Back to Journals » Cancer Management and Research » Volume 12
A Novel Epitope Quality-Based Immune Escape Mechanism Reveals Patient’s Suitability for Immune Checkpoint Inhibition
Authors Wessolly M ![]() , Stephan-Falkenau S, Streubel A, Werner R, Borchert S
, Stephan-Falkenau S, Streubel A, Werner R, Borchert S ![]() , Griff S, Mairinger E
, Griff S, Mairinger E ![]() , Walter RFH
, Walter RFH ![]() , Bauer T, Eberhardt WEE, Blum TG
, Bauer T, Eberhardt WEE, Blum TG ![]() , Schmid KW
, Schmid KW ![]() , Kollmeier J, Mairinger T, Mairinger FD
, Kollmeier J, Mairinger T, Mairinger FD ![]()
Received 30 April 2020
Accepted for publication 16 July 2020
Published 26 August 2020 Volume 2020:12 Pages 7881—7890
DOI https://doi.org/10.2147/CMAR.S258396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Michael Wessolly,1,2,* Susann Stephan-Falkenau,3,* Anna Streubel,3 Robert Werner,3 Sabrina Borchert,1,2 Sergej Griff,3 Elena Mairinger,1 Robert FH Walter,1,4 Torsten Bauer,5 Wilfried EE Eberhardt,4,6 Torsten G Blum,5 Kurt W Schmid,1 Jens Kollmeier,5 Thomas Mairinger,1,* Fabian D Mairinger1,2,*
1Institute of Pathology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany; 2German Cancer Consortium (DKTK), Partner Site University Hospital Essen, Essen, Germany; 3Department of Tissue Diagnostics, Helios Klinikum Emil Von Behring, Berlin, Germany; 4Ruhrlandklinik, West German Lung Center, University Hospital Essen, University of Duisburg-Essen, Essen, Germany; 5Lungenklinik Heckeshorn, Helios Klinikum Emil Von Behring, Berlin, Germany; 6Department of Medical Oncology, West German Cancer Center, University, Hospital Essen, Essen, Germany
*These authors contributed equally to this work
Correspondence: Michael Wessolly Research Group: Translational Cancer Research, Institute for Pathology
University Hospital Essen, Hufelandstraße 55, Essen D-45147, Germany
Tel +49 201 723 – 83035
Fax +49 201 723 – 3880
Email [email protected]
Background: Immune checkpoint inhibition, especially the blockade of PD-1 and PD-L1, has become one of the most thriving therapeutic approaches in modern oncology. Immune evasion caused by altered tumor epitope processing (so-called processing escapes) may be one way to explain immune checkpoint inhibition therapy failure. In the present study, we aim to demonstrate the effects of processing escapes on immunotherapy outcome in NSCLC patients.
Patients and Methods: Whole exome sequencing data of 400 NSCLC patients (AdC and SCC) were extracted from the TCGA database. The ICB cohort was composed of primary tumor probes from 48 NSCLC patients treated with nivolumab. Mutations were identified by targeted amplicon-based sequencing including hotspots and whole exomes of 22 genes. The effect of mutations on proteasomal processing was evaluated by deep learning methods previously trained on 1260 known MHC-I ligands. Cox regression modelling was used to determine the influence on overall survival.
Results: In the TCGA cohort, processing escapes were associated with decreased overall survival (p= 0.0140). In the ICB cohort, patients showing processing escapes in combination with high levels of PD-L1 (n=8/48) also showed significantly decreased overall survival, independently of mutational load or PD-L1 status.
Conclusion: The concept of altered epitope processing may help to understand immunotherapy failure. Especially when combined with PD-L1 status, this method can be used as a biomarker to identify patients not suitable for immunotherapy.
Keywords: massive parallel sequencing, NSCLC, immunotherapy, epitope, processing escape, deep learning
Plain Language Summary
Immune checkpoint inhibition has become a milestone in modern cancer therapy. The purpose of immune checkpoint inhibitors (ICBs), eg, PD-L1, is to counter the negative immune regulation induced by a tumor. By haltering its interaction, the anti-tumor immune activity increases.
Biomarkers (measurable clinical parameters) are clinically used to evaluate patient’s suitability for ICBs. One such biomarker, the tumor mutational burden (TMB), quantifies the amount of small tumor proteins (epitopes), which are presented on the cell surface. These proteins are changed by mutation and therefore recognized as foreign.
Shorter or extended fragments, caused by mutation, may impact the immune system in a negative way. Wessolly et al reported this deficient epitope quality in patients suffering from neuroendocrine lung tumors in 2018. The present study aims to verify this mechanism in patients diagnosed with non-small cell lung cancer (NSCLC).
Wessolly et al used bioinformatical methods to simulate the development of epitopes changed by mutation. In a second approach, their presence on the cell surface and immune activation capabilities were tested. Non-activators and non-presenters were considered of inferior quality. In the end, these findings were correlated to clinical data. Patients showing signs of inferior epitope quality in combination with high expression of PD-L1 had decreased survival rates.
In conclusion, patients would not respond well to immunotherapy when both mechanisms were active. Therefore, both parameters can be used to improve patient stratification for immunotherapy.
