")
Back to Journals » International Journal of Nanomedicine » Volume 15
A Novel Drug Delivery Carrier Comprised of Nimodipine Drug Solution and a Nanoemulsion: Preparation, Characterization, in vitro, and in vivo Studies
Authors Huang S, Huang Z, Fu Z , Shi Y, Dai Q , Tang S, Gu Y, Xu Y, Chen J, Wu X, Ren F
Received 9 August 2019
Accepted for publication 3 February 2020
Published 18 February 2020 Volume 2020:15 Pages 1161—1172
DOI https://doi.org/10.2147/IJN.S226591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Saixu Huang, 1–3,* Zhiyong Huang, 1–3,* Zhiqin Fu, 3,* Yamin Shi, 3, 4 Qi Dai, 1, 2 Shuyan Tang, 3 Yongwei Gu, 3 Youfa Xu, 3 Jianming Chen, 3, 4 Xin Wu, 3 Fuzheng Ren 1, 2
1Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai, People’s Republic of China; 2Engineering Research Centre of Pharmaceutical Process Chemistry, Ministry of Education, East China University of Science and Technology, Shanghai, People’s Republic of China; 3Shanghai Weier Biological Medicine Science and Technology Co. Ltd., Shanghai, People’s Republic of China; 4Department of Pharmacy, Fujian University of Traditional Chinese Medicine, Fujian, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fuzheng Ren
School of Pharmacy, East China University of Science and Technology, No. 130, Meilong Road, Shanghai 200237, People’s Republic of China
Tel/Fax +86 21 6425 3255
Email [email protected]
Xin Wu
Shanghai Weier Biological Medicine Science and Technology Co. Ltd., No. 358, Tian Chen Road, Shanghai 201799, People’s Republic of China
Tel/Fax +86 21 3119 8947
Email [email protected]
Purpose: Nimodipine (NIMO) is used clinically to treat ischemic damage resulting from subarachnoid hemorrhage. However, clinical application of NIMO is limited by poor aqueous solubility and low safety. To overcome these limitations, a novel two-vial NIMO-loaded nanoemulsion (NIMO-TNE) was designed in this study.
Methods: NIMO-TNE was prepared by mixing a nimodipine-polyethylene glycol 400 (NIMO-PEG400) solution and a commercially available 20% injectable blank nanoemulsion (BNE). Drug distribution in NIMO-TNE, physical stability, and dilution stability were evaluated in vitro, and pharmacokinetics and pharmacodynamics were evaluated in vivo. Safety was assessed using the hemolysis test and the intravenous irritation test, and acute toxicity of NIMO-TNE was compared with that of commercial Nimotop injection.
Results: Drug loading (DL) in NIMO-TNE was enhanced 5-fold compared with that in Nimotop injection. The mean particle size of NIMO-TNE was 241.53 ± 1.48 nm. NIMO-TNE and NIMO-TNE diluted in 5% glucose injection and 0.9% sodium chloride was stable for a sufficient duration to allow for clinical use. In addition, NIMO-TNE exhibited a similar pharmacokinetic profile and similar brain ischemia reduction in a rat middle cerebral artery occlusion (MCAO) model compared to Nimotop injection. Furthermore, NIMO-TNE did not induce hemolysis at 37°C, and NIMO-TNE induced less intravenous irritation than Nimotop injection. Moreover, NIMO-TNE could be injected at a 23-fold higher dose than the LD 50 of Nimotop injection with no obvious toxicity or side effects.
Conclusion: NIMO-TNE is a promising formulation suitable for intravenous injection, is easy to prepare, and exhibits excellent safety.
Keywords: nimodipine, nanoemulsion, pharmacokinetics, MCAO, LD50
Introduction
Nimodipine (NIMO), a dihydropyridine calcium antagonist, was originally developed by Bayer in 1982, and has been shown to dilate the cerebral arteries and increase cerebrovascular flow.1 Nimodipine is a preferred drug for treatment of ischemic cerebrovascular disease, and is also used to treat Alzheimer’s disease, stroke, migraine, and hypertension.2–4 However, NIMO exhibits limited efficacy due to low oral bioavailability and poor water solubility.5–7 Therefore, NIMO is administered intravenously, resulting in higher bioavailability than that observed following oral administration. Nimotop injection, produced by Bayer, is a commercially available injection that contains 23.7% (v/v) ethanol and 17% (v/v) polyethylene glycol 400 (PEG 400) to increase drug concentration in the formulation.8 The high concentration of organic solvent induces blood vessel irritation, resulting in pain and inflammation at the injection site. Furthermore, daily exposure to organic solvent is 101.7 mL according to the recommended NIMO dose, which may lead to phlebitis during infusion and poor compliance.6,9–13 In addition, drug crystallization may occur when Nimotop injection is diluted with glucose or saline solutions due to poor water solubility, which can result in increased patient health risk. Therefore, there is an urgent need to develop safe and effective injectable nimodipine formulations.
Nanoparticle drug delivery systems have been widely used for injection of poorly soluble drugs because they allow for high drug loading, good biocompatibility, and sustained release.14 Injectable nanoparticle formulations of NIMO have been developed previously, and include micelles, liposomes, and solid lipid nanoparticles.8,15–20 These nano-drug delivery systems have general advantages over Nimotop injection, but they typically exhibit poor stability, are subject to phagocytic effects in the reticuloendothelial system, and are difficult to produce. We initially designed a one-vial NIMO-loaded nanoemulsion, but this formulation exhibited poor stability. The drug content in the one-vial formulation decreased below 98% when stored at 25 °C ± 2 °C with a relative humidity of 60% ± 5% after only 72 hrs. To improve stability, a novel drug delivery carrier comprised of a commercially available injectable blank nanoemulsion (BNE) and a 4% (v/v) organic drug solution was designed based on our previous study.21 These two parts were designed to be mixed prior to clinical use to form an injectable drug-loaded nanoemulsion. Separation of the components of the formulation until use resulted in significantly improved stability. In addition, the amount of organic solvent was significantly lower than that in Nimotop injection due to high drug loading, which resulted in improved safety and less side effects. This novel two-vial drug-loaded nanoemulsion formulation may be an ideal drug carrier for intravenous injection of NIMO.
The objective of this study was to prepare a novel two-vial NIMO-loaded nanoemulsion (NIMO-TNE) that exhibited good safety and was easy to prepare. We evaluated drug distribution in NIMO-TNE, and physical and dilution stability of NIMO-TNE. Moreover, pharmacokinetic and pharmacodynamic properties of NIMO-TNE were compared with those of Nimotop injection in rats. Safety of NIMO-TNE was evaluated using the hemolysis test, assessment of intravenous irritation, and acute toxicity of NIMO-TNE was compared with that resulting from Nimotop injection.
Materials and Methods
Materials
Nimodipine was purchased from Kangmanlin Chemical Industry Co., Ltd (Nanjing, China). Nitrendipine, used as an internal standard, was purchased from the National Institute of Food and Drug Control (Beijing, China). Twenty percent (w/w) aqueous injectable emulsion (long-chain oil: medium-chain oil, 1:1, w/w) was purchased from Baxter Qiaoguang Healthcare Co., Ltd (Guangzhou, China). Nimotop injection (10 mg/50 mL) was purchased from Bayer Pharma AG. PEG400 was obtained from Well Chemical Co., Ltd (Nanjing, China). Five percent glucose solution injection and 0.9% sodium chloride injection were purchased from Tianrui Pharmaceutical Co., Ltd (Zhejiang, China). Two percent 2,3,5-triphenyl tetrazolium chloride, 4% paraformaldehyde solution, and 10% paraformaldehyde were purchased from Yuanye Biotechnology Co., Ltd (Shanghai, China). Heparin sodium salt, chloral hydrate, diethyl ether, and n-hexane were of AR grade and were purchased from Sinopharm Chemical Reagent Co., Ltd. Methanol (HPLC grade) and acetonitrile (HPLC grade) were purchased from Thermo Fisher (China) Co., Ltd. All other chemical reagents were of analytical grade or chromatographic grade.
