 April 2015 Volume 2015:11 Pages 635—638
DOI https://doi.org/10.2147/TCRM.S75544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor De Yun Wang
Wei-Xue Jia,1,2 Xue-Min Xiao,1,2 Jian-Bing Wu,1,2 Yi-Ping Ma,1,2 Yi-Ping Ge,1,2 Qi Li,1,2 Qiu-Xia Mao,1,2 Cheng-Rang Li1,2
1Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing, Jiangsu, China; 2Jiangsu Key Laboratory of Molecular Biology for Skin Diseases and STIs, Nanjing, Jiangsu, China
Abstract: Piebaldism is a rare autosomal dominant genodermatosis, manifesting as congenital and stable depigmentation of the skin and white forelock. It has been found to be associated with mutations in the KIT or SLUG genes. We report a Chinese piebaldism family including a 28-year-old woman and her 3-year-old son with characteristics of white patches and forelock associated with numerous brown macules and patches. Genomic DNA samples of the proband and her son were extracted from their peripheral blood. One hundred unrelated healthy individuals were used as controls. All coding regions of KIT, SLUG, and NF1 genes were amplified by polymerase chain reaction using exon flanking intronic primers and Sanger sequencings were performed. DNA sequencing revealed heterozygous missense c.2431T>G mutation in exon 17 of the KIT gene in the proband and the affected son. No potentially pathogenic variant was identified in SLUG or NF1 genes. The nucleotide substitution was not found in 100 unrelated control individuals. This study reveals a novel KIT mutation in piebaldism, and it further supports that café-au-lait macules and intertriginous freckling of piebaldism are parts of pigmented anomaly in piebaldism, which does not necessarily represent coexistence of neurofibromatosis type 1 (NF1).
Keywords: novel mutation, KIT gene, neurofibromatosis type 1
Introduction
Piebaldism (Online Mendelian Inheritance in Man [OMIM] #172800) is a rare autosomal dominant genodermatosis characterized by congenital leukoderma and poliosis. The depigmentation of skin has a characteristic distribution pattern that favors the central forehead, midfrontal portion of the scalp with a resultant triangular white forelock, central anterior trunk, and mid-extremities. Previous studies have revealed that most cases of piebaldism are caused by mutation of the KIT (mast cell/stem cell growth factor receptor, also known as c-kit or CD117) gene mapped on chromosome 4q12,1 which encodes KIT, a type 3 transmembrane receptor for mast cell growth factor (MGF, also known as stem cell factor). The mutation of the receptor is associated with defects in the proliferation or migration of melanoblasts from the neural crest during early embryonic development, which causes the absence of melanocytes from the nonpigmented patches of skin and from hairbulbs in the white patches of hair in piebaldism.1 Some cases of piebaldism result from mutation of the SLUG (also known as SNAI2) gene on chromosome 8q11.2 The SLUG gene encodes Slug, a zinc-finger neural crest transcription factor, which is critical for the development of human melanocytes. Several piebaldism patients with café-au-lait macules (CALMs) and intertriginous freckling have been previously reported,3–10 and the diagnosis of coexistence of neurofibromatosis type 1 (NF1) is controversial. Here, we report a Chinese family with piebaldism due to a novel heterozygous missense mutation in KIT, and investigate the relationships between piebaldism and manifestation of CALMs and intertriginous freckling.
All patients and normal individuals signed written informed consents. The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by our local ethics committee, the Institutional Ethical Review Boards of Peking Union Medical College.
Materials and methods
The proband of this family was a 28-year-old Chinese woman who presented with generalized white patches and brown macules since birth. The white patches remained stable and the brown macules gradually became larger and increased in frequency. Physical examination revealed symmetrical patches of depigmentation on the forehead, chin, central anterior trunk, and extremities, with islands of normal pigmentation within them. A white forelock was observed (Figure 1A). CALMs within the patches of depigmentation and normal skin were also noted (Figure 1B). Nine CALMs were greater than 15 mm in diameter. Numerous small 1–5 mm lentigines present in the bilateral axillary and inguinal regions (Crowe’s sign) were seen (Figures 1C and D). No neurofibromas were detected. The patient was otherwise healthy. Her parents were non-consanguineous and healthy. A skin biopsy was obtained from a depigmented lesion of the left upper arm. Basal hypopigmentation was present in the specimen (Figure 1G).