Introduction
Among available cancer therapies, immunotherapy has become a milestone in the treatment of many malignancies.1,2 This is especially true for lung cancer, still responsible for the highest number of cancer related deaths worldwide.3,4 Although the overall success of this therapeutic approach is impressive, oncologists face the problem that by far not all patients profit from immune checkpoint blockade. Predictive markers that allow precise patient selection for this therapeutic approach are therefore urgently needed.3
The cornerstone of all immunotherapeutical approaches is to take advantage of the immune system´s ability to identify tumor cells by cell surface autoantigens.
Tumor cells expressing such neoantigens are identified as foreign and subsequently obliterated by cytotoxic T-cells. The tumor cell itself tries to escape this auto aggression by using the PD-1/PD-L1 mechanism to switch off immune activity.
Subsequently two potential selection criteria seem appropriate for identifying patients potentially suitable for immune checkpoint blockade:
Patients with a high tumor mutational burden (TMB) may express more neoantigens and therefore could be more easily identified by the cellular immune system. High TMB therefore should result in high T-cell activity, especially during PD-1/PD-L1 blockade.
The second group consists of tumors expressing high amounts of PD-1 or PD-L1 on their cell surfaces. These tumors are obviously suitable for PD-1/PD-L1 inhibition.5,6
Both groups of tumors (TMB high with/without PD-1/PD-L1) showed statistically significant benefits from immune checkpoint inhibition in several studies.7–9 Nevertheless, the predictive power of both concepts is still not satisfying in times of personalized medicine.10
Up to now, prediction of ICB outcome was focused to the presence of the PD-1/PD-L1 molecules and the amount of potentially expressed neoantigens. Not much attention however has been paid on the presumed immunogenic quality of the latter until recently.
The activation of tumor specific T-cells occurs via presentation of small peptide fragments (epitopes) originating from tumor antigens.11,12 These originate from a complex intracellular pathway involved in the processing of the antigenic peptides starting with polyubiquitination of the protein, labeling it for proteasomal degradation. The resulting fragments are further trimmed down to an optimal amino acid (AA) length (eight to eleven AA), get translocated into the endoplasmic reticulum (ER) via TAP and are subsequently loaded on the HLA class I molecule. Finally, the complex is presented on the cell membrane.11,12
These neoantigens trigger the physiological T-cell based immune response against the tumor cell due to highlighting the tumor cell as a potential target for cytotoxic T-cells.
Consequently, the concept of tumor-driven immunologic reactions seems to be based not only on epitope amount but also on correct epitope processing. Any inconsistency within this complex protein cleavage process might subsequently result in an immune escape, independent from PD-1/PD-L1. This approach is supported by investigations of viral infections, in particular with the human immunodeficiency virus 1 (HIV-1) and hepatitis C virus (HCV), where a subset of mutations altered proteasomal processing of the viral proteins.13,14 The mutations lead to modified epitopes with different lengths and a decreased effectiveness in activating CTLs.13
In a previous study, we demonstrated that the presence of processing escapes was significantly associated with shortened OS as well as PFS.15 Within that analysis, around 35% of all registered mutations had a significant influence on proteasomal processing.
These results encouraged us to prove the value of this approach in a collective of ICB treated NSCLC patients with known outcome.
Patients and Methods
Demographic Data and Study Design
Our study is based on three patient cohorts. Two cohorts with whole exome-sequencing data available were obtained online from The Cancer Genome Atlas (TCGA). Cohort 1 consisted of 230 patients diagnosed with lung adenocarcinoma (AdC). In addition, 178 patients suffering from lung squamous-cell carcinoma (SCC) were acquired (Cohort 2).16,17 However, in order to compare all three cohorts, only genes analyzed in cohort 3 were extracted. Furthermore, mRNA expression profiles from whole-transcriptome analysis were available for both cohorts. Cohort 1 and cohort 2 are summarized under the umbrella-term TCGA-cohort.
The third collective consisted of 48 NSCLC patients, diagnosed with either AdC (n=23) or SCC (n=25). Patient data were collected at the Helios Klinikum Emil von Behring (Berlin) between 2012 and 2016. Inclusion criteria were the availability of both sufficient tumor material and a complete set of data concerning follow-up and treatment. Mutations were identified by targeted amplicon-based sequencing (0.7 Mb). Most patients were tested upfront for prominent NSCLC biomarkers including EGFR, ALK, ROS1 and KRAS. All AdCs were negative for EGFR mutations. PD-L1 expression was determined by immunohistochemistry using the QR-1 antibody (Quartett, Potsdam). A cell was determined as positive if membranous staining of any strength could be observed. PD-L1 status was reported as percentage of stained tumor cells in relation to all tumor cells. All patients received nivolumab. Most patients received multiple lines of chemotherapy and/or radiation before immunotherapy (supplementary Table 1). The third collective will from now on be coined as the ICB (immune checkpoint blockade) cohort.
Nucleic Acid Preparation
Genomic DNA was isolated on a Maxwell® 16 Research (Promega Corporation, Madison, USA) as recommended in the manufacturer’s protocol. Nucleic acid quantification was performed using Qubit (Life Technologies, Carlsbad, USA) and Nanodrop 1000 instrument (Thermo Fisher Scientific, Waltham, USA). To assess the exact amplifiable amount of DNA from FFPE samples, we measured intact DNA amounts by using the Applied Biosystems® TaqMan® RNase P Assay. Amplification was performed on the QuantStudio 5 (Thermo Scientific, Waltham, USA).
Next-Generation Sequencing
For this study, we used the Colon Lung v2 AmpliSeq Panel (Thermo Scientific, Waltham, USA) according to the manufacturer’s instructions.