Animals
Thirty-two male ICR mice (SPF, 20–25g), thirty-two female ICR mice (SPF, 20–25g), twelve male Sprague–Dawley rats (SPF, 250–300 g), twenty-four male Sprague–Dawley rats (SPF, 140–160g), and six male rabbits (2.5–3.0 kg) were purchased from The Second Military Medical University and were housed at a temperature of 25 °C ± 2 °C with relative humidity maintained at 47.5% ± 2.5%. All animal studies were performed in accordance with the Ethical Guidelines for Investigations in Laboratory Animals issued by the Second Military Medical university (Shanghai, People’s Republic of China). The animals were allowed to acclimate for 3 days prior to experimentation. Animals were fed standard feed (SPF level, Liaoning Qianmin Feedstuff Co., Ltd.) and provided autoclaved purified water. The animals were subjected to a 12 h light/12 h dark cycle. Following the experiments, the animal carcasses were frozen then collected by professional companies. The protocols and procedures were approved by the Institutional Animal Care and Use Committee of the Second Military Medical university (Shanghai, People’s Republic of China).
Preparation of NIMO-TNE
NIMO-TNE was prepared using NIMO-PEG400 solution and blank fat emulsion. The blank fat emulsion was a commercially available 20% injectable BNE. NIMO-PEG400 was prepared by dissolving NIMO (0.25 g) in PEG400 (10 mL) using ultrasound to aid dissolution. The NIMO-PEG400 solution was then sterilized at 121 °C for 15 min. NIMO-TNE (1 mg/mL) was prepared by combining 4 mL of NIMO-PEG400 solution with 96 mL of BNE, then shaking 30 times by rotating the vessel 180 degrees (about 30 seconds) at 25 °C ± 2 °C at a relative humidity of 60% ± 5%.
Characterization of NIMO-TNE
Analysis of NIMO Using HPLC
High performance liquid chromatography (HPLC) analysis was performed for drug quantitation using an Agilent 1260II HPLC system comprised of an Agilent G7111A pump and an Agilent G7114A-VWD detector. Data acquisition and processing were performed using Agilent openlab2.3 software.
Separation was performed on an Agilent Eclipse Plus C18 column (250 × 4.6 mm, 5 μm) maintained at 30 °C. The mobile phase consisted of methanol: acetonitrile: water (35:38:27 v/v/v). The flow rate was 1.0 mL/min and the injection volume was 10 μL. Absorbance was monitored at 235 nm.
Drug Distribution in NIMO-TNE
The drug distribution in NIMO-TNE was estimated as previously described.7,21–24 Drug content in the aqueous phase was determined using ultrafiltration and centrifugation. An appropriate amount of NIMO-TNE (4 mL) was transferred to an ultrafiltration centrifuge tube (100 k, Millipore Corporation, USA), and the tube was inserted into a 15-mL centrifuge tube. After centrifuging at 7000 rpm for 15 min at 4 °C (Kecheng Instrument Equipment Co., Ltd, Hunan, China), the aqueous phase was collected and analyzed using HPLC. The oil phase was separated by addition of inorganic salt. Three grams of sodium sulfate was added to 8 mL of NIMO-TNE and the resulting sample was placed in a 15-mL centrifuge tube. Centrifugation at 12,000 rpm for 90 min at 4 °C resulted in the following three layers from top to bottom: oil phase, phospholipid bilayer, and aqueous phase. The oil phase was placed in a 2-mL centrifuge tube and centrifuged at 10,000 rpm for 5 min at 4 °C. Fifty microliters of the oil phase was collected and diluted with diethyl ether-methanol (1:9) to 10 mL, and drug content was measured using HPLC. The drug content in the phospholipid layer (Cp) was calculated using the following formula:
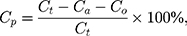
where Cp was the drug content in the phospholipid layer, Ct was the total drug content in NIMO-TNE, Ca was the drug content in the aqueous phase, and Co was the drug content in the oil phase.
Measurement of Particle Size and pH
The mean particle size and polydispersity index (PDI) of NIMO-TNE were measured using a Nano S Zetasizer (Malvern Instruments Ltd., UK) at 25 °C. The samples were analyzed following 20X dilution with double-distilled water. The pH values of NIMO-TNE were measured using a PB-10 pH detector (Sartorius Ltd., Germany).
Physical Stability Study
NIMO-TNE (1.0 mg/mL drug loading) was freshly prepared and stored at 25 °C ± 2 °C at a relative humidity of 60% ± 5%.21 Samples were analyzed for pH, drug content, particle size, and PDI at predetermined time intervals. Long-term stability of NIMO-PEG400 solution (25 mg/mL) was evaluated under the same experimental conditions for 12 months.21
Dilution Stability Study
NIMO-TNE was diluted to the same concentration as Nimotop injection (0.04 mg/mL in 5% glucose or 0.9% sodium chloride injection).25 The diluted samples were stored at 25 °C ± 2 °C at a relative humidity of 60% ± 5%. The samples were analyzed at the predetermined time intervals for drug content using HPLC. Drug content, pH, particle size, and PDI were evaluated to determine dilution stability.
In vivo Pharmacokinetic Study
Groups and Dosing
Twelve male Sprague–Dawley rats (250–300 g) were randomly divided into two groups with six rats in each group. NIMO-TNE was diluted with 0.9% sodium chloride to the same concentration as Nimotop injection. NIMO-TNE and Nimotop were injected intravenously via the tail vein at a single dose of 0.8 mg/kg. Blood samples (0.5 mL) were collected from the ocular vein at 2, 5, 10, 15, 30, 45, 60, 120, and 240 min following administration. Plasma samples were immediately centrifuged at 4000 rpm for 15 min at 4 °C. Two hundred microliters of plasma was collected and immediately stored at −20 °C until further analysis.
Sample Preparation
Plasma drug concentration was determined as follows. Forty microliters of internal standard (5 μg/mL nitrendipine in methanol) was added to each 200 μL plasma sample, and the samples were vortex-mixed for 30 s. Then, 100 μL of NaOH (1 mol/L) and 1.5 mL of N-hexane: ethyl ether (1:1) were added, and the mixture was vortexed for 5 min and sonicated for 5 min at room temperature.8,19,26 The mixture was then centrifuged at 10,000 rpm for 10 min at 4 °C, the supernatant was transferred to a 1.5-mL centrifuge tube, and the sample was evaporated using a RVC2-18CD plus vacuum desiccator (Christ Company, Germany) at 45 °C. The dried residue was dissolved in 100 μL of mobile phase, then centrifuged at 4000 rpm for 15 mins at 4 °C. Twenty microliters of the supernatant was injected for HPLC analysis.
Analysis of NIMO Using HPLC
Plasma samples were separated using an Agilent Eclipse Plus C18 column (250 × 4.6 mm, 5 μm) maintained at 30 °C. The mobile phase was a mixture of acetonitrile water (60:40, v/v), and the flow rate was 1.0 mL/min.8 Absorbance was monitored at 358 nm.
In vivo Pharmacodynamic Study
Groups and Dosing
Twenty-four Male Sprague–Dawley rats (140–160 g) were divided into the following three groups using a table of randomized numbers: Normal saline (NS) group (n=8); Nimotop injection group (n=8); and NIMO-TNE group (n=8). NIMO-TNE was diluted with 0.9% sodium chloride to the same concentration as Nimotop injection. NIMO-TNE and Nimotop were injected at a single dose of 0.8 mg/kg. Equal volumes of normal saline, Nimotop, and NIMO-TNE were injected intravenously via the tail vein.
