Back to Journals » The Application of Clinical Genetics » Volume 19
A Novel Deep-Intronic CFAP44 Variant Underlies Multiple Morphological Abnormalities of the Sperm Flagella
Authors Ma Y, Yang Y, Zhang T, Hu D, Zhao Y, Mai X, Ni J, Zhang J
Received 2 April 2026
Accepted for publication 8 June 2026
Published 24 June 2026 Volume 2026:19 609696
DOI https://doi.org/10.2147/TACG.S609696
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Yaxian Ma,1,2,* Yuecheng Yang,3,4,* Tong Zhang,1,2 Daoheng Hu,5,6 Yuancun Zhao,5,6 Xuancheng Mai,1,2 Junxue Ni,3 Jie Zhang5,6
1Department of Reproductive Medicine and NHC Key Laboratory of Healthy Birth and Birth Defect Prevention in Western China, The First People’s Hospital of Yunnan Province & The Affiliated Hospital of Kunming University of Science and Technology, Kunming, People’s Republic of China; 2KUST-YPFPH Reproductive Medicine Joint Research Center, Medical School of Kunming University of Science and Technology, Kunming, People’s Republic of China; 3Department of Pediatrics, The First People’s Hospital of Yunnan Province & The Affiliated Hospital of Kunming University of Science and Technology, Kunming, People’s Republic of China; 4Department of Pediatrics, The Second Affiliated Hospital of Kunming Medical University, Kunming, People’s Republic of China; 5Department of Medical Genetics, The First People’s Hospital of Yunnan Province & The Affiliated Hospital of Kunming University of Science and Technology, Kunming, People’s Republic of China; 6Yunnan Provincial Key Laboratory for Birth Defects and Genetic Diseases, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Junxue Ni, Email [email protected] Jie Zhang, Email [email protected]
Purpose: Multiple morphological abnormalities of the sperm flagella (MMAF), uncommonly causing primary infertility, are typical features of aberrant spermatozoa flagellum morphologies, which manifest as shortness, absence, bending, coiling, and irregularity of flagella. CFAP44, an important component of flagella assembly, has attracted significant interest due to its critical role in MMAF pathogenesis. Understanding the variants associated with CFAP44 can provide insights into the molecular mechanisms underlying MMAF.
Patients and Methods: A comprehensive clinical evaluation was conducted on an infertile Chinese male patient from a nonconsanguineous family with sever asthenozoospermia (no progressive sperm). By performing whole-exome sequencing (WES), a novel variant of CFAP44 was identified. Sanger sequencing was performed to confirm the variant. To better investigate its pathogenicity, In silico variant analyses, minigene splicing assays and RT-PCR in vivo were performed.
Results: As a result, a CFAP44 homozygous deep-intronic variant (NM_001164496.1:c.1890+5G>C) was detected in the proband by WES. Sanger sequencing confirmed this variant in this family. Splice site prediction suggested that this variant may be a disease-causing variant. Then, exon 15 skipping was identified through minigene assays and RT-PCR in vivo, resulting in a 111-bp deletion within the mutated sequence, thereby indicating a disruption in the normal splicing of the CFAP44 transcript.
Conclusion: This is the first study to detect a homozygous variant (c.1890+5G>C) within the CFAP44 gene causing MMAF in a Chinese family. Our results confirmed the pathogenicity of this deep-intronic variant and expanded the mutational spectrum of the CFAP44 gene. Consequently, this study may help elucidate the effect of CFAP44 on MMAF and provide a theoretical basis for MMAF.
Keywords: CFAP44, MMAF, male infertility, intronic variants
Introduction
Infertility is reported in 10–15% of married couples worldwide, with about 50% of cases caused by male factors.1 Typically, multiple morphological abnormalities of the sperm flagella (MMAF) constitute a major factor that induces male infertility, resulting in asthenozoospermia and almost no progressive sperm. MMAF typically features massive sperm cells with the absence, shortness, irregularity, and coiling of flagella.2 MMAF is a new concept that was first defined in 2014, at the beginning of the exploration of flagellar anomaly genetic pathogenicity.3 Researches have since rapidly expanded. Thus far, more than 40 MMAF-associated genes have been identified, some of which include DNAH1, DNAH2, DNAH6, DNAH17 (genes encoding dynein axonemal), CFAP43, CFAP44, CFAP65, CFAP69, CFAP70, CFAP75, and CFAP251 (cilia and flagella-associated proteins).4
Among the genes implicated in MMAF, CFAP44 (also known as WSR52, NM_001164496) is an important gene involved in flagellar anomaly pathogenesis. Located on chromosome 3, CFAP44 includes 35 exons and encodes a protein of about 1854 amino acids, called cilia and flagella-associated protein 44.5 The CFAP44 protein has one functional WD40 domain, which is involved in cytoskeleton assembly and possibly functions during centrosome formation.6 Coutton performed transmission electron microscopy (TEM) examination to investigate the sperm cell ultrastructure in CFAP44-mutated patients and detected no central pair complex (CPC) of flagella.5 Using CRISPR/Cas9 technology, Tang generated CFAP44-deficient male mice, and these knockout mice exhibited MMAF phenotypes.7 These consistent findings suggest that CFAP44 is important for MMAF pathogenesis.