The panel comprises 92 amplicons covering hotspot and targeted regions of 22 genes involved in colon and lung cancer tumorigenesis (KRAS, EGFR, BRAF, PIK3CA, AKT1, ERBB2, PTEN, NRAS, STK11, MAP2K1, ALK, DDR2, CTNNB1, MET, TP53, SMAD4, FBXW7, FGFR3, NOTCH1, ERBB4, FGFR1, FGFR2). We used 10ng DNA input for each NGS library generation following the AmpliSeq Library protocol Version E.0 (Thermo Scientific, Waltham, USA).
Filtering of and Identification of Mutations
Sequencing data were directly filtered after variant calling. First, variants with a coverage below 20 reads were discarded for subsequent analysis to overcome sequencing errors and fixation artefacts. Furthermore, variants showing an allelic frequency above 0.9 as well as known SNPs were filtered. Finally, a combined score, including both allelic frequency and tumor cell proportion was calculated. Mutations needed to be abundant in 50% of all tumor cells assuming a heterozygous mutation pattern.
All variants passing the filters were manually reviewed using the integrative genome viewer tool. Genomic alterations were translated to protein level changes. Only non-synonymous mutations were considered for further steps.
In silico Analysis of Altered Epitopes by Mutation
All bioinformatical, statistical and graphical analyses were performed using the R programming environment (v. 3.4.2).
Based on the number of cancer-related genes and a default epitope length of nine amino acids, databases were browsed for all available epitope information of analyzed genes, including the specific HLA-type for each epitope and the HLA binding affinity (IC50) (supplementary Table 2).
The machine learning tool NetChop 3.1 was utilized to predict the proteasomal processing of each antigen.18,19 All epitope sequences were extended by eight amino acids for each flanking region (N-and C-terminal, respectively). This resulted in a construct spanning 25 AS, which served as NetChop input. For every amino acid position, a cleavage probability was estimated. The absolute difference in cleavage probability between each wildtype and the respective mutated position was calculated. Any calculated difference above 50% was considered as a significant change in the cleavage pattern (supplementary Table 3).
The peptides, which cleavage patterns differed significantly according to NetChop were submitted to the prediction tool NetMHC (Version 4.0).20,21 For a given HLA-Type this tool predicts the HLA-Affinity (IC50 values, supplementary Table 4–6) of each peptide fragment. We focused primarily on 12 most prominent HLA supertypes.22–25 The IC50 values of mutated peptides and their corresponding wildtype variants were finally correlated against each other (supplementary Figure 5 and 6).
For in silico prediction of immune activation, wildtype and mutated epitope sequences were submitted to the Class I Immunogenicity tool provided by the Immune epitope data base (IEDB).26 The returned score indicated the potential TCR activation capability of each predicted epitope (supplementary Table 7).
Statistical Analysis in R
For each significant association double dichotomous contingency tables (DDCT) were created and positive and negative predictive values were calculated. DDCT were analyzed using Fisher’s Exact test. To test dependency of ranked parameters with more than two groups, the Pearson’s Chi-squared test was used. Correlations between metric variables were tested using the Spearman’s rank correlation test as well as the Pearson’s product moment correlation coefficient for linear modeling. Kaplan-Meier analysis was done for the assessment of associations between gene expression and progression-free survival (PFS) or overall survival (OS). Significant differences in PFS or OS between groups were verified by a COXPH-model using Wald-test, Likelihood-ratio test and Score (log-rank) test. Survival rate was defined as the number of patients in each group still alive at a certain time point. Median survival time was calculated in months. Due to the multiple statistical tests; all p-values were FDR adjusted (false discovery rate). The level of statistical significance was defined as p ≤ 0.05 after adjustment.
Results
Occurrence of Processing Escapes in Lung Adeno- and Squamous-Cell Carcinomas (TCGA Cohort)
Overall, 259 and 164 non-synonymous mutations were identified in the AdC-TCGA and the SCC-TCGA cohort, respectively (Table 1). Thirty-five percent to forty-five percent of all non-synonymous mutations were associated with altered proteasomal processing. They lead to 1245 and 624 affected epitope fragments, respectively. Of all predicted epitopes, only up to 20% were still binding to MHC-I in both collectives.
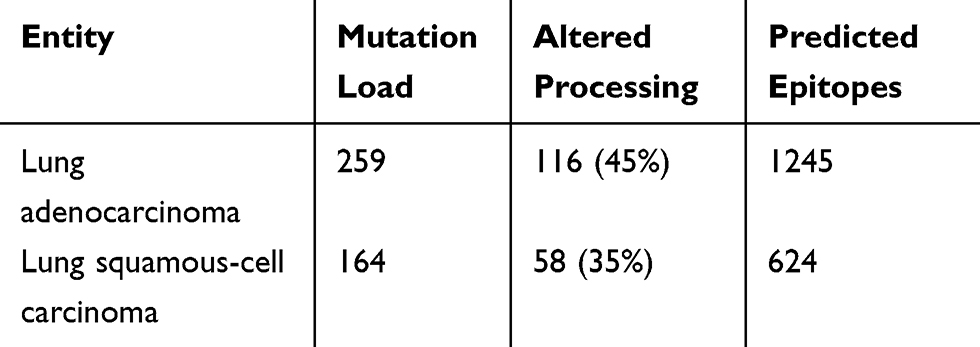 |
Table 1 TCGA validation cohort. The total number of non-synonymous mutations for both entities is displayed in the second column ("Mutation Load"). A proportion of those non-synonymous mutations is associated with altered proteasomal antigen processing ("Altered Processing" ). Furthermore, the number of predicted epitopes derived from altered processed antigens is displayed ("Predicted Epitopes"). |
Altered Proteasomal Processing Affects Hosts Immune Response and Thereby Dismals Patients’ Prognosis in the TCGA Cohort
For survival analysis, patients were separated into two groups: patients with the occurrence of mutations that result in altered processing and patients without them (Figure 1). COXPH regression analysis revealed significantly shortened overall survival for patients in the former group (p= 0.0140). Explorative data analysis revealed associations between the expression of specific immune factors (Granzyme K, CD20 and CD40L) and altered processing (supplementary Figure 13A-C, supplementary Table 8).