Middle Cerebral Artery Occlusion (MCAO) Model
Electrocoagulation was used to generate a rat middle cerebral artery occlusion (MCAO) model as previously described.27–30 Sprague-Dawley rats were anesthetized by intraperitoneal injection with 3% pentobarbital sodium at a dose of 30 mg/kg. The rats were placed in the lateral position on the operating table, and the left side of the face was shaved. An incision was made in the skin between the left orbit and the left ear following disinfection using 75% ethanol. The diaphragm and masseter muscle were separated and the tibia flap was exposed. A 2 mm × 2 mm hole was made in the bone using a Strong 102L skull drill (Precision Instrument Factory, Korea), and the skull was opened under an XTS-4A microscope (Zhongtian Optical Instrument Co., Ltd, Jiangsu, China). The middle cerebral artery was electrocauterized 1 mm below the intersection with the olfactory tract using a bipolar electrocautery (Jinbaiwei Photoelectric Technology Co., Ltd, Wuhan, China). The soft tissue was repositioned, and the skin was sutured. The rats were then dosed with the control solution or the drug products, then placed in their cages with enough food and water for 24 h.
The rats were anesthetized intraperitoneally with 3% pentobarbital sodium at a dose of 30 mg/kg 24 h after the MCAO procedure. The brains were immediately removed and sliced into 1-mm-thick coronal sections (Yuyan Scientific Instrument Co., Ltd, Shanghai, China). The slices were stained with a 1% solution of 2,3,5- triphenyl tetrazolium at 37 °C for 10 min. Viable brain tissue was stained red, and the infarcted area was unstained and appeared white. The slices were then transferred to a 10% formaldehyde solution and stored in the dark for 24 h for fixation. Images of the slices were acquired using a digital camera (Figure 1), and the infarcted area was measured using image analysis software (Image J, version: 1.4.3.67). The ratio of infarct volumes was obtained by calculating the percentage of infarct size compared to the total area of the infarcted cerebral hemisphere.28,30–32
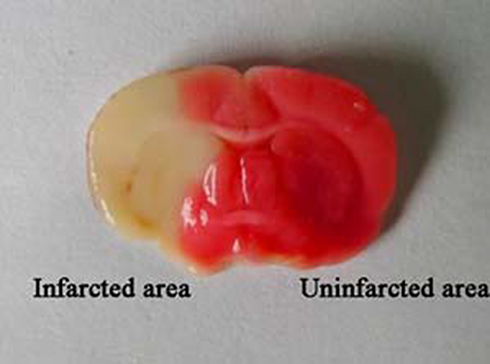 |
Figure 1 Stained coronal sections of rat brain following MCAO. Abbreviation: MCAO, Middle cerebral artery occlusion. |
Safety Assessment
Hemolysis Test
The hemolysis test was used to evaluate the safety of NIMO-TNE for clinical use. Fresh rabbit blood (10 mL) was collected, and the fibrinogen fraction was removed by stirring with a glass rod. The erythrocytes in the defibrinated blood were washed three times with 10 mL of 0.9% saline solution. Each wash step included centrifugation at 2500 rpm for 5 min, followed by removal of the supernatant. A 2% (v/v) erythrocyte suspension was prepared by diluting the erythrocytes with 0.9% saline solution.
Nine glass test tubes containing 2.5 mL of 2% (v/v) erythrocyte suspension were labeled numerically. Various amounts of NIMO-TNE (0.1, 0.2, 0.3, 0.4, and 0.5 mL) were added to tubes 1–5. Tube 6 contained only 2.5 mL of 0.9% saline, and served as a negative control. Tube 7 contained 2.5 mL of distilled water and served as a positive control. Tube 8 contained 2.5 mL of Nimotop injection, and served as a reference control. Blank nanoemulsion (0.5 mL) was added to tube 9. Saline solution was added to each tube to a final volume of 5 mL. The samples were incubated at 37 °C following mixing, and were observed at 15 min, 30 min, 45 min, 1 h, 2 h, 3 h, and 4 h.33,34
Intravenous Irritation Evaluation
Six male rabbits weighing 2.5–3.0 kg were randomly divided into two groups that received either Nimotop injection or NIMO-TNE. Nimotop injection and NIMO-TNE were administered at a dose of 0.4 mg/kg via the marginal vein of the right ear, and an equivalent volume of 0.9% saline was injected via the marginal vein of the left ear.8,34,35 All injections were administered at a rate of 1 mL/min once per day for three consecutive days. The appearance of the injection sites and the surrounding tissue was visually observed during the experiment. All rabbits in the study were euthanized 24 h after the final injection, and tissue samples located 0.5 cm and 3 cm from the injection site were cut. The tissue samples were fixed in 10% formalin, dehydrated using an ethanol gradient, embedded in paraffin, and stained with hematoxylin and eosin. All samples were examined using a BX43-DP21 light microscope (Olympus ltd. Japan) and pathological changes were evaluated.
Acute Toxicity
The median lethal dose (LD50) of Nimotop injection was evaluated. The maximal feasible dose (MFD) of NIMO-TNE was evaluated to assess acute toxicity, based on excellent safety observed in preliminary studies.
The LD50 of Nimotop injection was determined as follows.36–38 Forty ICR mice (20–25 g) were assigned randomly to five groups, with 4 males and 4 females in each group. Nimotop was injected intravenously via the tail vein at single doses of 1.90, 2.10, 2.32, 2.56, or 2.83 mg/kg. The mice were observed for general behavior and signs of toxicity for 14 d.
Twenty-four ICR mice (20–25 g) were divided randomly into a NIMO-TNE group and a control group comprised of 6 males and 6 females in each group to evaluate the maximum feasible dose of NIMO-TNE. Mice in the NIMO-TNE group were injected with NIMO-TNE at a dose of 18 mg/kg via the tail vein three times in a 24 h period (54 mg/kg cumulative dose). The mice in the control group were not subjected to any experimental procedures. The mice were observed for general behavior and signs of toxicity for 14 d. Changes in body weight were also monitored.
Statistical Analysis
All the results are presented as means ± standard deviations (SD). Plasma concentration data were subjected to non-linear regression analysis using Drug and Statistics Software (DAS, version 2.0, Mathematical Pharmacology Professional Committee of China). Data from the MCAO study were subjected to analysis of variance using Excel 2010 and SPSS software (version 18.0). The LD50 and the associated 95% confidence interval of Nimotop injection were calculated using SPSS software (version 18.0) with the Bliss method. Differences between the different groups were determined using unpaired t-tests. P values < 0.05 were considered statistically significant.