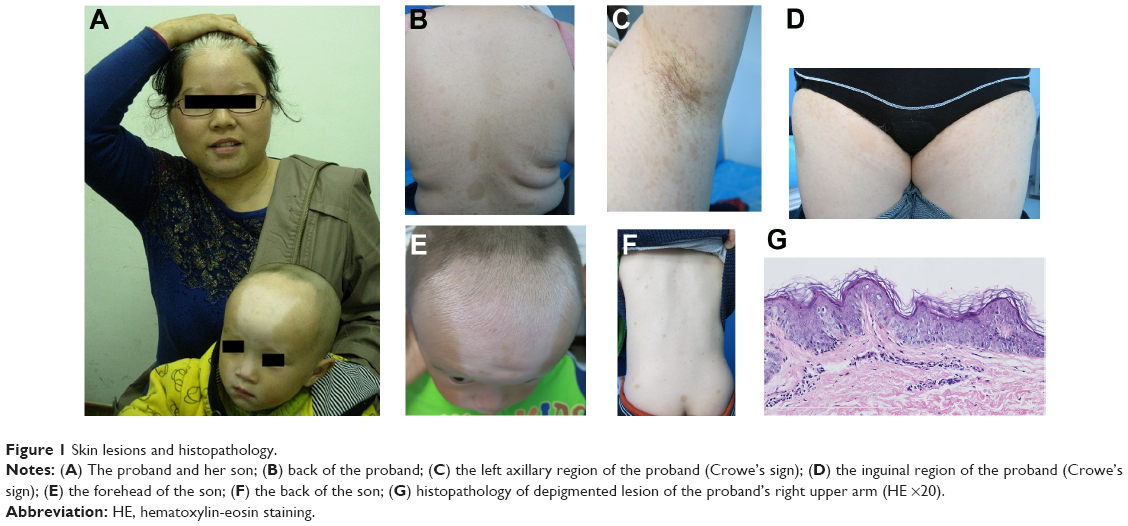 | Figure 1 Skin lesions and histopathology. |
A similar phenotype of a typical triangular white patch on the forehead and multiple CALMs in the patient’s 3-year-old son was confirmed (Figures 1A, E, and F). He was developing normally. His parents were non-consanguineous, and his father was healthy. There were no other relatives with a similar condition. There were no reported findings such as facial dysmorphism, deafness, iris heterochromia, mental retardation, or other features of Waardenburg syndrome in the family history.
Genomic DNA samples of the proband and her son were extracted from their peripheral blood leukocytes using the Wizard® Genomic DNA Purification Kit (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s protocol. In addition, genomic DNA of 100 unrelated healthy individuals were used as controls. The primers flanking all exons of the KIT, SLUG, and NF1 genes were designed using the web-based version of the Primer 3.0 program (http://www.genome.wi.mit.edu/cgibin/primer/primer3_www.cgi). Primer sequences are available on request. The primer sequence of the 17th exon of the KIT gene was designed as follows: FOR, 5′>TGTGAACATCATTCAAGGCG<3′ and REV, 5′> TGACTGCTAAAATGTGTGATATCCC<3′. Genomic DNA was amplified by polymerase chain reaction (PCR). The PCR conditions were: Hotstar Taq activation at 95°C for 3 minutes, followed by 14 cycles, each having denaturation at 95°C for 35 seconds, annealing at 63°C for 45 seconds and extension at 72°C for 75 seconds, then followed by 27 cycles, each having denaturation at 95°C for 30 seconds, annealing at 56°C for 45 seconds and extension at 72°C for 75 seconds, and the final extension was 72°C for 5 minutes. After the amplification, the PCR products were purified using the QIA-quick PCR Purification Kit (Promega) and sequenced using ABI Prism® 3730 automated sequencer (Thermo Fisher Scientific, Waltham, MA, USA). Sequence comparisons and analyses were performed using Phred-Phrap-Consed v12.0 program (http://www.phrap.org).
Results
No potentially pathogenic variant was identified in the SLUG or NF1 genes in the proband and her son. We detected a heterozygous missense c.2431T>G mutation in exon 17 of the KIT gene in the proband (Figure 2A). The same mutation was also found in the affected son (Figure 2B). In addition, the nucleotide substitution was not found in 100 unrelated control individuals (Figure 2C).
 | Figure 2 Mutation of the KIT gene. |
Discussion
The KIT gene encodes the human homologue of the proto-oncogene c-kit. C-kit was first identified as the cellular homologue of the feline sarcoma viral oncogene v-kit. This protein is a type 3 transmembrane receptor for MGF.
Genetic testing demonstrated a novel heterozygous missense mutation of the KIT gene, c.2431T>G, which results in the substitution of Phe (TTT) at position 811 to Val (GTT) in the intracellular tyrosine kinase domain in KIT protein. The nucleotide substitution was not found in 100 unrelated control individuals, suggesting that the mutation was not likely to be neutral polymorphism.
Using the tool of PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), it is reported that this mutation is predicted to be probably damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00).