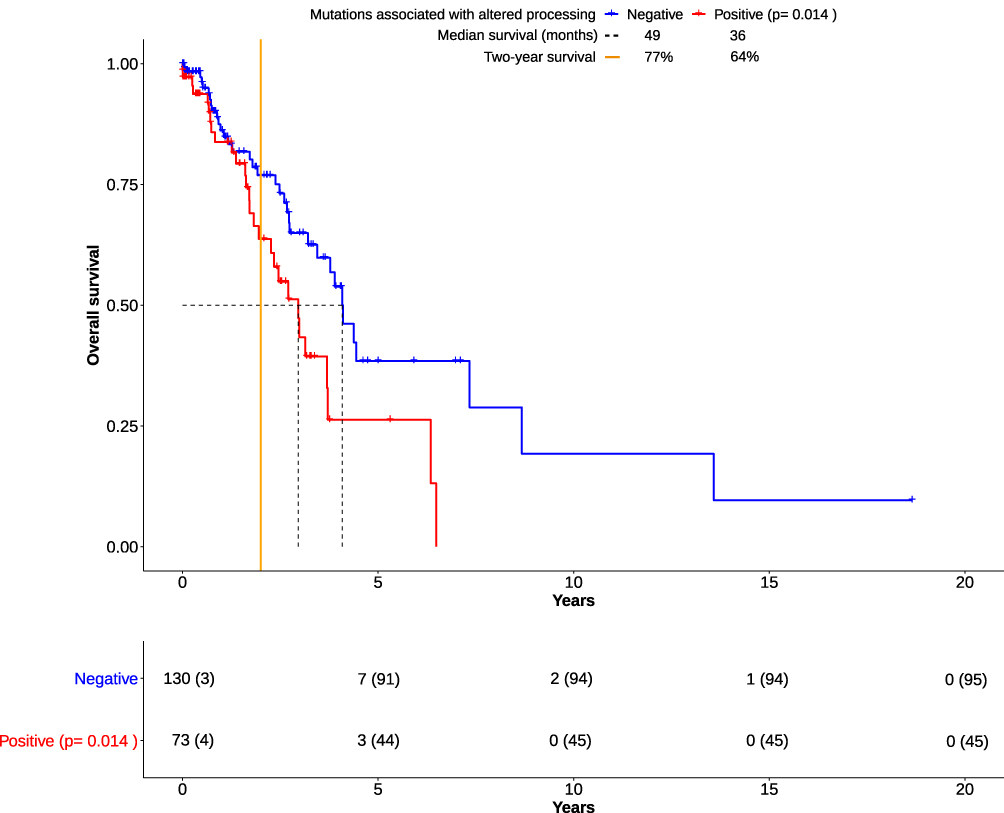 |
Figure 1 Overall survival (OS) in lung adenocarcinomas derived from the TCGA cohort. Kaplan-Meier plots show the course of overall survival for patients with presence (n= 73) or absence (n=130) of mutations associated with altered processing. The number at risk for each group of patients was displayed in a table below. The number of censored patients at specific time points was also added (parentheses). Over the course of 15+ years, both groups have become clearly distinguishable. Though, the overall survival of the “Positive” group was significantly impaired in comparison to the “Negative” group (p=0.0140, Score (log-rank) test), two long-time survivors in the “Negative” group were seemingly outliers, thereby skewing the calculation. The “Negative” group had a survival benefit of one year according to median survival. However, the number of patients living past two years (“Two-year survival”) differed from 77% to 64%. This hinted towards an association of altered processing with impaired overall survival by deficient immune response. All data were based on cohort 1 (see Material and Methods), and downloaded from the TCGA database. |
Verification of Mutational Load and Altered Proteasomal Epitope Processing in I/O Treated NSCLCs
Eighty-five non-synonymous mutations were identified in the ICB cohort. TP53 (n=22) and KRAS (n=15) were most frequently mutated (supplementary Figure 4A). Other prominent genes were EGFR (n=4), FGFR3 (n=6), SMAD4 (n=5) and STK11 (n=5).