Results and Discussion
Characterization of NIMO-TNE
Drug Distribution in NIMO-TNE
Drug distribution of NIMO-TNE is summarized in Table 1. The average drug content was 35.35% ± 1.26% in the oil phase, 0.10% ± 0.03% in the aqueous phase, and 64.56% ± 1.26% in the phospholipid bilayer. The majority of NIMO in NIMO-TNE was dispersed in the phospholipids layer and the oil phase because NIMO is poorly water soluble.7,19
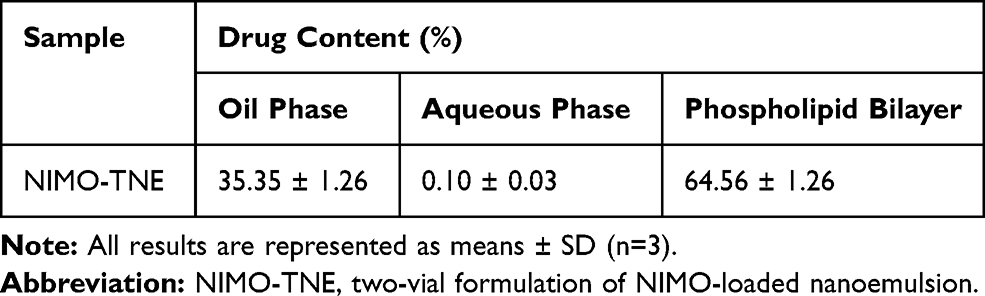 |
Table 1 Drug Distribution in NIMO-TNE at a Concentration of 1.0 mg/mL |
Physical Stability Study
Physical stability of NIMO-TNE and NIMO-PEG400 solution was evaluated, and the results are summarized in Table 2. The results showed that NIMO-TNE had an average particle size of 241.53 nm, an average PDI of 0.11, and a pH value of 7.23. These results were consistent throughout the experiments. Drug content of NIMO-TNE (1.0 mg/mL drug load) remained stable for 24 h. In the clinic, NIMO-TNE is prepared by mixing the blank fat emulsion and NIMO-PEG400 solution immediately prior to use. Therefore, 24 h stability was appropriate for clinical use. In addition, no significant changes were observed during long-term stability evaluation of NIMO-PEG400, as shown in Table 3. These results showed that NIMO-TNE was stable at 25 °C ± 2 °C at a relative humidity of 60% ± 5% for at least 24 h, which is appropriate for clinical use.
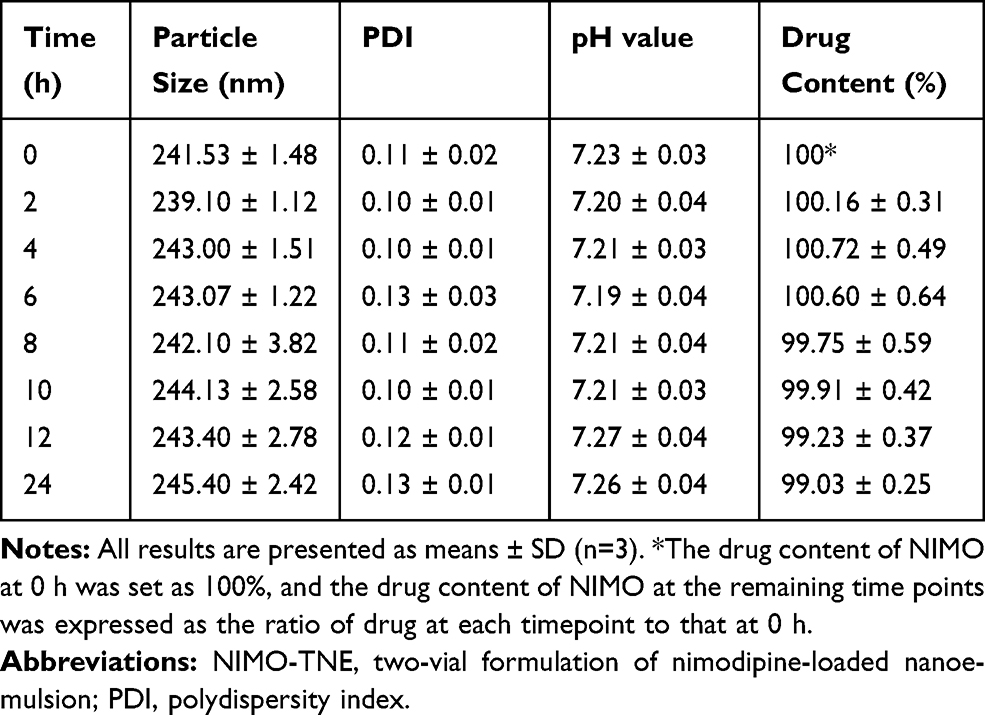 |
Table 2 Physical Stability of NIMO-TNE (1.0 mg/mL) |
 |
Table 3 Long-Term Stability of NIMO-PEG400 |
Dilution Stability Study
Stability of diluted NIMO-TNE was evaluated because this formulation must be diluted prior to clinical use. When the emulsion was diluted to 0.04 mg/mL, the concentration of Nimotop injection used clinically, no significant changes in particle size, PDI, pH, or drug content were observed (Tables 4 and 5). The pH of diluted injection solutions increased slightly over time, but the changes were not statistically significant (p > 0.5; Tables 4 and 5). A previous study showed that dilution of Nimotop injection resulted in precipitation of drug crystals due to the reduced ratio of organic solvent, which could result in increased risk to the patient.8 In contrast, the drug in NIMO-TNE was dissolved in the phospholipid layer and oil phase because nimodipine is highly lipophilic, which resulted in excellent stability following dilution. These results showed that NIMO-TNE could be diluted to 0.04 mg/mL and remain stable within 24 h at the same concentration of Nimotop injection used clinically.
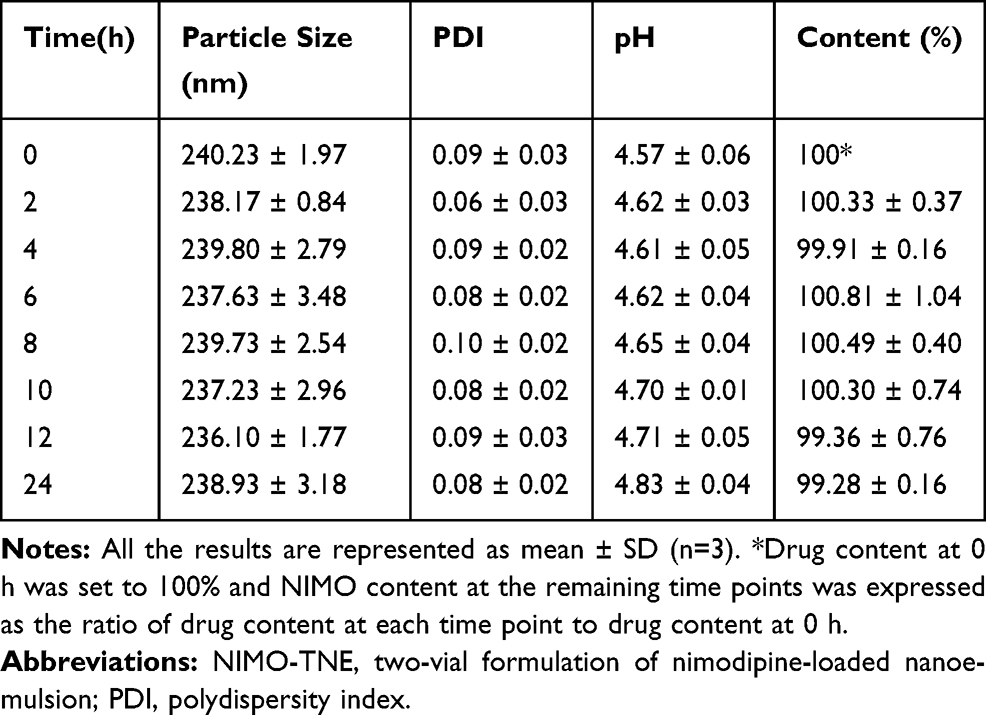 |
Table 4 Physical Stability of NIMO-TNE Following Dilution to 0.04 mg/mL in 5% Glucose |
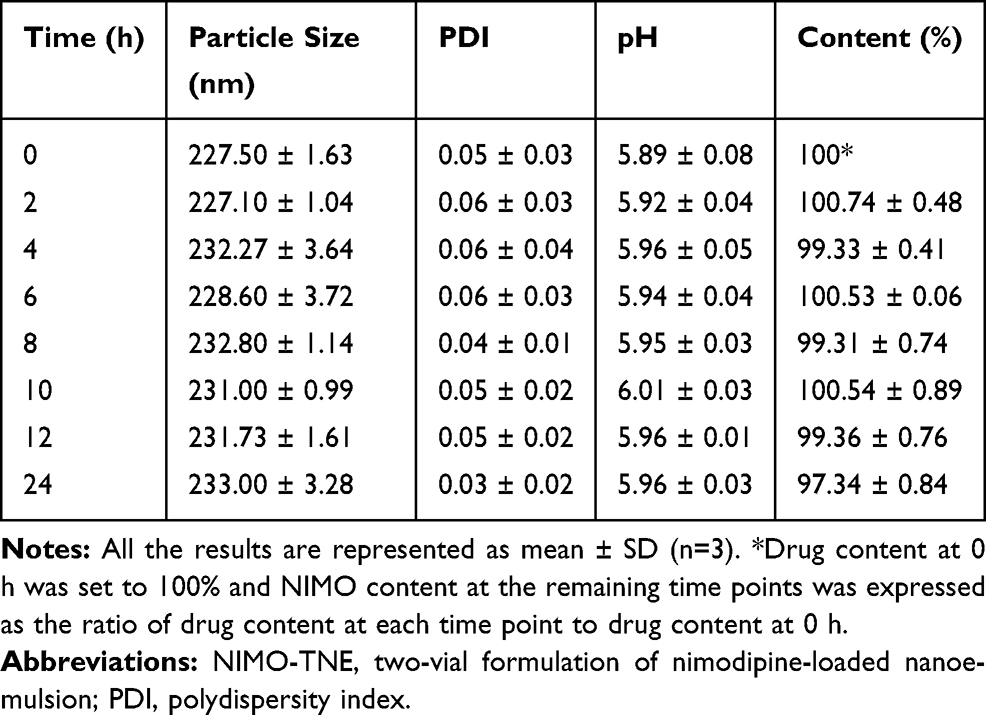 |
Table 5 Physical Stability of NIMO-TNE Following Dilution to 0.04 mg/mL in 0.9% Sodium Chloride |