However, only three articles reported CFAP44-related MMAF. The guidelines for clinical practice are still very limited. Whole-exome sequencing (WES) has recently achieved progress, revolutionizing the diagnosis of rare diseases and the identification of disease-causing genes. In this study, a novel deep-intronic variant, CFAP44:NM_001164496.1:c.1890+5G>C, was identified in a Chinese male via WES. We also conducted minigene splicing assays and in vivo RT-PCR assays to verify the pathogenicity of the novel CFAP44 variant in this proband. Our results may help elucidate the role of CFAP44, pave the way for further studies, and optimize treatments for patients with CFAP44-induced MMAF.
Materials and Methods
Clinical Evaluation of the Proband
The patient visited the First People’s Hospital of Yunnan Province due to infertility, and underwent a detailed physical examination, including an examination of secondary sexual characteristics. The patient was also subjected to routine semen analysis, sex hormone tests, testicular puncture biopsy, chromosome karyotype analysis, and AZF region testing.
WES and Sanger Sequencing
Peripheral venous blood samples were collected from this proband to isolate genomic DNA. Next, WES was performed on the MGISEQ-2000 DNA sequencer (BGI, China) using the MGIEasy Exome Enrichment Kit V4 (Probe-based) kit. Reads were aligned to GRCh37 (hg19). Variant annotation and screening were performed based on clinical data, disease and population databases, and biological information prediction procedures. Following ACMG guidelines, variant pathogenicity was analyzed. Moreover, Sanger sequencing was conducted to validate the potential CFAP44 variant. The following primers were used for PCR amplification: 5`-CAAAAGTCCCGTTGGAGCAA-3` (forward) and 5`-TCGTCGGTGTGTCTCTTTTTC-3` (reverse).
Kinship Analysis
Kinship among family samples was analyzed using WES data. We first applied strict quality control to all sample variant data and extracted high-quality single-nucleotide variant (SNV) sites for analysis. Similarity metrics for each pair of samples, including genotype correlation and the percentage of overlapping variant sites (overlap percentage), were analyzed using PLINK. Genotype correlation refers to the Pearson correlation coefficient between genotypes (coded as 0/1/2) at shared SNV positions. The overlap percentage (overlap_percent) is defined as the number of SNV sites detected in both samples divided by the total number of unique SNV sites detected in either sample. This metric intuitively reflects the degree of overlap in their variant profiles.
Each pair of samples was plotted on a two-dimensional scatter plot (correlation on one axis and overlap percentage on the other) to visualize clustering patterns corresponding to different kinship relationships. Based on internal historical WES sample data, our platform established empirical boundaries for relationship clusters. Specifically, first-degree relatives (full siblings, parent-child) typically present genotype correlation values between 0.50 and 0.60 and overlap_percent values between 70% and 80%. In contrast, unrelated individuals (eg, spouses) tend to fall between 0.40 and 0.50 for correlation and between 55.5% and 70% for overlap_percent. Based on these observed patterns, we established threshold values to tentatively distinguish first-degree relatives, more distant relatives, and unrelated individuals.
In silico Variant Analyses
Rare Disease Data Center (RDDC),8 NNSPLICE 0.9,9 and NetGene210 in silico splice site prediction software were used for predicting splice variants to evaluate variant pathogenicity.