Altered Proteasomal Epitope Processing is Linked to Reduced MHC-I Presentation of Mutated Epitopes
Forty-four percent of all mutations could be linked to altered proteasomal processing (Table 2). On a mechanical level, processing escapes could be associated with reduced MHC-I presentation. Of all predicted 366 altered epitopes, only 35 were presented by MHC-I. Roughly 11% of these presented epitopes might trigger an actual immune response according to immunogenicity scoring.
 |
Table 2 I/O treated NSCLC cohort. In addition to the characteristics displayed in Table 1, NetMHC 4.0 was used to determine the affinity of predicted epitopes for MHC Class I. Some of the mutated epitopes had a higher chance to trigger an immune response by cytotoxic lymphocytes according to their immunogenicity score. |
The Presence of Altered Processing Combined with PD-L1 Expression Negatively Impacts Overall Survival in I/O Treated NSCLC Patients
COXPH-modeling was used to ascertain the influence of single and multiple clinical covariates on OS. Important variables weighted to the model were the "Mutational Load" (all non-synonymous mutations identified by sequencing), "Processing Mutations" (mutations linked to altered proteasomal processing) and the "Ratio" between both variables. None of these showed a significant impact on OS (Table 3). Furthermore, neither the histological subtype (adenocarcinoma, squamous cell-carcinoma) nor the PD-L1 status had any significant influence (Table 3). In multivariate COXPH analyses, processing mutations in combination with PD-L1 expression seemed to significantly shorten OS (Score (log-rank) test, p= 0.0437). Both covariates synergistically affected OS (p=0.0246). The survival impact of all important covariates was additionally visualized via forest plot (Figure 2A). Based on hazard ratios and confidence intervals, altered processing in combination with PD-L1 expression had the most negative impact on OS.
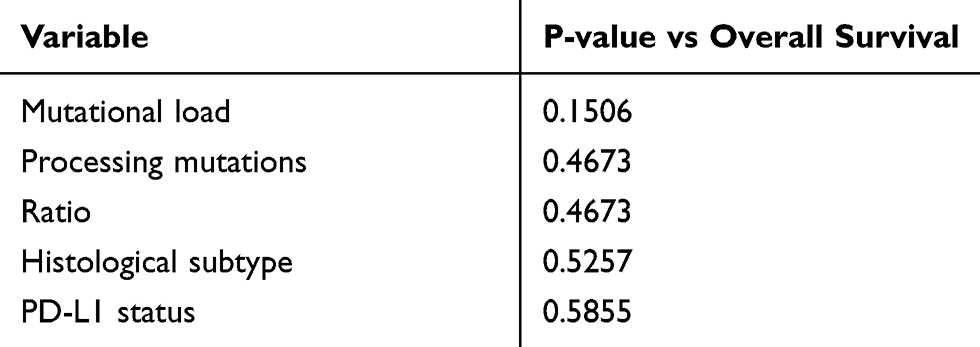 |
Table 3 Single covariates were tested against overall survival. Significant associations are reflected by p-value < 0.05, which was calculated by the Score (log-rank) Method. “Ratio” represents a combined score from “Mutational Load” and “Processing Mutations” (number of processing Mutations/Overall Mutation Load). |
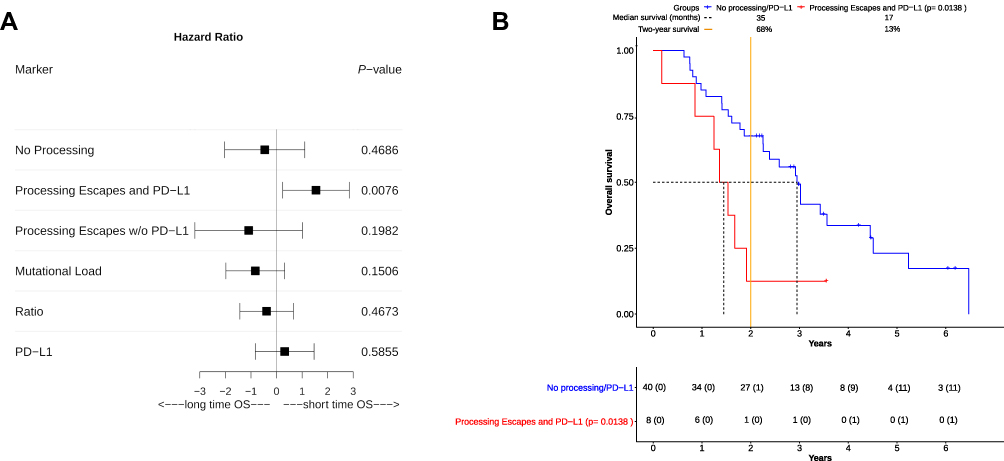 |
Figure 2 Overall survival (OS) in NSCLC patients based on calculated risk groups. (A) A risk estimation for overall survival benefit was performed for all patients in the risk groups “No Processing” (nPnP), “Processing Escapes and PD-L1” (PnP), and “Processing Escapes w/o PD-L1” (PoP). Three separate biomarkers in patients (“Mutational Load”, “Ratio”, and “PD-L1”) were also added. Their beneficial or detrimental impact on OS was visualized via forest plot. For each marker, the hazard ratio (square), the upper-and lower confidence interval was calculated. “PD-L1” and PnP were both detrimental to overall survival, while other markers were apparently more beneficial. On the one hand PnP significantly (p=0.0076) impaired overall survival, on the other hand the 95% CI of hazard ratios indicated detrimental effects only, which could not be observed from other markers. (B) As in Figure 1 the course of OS was visualized via Kaplan-Meier plot. Patients were separated into two groups, they had either no processing escapes with or without an additional PD-L1 overexpression (“No processing/PD-L1”) or they harbored mutations associated with altered proteasomal epitope processing combined with PD-L1 overexpression (“Processing Escapes and PD-L1”). The number at risk for each group of patients was displayed in the table below the plot. The number of censored patients at specific time point was also added (parentheses). Patients with PD-L1 expression and processing escapes showed significantly impaired OS (p= 0.0138). No patient survived past four years and only one long-time survivor lived past two years. Both median survival (17 vs 35 months) and two-year survival (13% vs 68%) indicated detrimental effects for this group by altered processing in combination with PD-L1 overexpression. Although a small group of patients (around 17%) was affected, they were clearly identifiable according to their survival course. This course also lead to dismal survival prognosis in comparison to all other patients. |
After COXPH analysis patients were separated into three different groups: Patients without altered processing (nPnP, 29 patients), patients without PDL-1 expression but altered processing (PoP, five patients) and patients with altered processing and PD-L1 expression (PnP, eight patients). To ascertain survival differences between all patient groups, Kaplan-Maier plots were generated (Figure 2B). PnP patients were showing significantly shortened overall survival (Score (log-rank) test, p=0.0138), compared to nPnP patients. Strikingly, this association had a positive predictive value of 88% and a high specificity (96%, Table 4). PnP patients had both the lowest median survival (17 vs 35 months) and two-year survival rate (13% vs 68%). The PoP group apparently consisted of many long-time survivors.