In vivo Pharmacokinetic Study
Mean plasma concentration-time profiles following single intravenous injections of Nimotop and NIMO-TNE are shown in Figure 2. The results showed that Nimotop injection and NIMO-TNE had similar pharmacokinetic profiles. The drug was eliminated rapidly in the first 60 min, after which elimination occurred more slowly. The drug was not detectable at 240 min for either Nimotop or NIMO-TNE. The main pharmacokinetic parameters of both groups are summarized in Table 6. The results showed that the area under the plasma-time curve, mean residence time, and half-life were similar between the two groups (p > 0.05), which indicated that Nimotop and NIMO-TNE were bioequivalent. The rapid elimination rate of NIMO-TNE may have been due to rapid release of drug from NIMO-TNE as a result of high drug content (64.56% ± 1.26%) in the phospholipid bilayer.8,21 In addition, a previous study showed that the plasma protein-binding rate of nimodipine was 97–99% in blood.7 The released drug might interact with plasma proteins to promote further drug release. An MCAO model was used to evaluate the potential for treatment of ischemic cerebrovascular disease using NIMO-TNE.
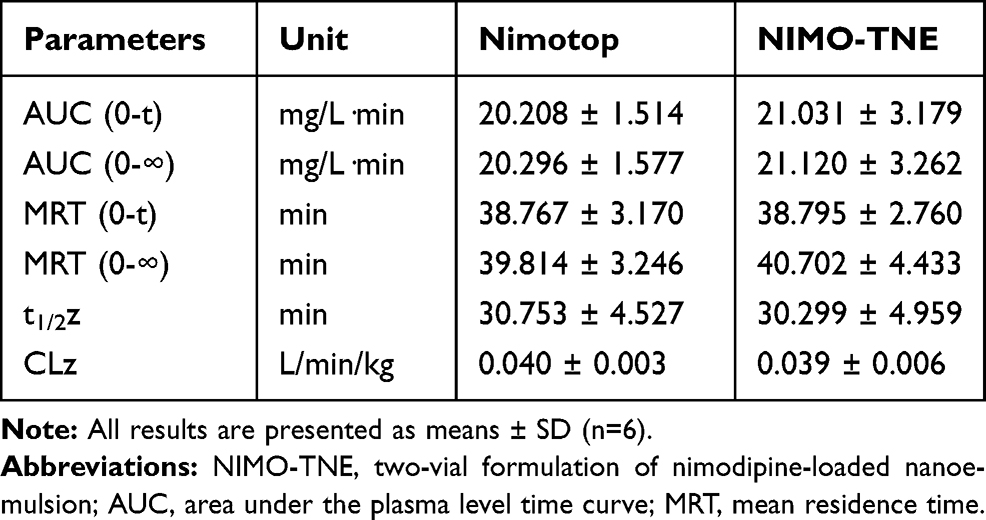 |
Table 6 Pharmacokinetic Parameters of Nimotop Injection and NIMO-TNE in Rats |
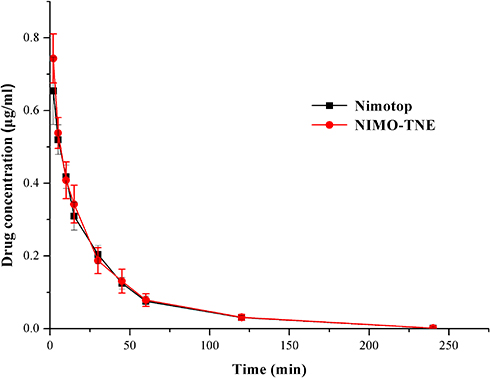 |
Figure 2 Mean plasma concentration-time profiles following a single intravenous injection of Nimotop or NIMO-TNE in rats (n=6). Abbreviation: NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion. |
In vivo Pharmacodynamic Study
No deaths occurred during the experimental procedures. The infarction areas in the Nimotop injection group and the NIMO-TNE group were significantly smaller than those in the normal saline group, and no significant differences in infarct size were observed between the NIMO-TNE injection group and the Nimotop injection group (Figure 3A). These results were consistent with the quantitative results summarized in Figure 3B, in which the average rate of infarctionin the NS group was 37.70% ± 4.08%, the half-cerebral infarction ratio in the Nimotop injection group was 17.34% ± 3.24%, and the half-cerebral infarction ratio in the NIMO-TNE group was 17.51% ± 3.42%. The reduction in rate of half-cerebral infarction in response to Nimotop injection and NIMO-TNE compared to the NS group was 54.03% ± 8.60% and 53.56% ± 9.08%, respectively (Figure 3C). There was no difference in the rate of reduction of half-cerebral infarction between the Nimotop injection group and the NIMO-TNE group (p > 0.05). These results indicated that a single intravenous injection of NIMO-TNE significantly reduced infarct size in a rat MCAO model. Furthermore, NIMO-TNE reduced infarct size to a similar degree as Nimotop injection at the same dose.