Clinical manifestations and phenotypic severity of piebaldism strongly correlates with the mutation site within the KIT gene. The most severe phenotypes usually result from mutations involving the intracellular tyrosine kinase domain, whereas the mildest phenotypes are usually caused by mutations involving the amino-terminal extracellular ligand-binding domain.11,12 The mutation reported here confirms the involvement of the KIT gene in human piebaldism and provides supportive evidence that a missense mutation within the intracellular tyrosine kinase domain of KIT produces a severe phenotype.11
Several piebaldism patients that is associated with CALMs and intertriginous freckling have been reported. Some researchers considered it as an overlap of piebaldism and NF1, for the diameters and numbers of CALMs and freckling satisfy the diagnostic criteria for NF1, established by the National Institute of Health (NIH) Consensus Conference in 1988.13 However, none of the reported cases had cutaneous neurofibromas or other non-pigmentary features of NF1; one patient, however, had Lisch nodules in the iris.4 Both piebaldism and NF1 are autosomal dominant conditions, while their genes have been localized to different chromosomes, 4q12 or 8q11 and 17q11.2, respectively. Therefore, there is only a small chance for an association, especially in cases with an absence of family histories of NF1 with non-pigmentary features. Furthermore, all piebaldism patients that is associated with CALMs and intertriginous freckling who underwent comprehensive gene testing showed mutations in the KIT gene,6,7,9 and the patients who underwent gene testing of NF1 showed no mutation.7,8 Surprisingly, those piebaldism patients with mutations in the intracellular tyrosine kinase domain of KIT, G610D,7 E640D,9 R791G,6 or G812V,6 all showed CALMs and freckling, which is similar to our patient. This supports the theory that mutation in the intracellular tyrosine kinase domain of KIT results in severe piebaldism and may be related to the appearance of CALMs and freckling. It also suggests that the unusual phenotype is related to the loss of KIT tyrosine kinase function and subsequent effect on SPRED1 and the Ras/MAPK pathway.9
To conclude, we demonstrate a family with piebaldism due to a novel KIT missense mutation. We consider the CALMs and intertriginous freckling seen in some piebaldism patients are parts of pigmented anomalies in piebaldism, which does not necessarily represent coexistence of NF1. Careful clinical evaluation, especially of the non-pigmentary features of NF1 and molecular testing if necessary, should be used to identify NF1.
Acknowledgments
We thank the patients for their participation in this study. This work was supported by PUMC Innovation Team Fund, and National Natural Science Foundation of China (Grant No.81472872).
Disclosure
The authors report no conflicts of interest in this work.
References
Giebel LB, Spritz RA. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci U S A. 1991;88(19):8696–8699. | ||
Sánchez-Martín M1, Pérez-Losada J, Rodríguez-García A, et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A. 2003;122A(2):125–132. | ||
Chang T, McGrae JD Jr, Hashimoto K. Ultrastructural study of two patients with both piebaldism and neurofibromatosis 1. Pediatr Dermatol. 1993;10(3):224–234; discussion 288. | ||
Tay YK. Neurofibromatosis 1 and piebaldism: a case report. Dermatology. 1998;197(4):401–402. | ||
Angelo C, Cianchini G, Grosso MG, Zambruno G, Cavalieri R, Paradisi M. Association of piebaldism and neurofibromatosis type 1 in a girl. Pediatr Dermatol. 2001;18(6):490–493. | ||
Spritz RA, Holmes SA, Itin P, Küster W. Novel mutations of the KIT (mast/stem cell growth factor receptor) proto-oncogene in human piebaldism. J Invest Dermatol. 1993;101(1):22–25. | ||
Duarte AF, Mota A, Baudrier T, et al. Piebaldism and neurofibromatosis type 1: family report. Dermatol Online J. 2010;16(1):11. | ||
Stevens CA, Chiang PW, Messiaen LM. Cafe-au-lait macules and intertriginous freckling in piebaldism: clinical overlap with neurofibromatosis type 1 and Legius syndrome. Am J Med Genet A. 2012;158A(5): 1195–1199. | ||
Chiu YE, Dugan S, Basel D, Siegel DH. Association of Piebaldism, multiple cafe-au-lait macules, and intertriginous freckling: clinical evidence of a common pathway between KIT and sprouty-related, ena/vasodilator-stimulated phosphoprotein homology-1 domain containing protein 1 (SPRED1). Pediatr Dermatol. 2013;30(3):379–382. | ||
Park SY, Kim HJ, Ahn SK. Piebaldism with neurofibromatosis type I: a familial case. Ann Dermatol. 2014;26(2):264–266. | ||
Spritz RA, Holmes SA, Ramesar R, Greenberg J, Curtis D, Beighton P. Mutations of the KIT (mast/stem cell growth factor receptor) proto-oncogene account for a continuous range of phenotypes in human piebaldism. Am J Hum Genet. 1992;51(5):1058–1065. | ||
Ward KA, Moss C, Sanders DS. Human piebaldism: relationship between phenotype and site of kit gene mutation. Br J Dermatol. 1995; 132(6):929–935. | ||
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45(5):575–578. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.