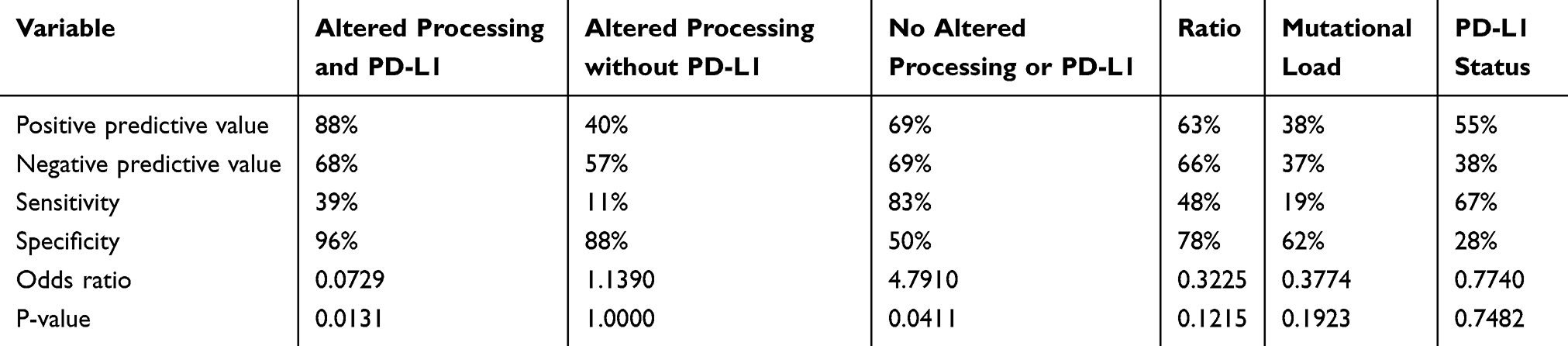 |
Table 4 Evaluation of impact on two-year survival by risk groups (column 2–4) and the single variates shown in Table 3 (column 5–7). In general, no impact on survival was assumed according to null hypothesis. Statistical evaluation was conducted by calculation of predictive values (row 1–4) and the application of Fisher´s Exact Test (Row 6–7) |
Additional results and visualizations are given in supplementary Figures 1–13.
Discussion
The T-cell based immune reaction is by far the most effective guardian of keeping malignancies down. After identifying a tumor cell as being foreign, the neoantigen-harboring cells are attacked by cytotoxic T-cells. This process is influenced by some key features harbored by the tumor cell. The anti-tumorigenic potential of the T-cells increases when tumor cells are easily recognizable as foreign. This effect depends on the amount and structure of the aberrant antigens presented on the cell surface, which are caused by a high number of mutations. This high tumor mutational burden has been proven to induce T-cell mediated tumor cell destruction. One of the strategies against this induced suicide is silencing the T-cell attack using the PD-1/PD-L1 system.5,6
Deductive rethinking of the concept described above, easily explains why immune checkpoint inhibition is such a powerful tool within anti-cancer therapies and why the concept of TMB as well as cell surface PD-L1 detection has attracted utmost attention in identifying patient collectives, suitable for ICB.
Beyond all obvious rationale, clinical experiences however show that by far not all patients profit from cancer immunotherapy.27 A reasonable proportion of patients show tumor progression under immune checkpoint blockade, even when expressing high amounts of PD-L1. The same is true for high TMB cases. Consequently, we may assume additional factors influencing this autoimmune approach beside PD-1/PD-L1 expression or high load of TMB-associated neoantigens. One promising idea in this context may be altered proteasomal processing of those neoantigens, resulting in an immune escape of tumor cells. The effect of such processing escapes has been first described in viral infections.13,14 If antigen processing in the cell is deteriorated, antigens suitable to highlight the cancer cell for the immune system may not be produced or not be designed to work effectively.
This process of escaping the immune system by altered epitope processing in malignancies has now been discovered analogous to the findings in viral infections.13,14 In addition, we were able to demonstrate the effect of processing escapes in neuroendocrine lung cancers.28 The background behind processing escapes was outlined in previous works.15,28 The data presented, guides the predictive power of processing escapes in ICB treated patients.
The results presented were found in a three-step process. First deep learning algorithms were used to identify epitopes influenced by altered proteasomal cleavage. Secondly, their binding to MHC/HLA molecules was verified by another deep learning-based tool. Thirdly, it was tested whether bound fragments can trigger an immune response.