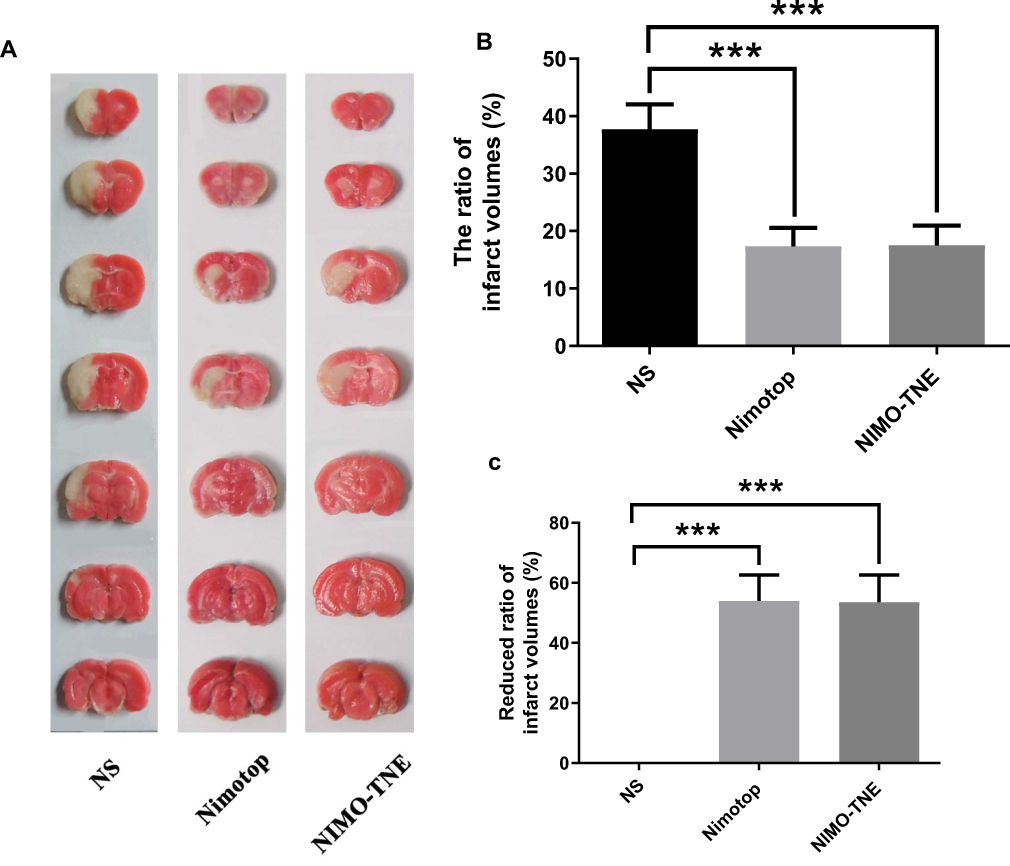 |
Figure 3 Brain infarction in a rat MCAO model. (A) Representative photographs of TTC stained brain slices from different groups. (B). The ratio of infarct volumes following MCAO in rats. (C) Reduced ratio of infarct volumes following Nimotop injection or NIMO-TNE injection versus the NS group. Notes: Normal saline (NS) group (n=8); Nimotop injection group (n=8); NIMO-TNE group (n=8). ***p < 0.001 vs NS group. Abbreviations: NS, Normal saline; NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion; MCAO, Middle cerebral artery occlusion. |
Intravenous Injection Safety
Evaluation of Hemolysis
The results of the hemolysis test were shown in Figure 4. Complete hemolysis was observed in the positive control tube (No. 7) at 0 min, as evidenced by a clear red solution with no erythrocytes at the bottom of the tube.33,39 In contrast, no hemolysis was observed in the negative control (No. 6) or in the Nimotop injection tube (No. 8), and erythrocytes precipitated at the bottom of each tube. The NIMO-TNE (No. 1–5) and BNE (No.9) tubes were divided into three layers, including an emulsion layer, a mixed emulsion and erythrocyte layer, and an erythrocyte layer. The emulsion layer thickened over time, and the layer containing emulsion and erythrocytes thinned over time, and erythrocytes precipitated at the bottoms of the tubes. These results indicated that NIMO-TNE (1 mg/mL) did not induce hemolysis at 37 °C.
 |
Figure 4 Hemolysis results for NIMO-TNE at 4 h. Abbreviation: NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion. |
Intravenous Irritation Assessment
The intravenous irritation test was performed to compare irritation resulting from saline injection and NIMO-TNE to that resulting from Nimotop injection. Mice in the Nimotop group experienced more irritation than those in the NS or NIMO-TNE groups during administration. In addition, Nimotop injection induced edema, which indicated severe irritation during administration.40 In contrast, no significant changes were observed in the ears of rabbits in the NS or NIMO-TNE groups following infusion.
Images of the rabbit ear tissue sections from the injection site are shown in Figure 5. The rabbit ear marginal vein in the Nimotop injection group showed vasodilation, hemorrhage, and edema around the vein. In addition, these tissue sections showed inflammatory cell infiltration in the vessel wall and surrounding tissues, and endothelial cell swelling, which indicated severe vascular irritation. In contrast, no inflammatory cell infiltration was observed in the surrounding tissues in the NS or NIMO-TNE groups, which indicated that no irritation had occurred. These differences may have been due to high ethanol content in Nimotop injection. The high concentration of ethanol may have induced vasodilation and increased vascular permeability, which results in leakage of red blood cells, drug, and organic solvent.8,40 These results indicated that Nimotop injection may induce significant pain and serious vascular irritation. The results showed that NIMO-TNE did not induce adverse effects such as pain and vascular irritation, which represented a marked improvement compared to Nimotop injection.
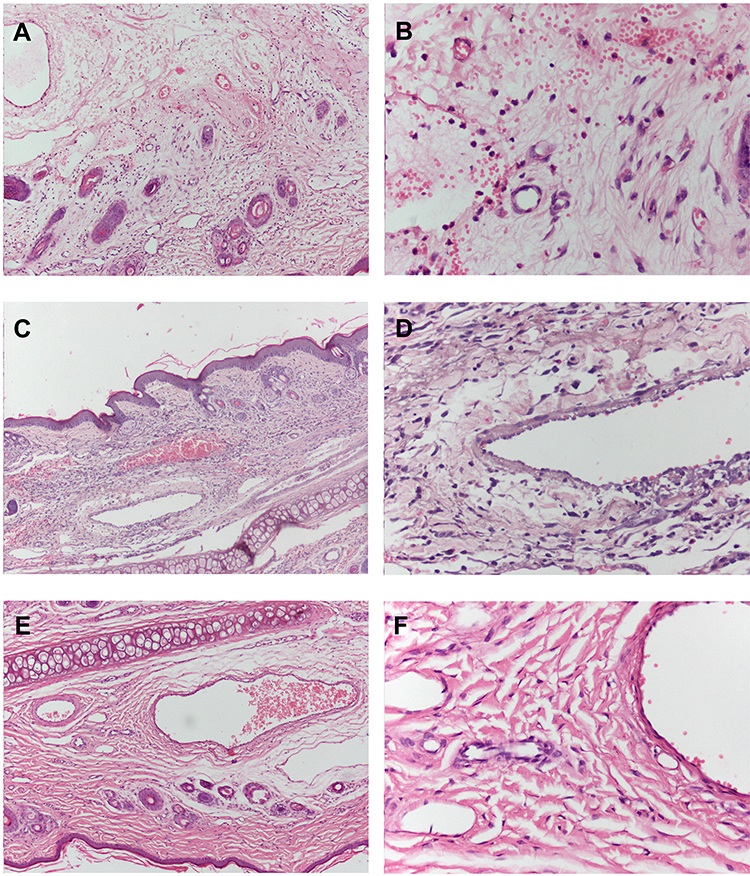 |
Figure 5 Images of rabbit ear pathological sections. (A) Nimotop injection group (100×); (B) Nimotop injection group (400×); (C) NIMO-TNE group (100×); (D) NIMO-TNE group (400×); (E) NS group (100×); (F) NS group (400×). Abbreviations: NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion; NS, Normal saline. |
Acute Toxicity
As shown in Table 7, acute toxicity of Nimotop injection increased in a dose-dependent manner from 1.90 to 2.83 mg/kg. The clinical signs of toxicity were asthenia, spasticity of the hind limbs, and tics. In addition, urinary incontinence was observed in mice that died as a result of the injection. All the surviving mice recovered after 14 days. The LD50 of Nimotop injection was estimated to be 2.35 mg/kg, and the 95% confidence interval was 2.20–2.51 mg/kg.
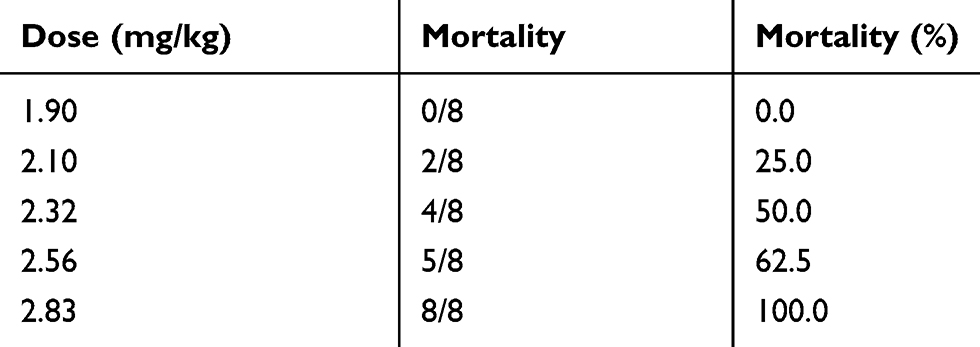 |
Table 7 Acute Toxicity of Nimotop Injection in Mice |
In the maximum feasible dose (MFD) test of NIMO-TNE, no abnormal behavior or obvious signs of toxicity were observed following injection. Changes in body weight were similar between control mice and those treated with NIMO-TNE (Figure 6). The MFD of NIMO-TNE was at least 54 mg/kg.
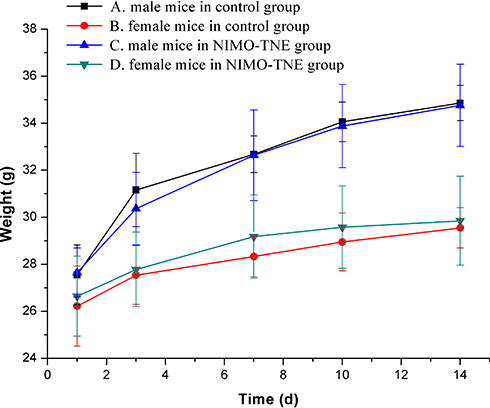 |
Figure 6 Weight change curve. A. Male mice in the control group (n=6). B. Female mice in the control group (n=6). C. Male mice in the NIMO-TNE group (n=6). D. Female mice in the NIMO-TNE group (n=6). Abbreviation: NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion. |
Given that the LD50 of Nimotop injection was 2.35 mg/kg, and NIMO-TNE could be dosed at 54 mg/kg with no obvious toxicity or side effects, we concluded that NIMO-TNE was significantly safer than Nimotop injection. These results were consistent with those from the intravenous irritation test.
Conclusion
In summary, NIMO-TNE was successfully prepared using a simple method. The drug loading of NIMO-TNE reached 1 mg/mL, and 64.56% ± 1.26% and 35.35% ± 1.26% of the drug was distributed in the phospholipid bilayer and oil phase, respectively. NIMO-TNE was stable for a sufficient duration to allow for clinical application. In addition, NIMO-TNE was physically compatible with 5% glucose and 0.9% sodium chloride, as indicated by stability for 24 h in these solutions. NIMO-TNE showed similar pharmacokinetic properties to Nimotop injection. Moreover, there were no significant differences in reduction of MCAO-induced brain ischemia following NIMO-TNE or Nimotop injection in rats. The hemolysis test showed that NIMO-TNE did not induce hemolysis at 37 °C. Furthermore, NIMO-TNE did not induce irritation during or after intravenous infusion, which represented a significant improvement over Nimotop injection. In addition, NIMO-TNE could be dosed at 54 mg/kg with no obvious toxicity or side effects, whereas the LD50 of Nimotop injection was 2.35 mg/kg. Therefore, NIMO-TNE may be a promising drug carrier for NIMO. Furthermore, this novel two-vial formulation of a drug-loaded nanoemulsion may be a promising method for the intravenous administration of lipophilic drugs.
Abbreviations
NIMO, Nimodipine; NIMO-TNE, two-vial formulation of nimodipine-loaded nanoemulsion; PEG400, Polyethylene glycol 400; NIMO-PEG400, Nimodipine-polyethylene glycol 400; BNE, Blank nanoemulsion; DL, Drug loading; HPLC, High performance liquid chromatography; PDI, Polydispersity index; NS, Normal saline; MCAO, Middle cerebral artery occlusion; LD50, Median lethal dose; MFD, Maximal feasible dose; SD, Standard deviation; AUC, Area under the plasma level time curve; MRT, Mean residence time.
Acknowledgments
This study was supported by Shanghai Key Laboratory of New Drug Design (Grant No. 17DZ2271000), the National Natural Science Foundation of China (NO. 81772749), Shanghai Rising-Star Program (NO. 18QB1400400), Shanghai Qingpu District Industry-University-Research Cooperation Development Fund Project (QIUR 2019-5), and Shanghai Science and Technology Project of Little Giant (1902HX76600).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhao Y, Xin T, Ye T, Yang X, Pan W. Solid dispersion in the development of a nimodipine delayed-release tablet formulation. Asian J Pharm Sci. 2014;9(1):35–41. doi:10.1016/j.ajps.2013.11.006
2. Barmpalexis P, Kanaze FI, Kachrimanis K, Georgarakis E. Artificial neural networks in the optimization of a nimodipine controlled release tablet formulation. Eur J Pharm Biopharm. 2010;74(2):316–323. doi:10.1016/j.ejpb.2009.09.011
3. Gelmers HJ. Calcium-channel blockers in the treatment of migraine. Am J Cardiol. 1985;55:3. doi:10.1016/0002-9149(85)90622-8
4. Caceres JF, Blangy D, Glikin GC. Requirement of DNA topoisomerases for in vitro chromatin assembly by 3T6 mouse cell extracts. Eur J Biochem. 1989;181(2):531–537. doi:10.1111/ejb.1989.181.issue-2
5. Sun Y, Rui Y, Wenliang Z, Tang X. Nimodipine semi-solid capsules containing solid dispersion for improving dissolution. Int J Pharm. 2008;359(1–2):144–149. doi:10.1016/j.ijpharm.2008.03.040
6. Soliman GM, Sharma R, Choi AO, et al. Tailoring the efficacy of nimodipine drug delivery using nanocarriers based on A 2 B miktoarm star polymers. Biomaterials. 2010;31(32):8382–8392. doi:10.1016/j.biomaterials.2010.07.039
7. Hu FQ, Zhang Y, Du YZ, Yuan H. Nimodipine loaded lipid nanospheres prepared by solvent diffusion method in a drug saturated aqueous system. Int J Pharm. 2008;348(1–2):146–152. doi:10.1016/j.ijpharm.2007.07.025
8. Song X, Jiang Y, Ren C, et al. Nimodipine-loaded mixed micelles: formulation, compatibility, pharmacokinetics, and vascular irritability study. Int J Nanomedicine. 2012;7:3689–3699. doi:10.2147/IJN.S33228
9. Li J, Fan L, Yuan M, Xing M. Salidroside inhibits lipopolysaccharide-ethanol-induced activation of proinflammatory macrophages via notch signaling pathway. Curr Med Sci. 2019;39(4):526–533. doi:10.1007/s11596-019-2069-4
10. Shirpoor A, Gharalari FH, Rasmi Y, Heshmati E. Ginger extract attenuates ethanol-induced pulmonary histological changes and oxidative stress in rats. J Biomed Res. 2017;31(6):521–527.