It must be outlined that the data set investigated was taken out of the raw data analyzed during our routine NGS panel diagnostics. No additional new or unknown DNA-regions had to be investigated to find out tumors affected by altered epitope processing. This makes the method especially interesting for being included in routine diagnostics. Although one may term the methodology chosen using a deep learning-based approach theoretical, the practical application on real-life patients’ outcomes are obvious, as we found several strong signals.
Our results suggest that altered epitope processing is not a continuum from low and intermediate to high levels of alternatively processed epitopes. Instead altered processing is either present (“On”) or absent (“Off”) in a tumor. One may explain this finding as the final stage of a selection within the tumor cells. In patients with a high selection pressure due to high immune activity by CTLs, the tumor might survive by selecting mutations leading to altered epitope processing. Non- or faint escapers simply do not survive, explaining the missing intermediate group. Though it may be argued that problems may arise from the different panel sizes of the TCGA cohort (whole-exome sequencing) and ICB cohort (targeted-amplicon based sequencing). An increase in panel size may possibly lead to a change of patient classification. Due to the binary “on-off” nature of the underlying mechanism (No mutations or all mutations are affected), small-sized panels would already lead to consisted results. Therefore, the approach to identify altered processing is rather attractive for institutions not equipped with whole-exome or whole genome technology.
Altered processing is present in the whole exome data of AdC as well as SCC in more than one-third of cases. This in silico findings could also be confirmed within the real-life data of the I/O treated cancers. Interestingly about 90% of the alternatively processed epitopes are not presented on the cell surface. Thus, altered processing may be one explanation for cancer immunoediting. In this case, selection pressure by the immune system forces the tumor entities to reduce its antigenic variety.29–31 This automatically results in lower immunogenic activity on the tumor cell surface. To our understanding these cases are not profiting from PD-L1 blockade. In our real-world cohort, we can see a clear trend towards better survival in I/O treatment when altered processing is low (p=0.0138). This may serve as indicative for our hypothesis.
It must be highlighted that very impressively we could demonstrate that the findings of the in silico analysis are more or less completely present in the mutation analysis of the cancer cohort treated in our institution, showing the identical mutational distribution (supplementary Figure 4A). In terms of survival analysis, we found just small differences in the real life collective when looking at PD-L1 expression, TMB or processing escapes as single predictors of survival under therapy. It is not surprising that PD-L1 expressers with intact epitope processing are best survivors, as the tumor cells are recognized by cytotoxic T-cells, but these are silenced by the PD-L1 pathway. Activating the Cytotoxic T-Cells by PD-1 or PD-L1 blockade leads to tumor cell death. For the group of long-term survivors without PD-L1 expression one may assume that their immune system is per se not very effective in tumor cell elimination, therefore the PD-L1 pathway is not important enough to stay switched on. So, their altered epitope processing is not important due to basically low or ineffective T-cell answer.
However, these results must be weighted with caution due to the small number of patients and should be proved in further studies.
The most impressive result however is the clearly detrimental effect of PD-L1 expression and simultaneous insufficient epitope processing. Although this finding seems inconclusive at the first glance one, on the background of what has been discussed above, may assume that in these patients the tumors are the most aggressive ones in the collective. They use a combination of both hiding from cytotoxic T-cells by non-expression of MHC-binding antigens and, to overcome the rest of tumor immunogenicity, expressing PD-L1 on the surface, resulting in a complete knock-out of the autoimmune reaction against the tumor.
Conclusion
The study presented opens a new dimension for understanding why prediction of therapy effectiveness in immune checkpoint inhibition is unprecise in a reasonable number of cases up to now. Although PD-L1 blockade and TMB-driven neoantigen induction seem to be sufficiently explaining the therapy rationale, mutations easily detectable by routine NGS analysis may affect correct epitope processing in NSCLC, resulting in tumor cells that cannot be identified by the immune system due to a lack of MHC-binding tumor antigens.
To our opinion these findings would deserve to be further investigated and clarified in larger cohorts in the near future.
Abbreviations
AdC, adenocarcinoma; ANOVA, analysis of variance; AS, amino acid; COXPH, Cox proportional-hazards model; CTL, cytotoxic T-lymphocyte; CTLA-4, cytotoxic T-lymphocyte-associated Protein 4; DDCT, double dichotomous contingency tables; DNA, deoxyribonucleic acid; ER, endoplasmic reticulum; FDR, False discovery rate; FFPE, Formalin-fixed paraffin-embedded tissue; HLA, human leucocyte antigen; H&E stain, Hematoxylin, and eosin stain; IC50, half maximal inhibitory concentration; IEDB, Immune epitope data base; I/O, Immune-Oncology; Mb, Megabase; MHC, major histocompatibility complex; mRNA, messenger ribonucleic acid; NGS, Next-Generation Sequencing; nPnP, patients without altered processing or PDL-1 expression; NSCLC, Non-small cell lung cancer; OS, overall survival; PD-1, programmed cell death protein 1; PD-L1, programmed cell death 1 ligand 1; PFS, progression-free survival; PnP, patients with altered processing and PD-L1 expression; PoP, patients without PD-L1 expression but altered processing; SCC, Squamous-cell carcinoma; TAP, transporter associated with antigen processing; TCGA, The Cancer Genome Atlas; TCR, T-cell receptor; TMB, tumor mutational burden; UICC, union internationale contre le cancer.
Data Sharing Statement
For inquiries regarding the original data and workflow underlying this work, please contact the corresponding author.
Ethics Approval and Informed Consent
The study was conducted retrospectively and was approved by the Ethics Committee of the Medical Faculty of the University Duisburg-Essen (identifier: 13-5382-BO). The investigations conform to the principles of the declaration of Helsinki. All patient data were anonymized to make sure that their identity cannot be assumed. According to the ethics committee, a written consent was no longer necessary since the majority of patients was deceased at the time of data collection. Furthermore, it was no longer possible to obtain the consent retrospectively, as all patient data have been anonymized.
Acknowledgment
Robert, Werner’s current affiliation is the Institute of Pathology, University Hospital Charité Berlin, Berlin, Germany.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
Wilfried EE Eberhardt reports grants and personal fees from BMS and Astra Zeneca and personal fees from MSD/Merck and Roche, outside the submitted work. Jens Kollmeier reports being an Advisory Board Member (no personal fees) for Roche, Boehringer Ingelheim, Bristol-Meyers Squibb, MSD, and Takeda, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–e551. doi:10.1016/S1470-2045(16)30406-5
2. Keung EZ, Wargo JA. The current landscape of immune checkpoint inhibition for solid malignancies. Surg Oncol Clin N Am. 2019;28(3):369–386. doi:10.1016/j.soc.2019.02.008
3. Doroshow DB, Sanmamed MF, Hastings K, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res. 2019;25(15):4592–4602. doi:10.1158/1078-0432.CCR-18-1538
4. Herzberg B, Campo MJ, Gainor JF. Immune checkpoint inhibitors in non-small cell lung cancer. Oncologist. 2017;22(1):81–88. doi:10.1634/theoncologist.2016-0189
5. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi:10.1038/ni.2703
6. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
7. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. Ne Eng J Med. 2018;378(22):2078–2092. doi:10.1056/NEJMoa1801005
8. Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. Ne Eng J Med. 2018;379(21):2040–2051. doi:10.1056/NEJMoa1810865
9. Reck M, Rodríguez–Abreu D, Robinson AG, et al. Updated Analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non–small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. 2019;37(7):537–546. doi:10.1200/JCO.18.00149
10. Yi M, Jiao D, Xu H, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17(1):129. doi:10.1186/s12943-018-0864-3
11. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31(1):443–473. doi:10.1146/annurev-immunol-032712-095910
12. Kloetzel PM. The proteasome and MHC class I antigen processing. Biochim Biophys Acta. 2004;1695(1–3):225–233. doi:10.1016/j.bbamcr.2004.10.004
13. Milicic A, Price DA, Zimbwa P, et al. CD8+ T cell epitope-flanking mutations disrupt proteasomal processing of HIV-1 Nef. J Immunol. 2005;175(7):4618–4626. doi:10.4049/jimmunol.175.7.4618
14. Seifert U, Liermann H, Racanelli V, et al. Hepatitis C virus mutation affects proteasomal epitope processing. J Clin Invest. 2004;114(2):250–259. doi:10.1172/JCI200420985
15. Wessolly M, Mairinger F. Processing escapes: a new perspective on immune escape mechanisms. Canc Therapy Oncol Int J. 2017;5(3):154.
16. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi:10.1038/nature13385
17. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi:10.1038/nature11404
18. Kesmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002;15(4):287–296. doi:10.1093/protein/15.4.287
19. Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57(1–2):33–41. doi:10.1007/s00251-005-0781-7
20. Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–1017. doi:10.1110/ps.0239403
21. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi:10.1093/bioinformatics/btv639
22. Del Guercio MF, Sidney J, Hermanson G, et al. Binding of a peptide antigen to multiple HLA alleles allows definition of an A2-like supertype. J Immunol. 1995;154(2):685.
23. Kangueane P. HLA supertypes. 2009;131–139.
24. Sette A, Sidney J. HLA supertypes and supermotifs: a functional perspective on HLA polymorphism. Curr Opin Immunol. 1998;10(4):478–482. doi:10.1016/S0952-7915(98)80124-6
25. Sidney J, Southwood S, Pasquetto V, Sette A. Simultaneous prediction of binding capacity for multiple molecules of the HLA B44 supertype. J Immunol. 2003;171(11):5964–5974. doi:10.4049/jimmunol.171.11.5964
26. Calis JJA, Maybeno M, Greenbaum JA, et al. Properties of MHC class i presented peptides that enhance Immunogenicity. PLoS Comput Biol. 2013;9(10):e1003266. doi:10.1371/journal.pcbi.1003266
27. Wojas-Krawczyk K, Kalinka E, Grenda A, Krawczyk P, Milanowski J. Beyond PD-L1 markers for lung cancer immunotherapy. Int J Mol Sci. 2019;20(8). doi:10.3390/ijms20081915
28. Wessolly M, Walter RFH, Vollbrecht C, et al. Processing escape mechanisms through altered proteasomal cleavage of epitopes affect immune response in pulmonary neuroendocrine tumors. Technol Cancer Res Treat. 2018;17:1533033818818418. doi:10.1177/1533033818818418
29. Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7(3):264–276. doi:10.1158/2159-8290.CD-16-0828
30. Nejo T, Matsushita H, Karasaki T, et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res. 2019;7(7):1148–1161. doi:10.1158/2326-6066.CIR-18-0599
31. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. 2013;1284(1):1–5. doi:10.1111/nyas.12105
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.