11. Le Daré B, Victoni T, Bodin A, et al. Ethanol upregulates the P2X7 purinergic receptor in human macrophages. Fundam Clin Pharmacol. 2019;33(1):63–74. doi:10.1111/fcp.2019.33.issue-1
12. Schneider ACR, Gregório C, Uribe-Cruz C, et al. Chronic exposure to ethanol causes steatosis and inflammation in zebrafish liver. World J Hepatol. 2017;9(8):418–426. doi:10.4254/wjh.v9.i8.418
13. Zamani E, Mohammadbagheri M, Fallah M, Shaki F. Atorvastatin attenuates ethanol-induced hepatotoxicity via antioxidant and anti-inflammatory mechanisms. Res Pharm Sci. 2017;12(4):315–321. doi:10.4103/1735-5362.212049
14. Garg NK, Tyagi RK, Singh B, et al. Nanostructured lipid carrier mediates effective delivery of methotrexate to induce apoptosis of rheumatoid arthritis via NF-κB and FOXO1. Int J Pharm. 2016;499(1–2):301–320. doi:10.1016/j.ijpharm.2015.12.061
15. Rashed HM, Shamma RN, Basalious EB. Contribution of both olfactory and systemic pathways for brain targeting of nimodipine-loaded lipo-pluronics micelles: in vitro characterization and in vivo biodistribution study after intranasal and intravenous delivery. Drug Deliv. 2017;24(1):181–187. doi:10.1080/10717544.2016.1236848
16. Ji B, Wang M, Gao D, et al. Combining nanoscale magnetic nimodipine liposomes with magnetic resonance image for Parkinson’s disease targeting therapy. Nanomedicine-UK. 2017;12(3):237–253. doi:10.2217/nnm-2016-0267
17. Moreno LCGEAI, Cavalcanti IMF, Satyal P, Santos-Magalhães NS, Rolim HML, Freitas RM. Acute toxicity and anticonvulsant activity of liposomes containing nimodipine on pilocarpine-induced seizures in mice. Neurosci Lett. 2015;585:38–42. doi:10.1016/j.neulet.2014.11.025
18. Xiong R, Lu W, Li J, Wang P, Xu R, Chen T. Preparation and characterization of intravenously injectable nimodipine nanosuspension. Int J Pharm. 2008;350(1–2):338–343. doi:10.1016/j.ijpharm.2007.08.036
19. Ding Y, Teng Z, Zhou J, et al. Preparation and characterization of nimodipine-loaded nanostructured lipid systems for enhanced solubility and bioavailability. Int J Nanomedicine. 2018;14:119–133. doi:10.2147/IJN.S186899
20. Guada M, Lasa-Saracíbar B, Lana H, Del Carmen Dios-Viéitez M, Blanco-Prieto MJ. Lipid nanoparticles enhance the absorption of cyclosporine A through the gastrointestinal barrier: in vitro and in vivo studies. Int J Pharm. 2016;500(1–2):154–161. doi:10.1016/j.ijpharm.2016.01.037
21. Jing X, Deng L, Gao B, et al. A novel polyethylene glycol mediated lipid nanoemulsion as drug delivery carrier for paclitaxel. Nanomed Nanotechnol Biol Med. 2014;10(2):371–380. doi:10.1016/j.nano.2013.07.018
22. Chen L, Chen B, Deng L, et al. An optimized two-vial formulation lipid nanoemulsion of paclitaxel for targeted delivery to tumor. Int J Pharm. 2017;534(1–2):308–315. doi:10.1016/j.ijpharm.2017.10.005
23. Chalikwar SS, Belgamwar VS, Talele VR, Surana SJ, Patil MU. Formulation and evaluation of nimodipine-loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloid Surface B. 2012;97:109–116. doi:10.1016/j.colsurfb.2012.04.027
24. Guan T, Miao Y, Xu L, et al. Injectable nimodipine-loaded nanoliposomes: preparation, lyophilization and characteristics. Int J Pharm. 2011;410(1–2):180–187. doi:10.1016/j.ijpharm.2011.03.009
25. Tian S, Gao W, Liu Y, Kang W. Study on the stability of heavy crude oil-in-water emulsions stabilized by two different hydrophobic amphiphilic polymers. Colloids Surfaces a Physicochem Eng Asp. 2019;572:299–306. doi:10.1016/j.colsurfa.2019.04.017
26. Sun C, Wang J, Liu J, Qiu L, Zhang W, Zhang L. Liquid proliposomes of nimodipine drug delivery system: preparation, characterization, and pharmacokinetics. AAPS PharmSciTech. 2013;14(1):332–338. doi:10.1208/s12249-013-9924-6
27. Liu DZ, Jickling GC, Ander BP, et al. Elevating microRNA-122 in blood improves outcomes after temporary middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2016;36(8):1374–1383. doi:10.1177/0271678X15610786
28. Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischaemia in the rat: I. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1(1):53–60. doi:10.1038/jcbfm.1981.6
29. Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. J Am Heart Assoc. 1986;17(3):472–476.
30. Xie HH, Zhang XF, Chen YY, Shen FM, Su DF. Synergism of hydrochlorothiazide and nifedipine on blood pressure variability reduction and organ protection in spontaneously hypertensive rats. Hypertens Res. 2008;31(4):685–691. doi:10.1291/hypres.31.685
31. Yu J, Li X, Matei N, et al. Ezetimibe, a NPC1L1 inhibitor, attenuates neuronal apoptosis through AMPK dependent autophagy activation after MCAO in rats. Exp Neurol. 2018;307(5):12–23. doi:10.1016/j.expneurol.2018.05.022
32. Wu JY, Li M, Cao LJ, et al. Protease Omi cleaving Hax-1 protein contributes to OGD/R-induced mitochondrial damage in neuroblastoma N2a cells and cerebral injury in MCAO mice. Acta Pharmacol Sin. 2015;36(9):1043–1052. doi:10.1038/aps.2015.50
33. Li L, Li W, Sun J, et al. Preparation and evaluation of progesterone nanocrystals to decrease muscle irritation and improve bioavailability. AAPS PharmSciTech. 2018;19(3):1254–1263. doi:10.1208/s12249-017-0938-3
34. Yang F, Yu XH, Qiao F, et al. Formulation and characterization of Brucea javanica oil microemulsion for improving safety. Drug Dev Ind Pharm. 2014;40(2):266–277. doi:10.3109/03639045.2012.756887
35. Harpur ES, Worah D, Hals PA, Holtz E, Furuhama K, Nomura H. Preclinical safety assessment and pharmacokinetics of gadodiamide injection, a new magnetic resonance imaging contrast agent. Invest Radiol. 1993;28:S28–S43. doi:10.1097/00004424-199303001-00004
36. Torii Y, Goto Y, Nakahira S, Kozaki S, Kaji R, Ginnaga A. Comparison of systemic toxicity between botulinum toxin subtypes A1 and A2 in mice and rats. Basic Clin Pharmacol Toxicol. 2015;116(6):524–528. doi:10.1111/bcpt.2015.116.issue-6
37. Misik J, Pavlikova R, Cabal J, Kuca K. Acute toxicity of some nerve agents and pesticides in rats. Drug Chem Toxicol. 2015;38(1):32–36. doi:10.3109/01480545.2014.900070
38. Zhang Q, Li J, Zhang W, et al. Acute and sub-chronic toxicity studies of honokiol microemulsion. Regul Toxicol Pharmacol. 2015;71(3):428–436. doi:10.1016/j.yrtph.2014.11.007
39. Li G, Zhao M, Zhao L. Well-defined hydroxyethyl starch-10-hydroxy camptothecin super macromolecule conjugate: cytotoxicity, pharmacodynamics research, tissue distribution test and intravenous injection safety assessment. Drug Deliv. 2016;23(8):2860–2868. doi:10.3109/10717544.2015.1110844
40. Lazarewicz JW, Pluta R, Saliriska E, Puka M. Beneficial effect of nimodipine on metabolic and functional disturbances in rabbit hippocampus following complete cerebral ischemia. Stroke. 1989;20(1):70–77. doi:10.1161/01.STR.20.1.70
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.