Minigene Assay
The proband-derived genomic DNA was used as the template, and the CFAP44 gene was subsequently amplified through primer design, which consisted of exons 14–16 and introns 14–15. Primers were designed via seamless cloning to amplify heterozygous genomic DNA carrying the c.1890+5G>C variant site to obtain two target insertion gene fragments, CFAP44-A and CFAP44-B. The sequences of the primers used were as follows: AF: AAGCTTGGTACCGAGCTCGGATCCGTAAACTTCACTGGAGCACAAATTATTG; AR: CTGTTAGCCGGGGATGGACAAGCTTGGTCTACACC; BF: TGTCCATCCCCGGCTAACAGATTTCACAACCACATATATTC; and BR: TTAAACGGGCCCTCTAGACTCGAGCAGAATCTTAGATTTGACACTTGAAAAATGG.
The double restriction enzyme BamHI/XhoI was used to digest the pMini-CopGFP vector. Next, the digested products were recombined with the CFAP44-A and CFAP44-B fragments. Correct mutant-type (MT) and wild-type (WT) minigene plasmids were selected and transfected into 293T cells using Lipofectamine 3000 as recommended. After 48 h, RT-PCR was conducted to analyze the transcripts. The primers used for RT-PCR were as follows: F: GGCTAACTAGAGAACCCACTGCTTA; R: CAGAATCTTAGATTTGACACTTGAA. For the MT and WT variants, the PCR products were subjected to 1.0% agarose gel electrophoresis and then to Sanger sequencing.
RT-PCR Assay in vivo
EasyPure Blood RNA Kit (Lot# R21708) was used to extract total RNA from peripheral blood leukocytes of the family members, and one healthy individual without this variant was selected from our internal biobank as the control subject. The RNA was subsequently transformed into cDNA using HiScript III 1st Strand cDNA Synthesis (LOt:7E77213) through reverse transcription for RT-PCR analysis. Target sequence amplification was performed using 2Taq Master Mix (Lot. No:05230003) with the following thermal cycling parameters: 5 min of initial denaturation at 95 °C; 15s of denaturation at 95 °C, 10s of annealing at 62 °C, and 40s of extension at 72 °C for 30 cycles; and 7 min of extension at 72 °C.
The sequences of primers used were as follows: 5’-CGAATGGTAAACTTCACTGGAGCAC-3’ (forward) and 5’-CTACATCATGATCATCCTCTTCTTGC-3’ (reverse). The PCR products were analyzed by 1% agarose gel electrophoresis and subsequently sequenced by Sanger sequencing. The resulting sequences were aligned and analyzed using SnapGene software for verification.
CFAP44 Related Genotype and Phenotype Review
The PubMed, Embase, and CNKI databases were searched to summarize presentations of CFAP44-induced spermatogenic failure and pathogenic variant-related clinical phenotypes.
Results
Case Report
The 30-year-old remarried male patient had no history of fathering children. He and his ex-wife did not conceive after two years of unprotected intercourse. Similarly, he and his current wife have not been pregnant for more than two years. Twice semen analyses revealed severe oligospermia combined with 100% asthenozoospermia. The first analysis revealed a concentration of 2.5 million/mL, total motility of 0%, and a viability rate of 15.4%; the second analysis revealed a concentration of 2.6 million/mL, total motility of 0%, and a viability rate of 44.2%. The morphology of his sperm is shown in Figure 1. Hormonal tests for testosterone (T), luteinizing hormone (LH), and follicle-stimulating (FSH) hormone were normal: FSH 4.3 U/L, LH 8.8 U/L, and T 13.5 nmol/L.
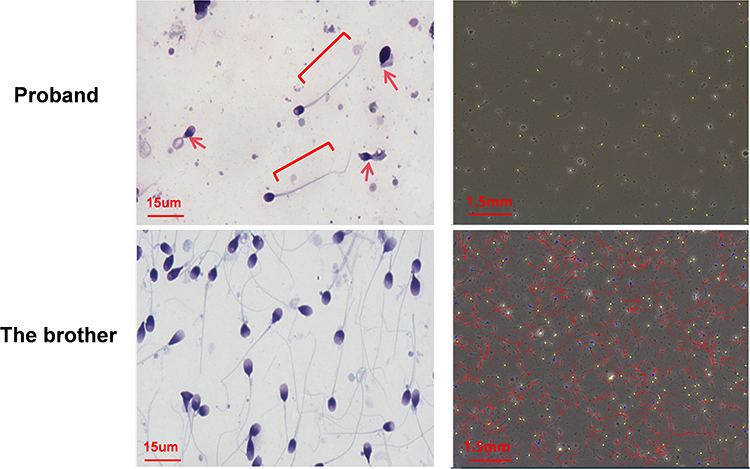 |
Figure 1 The patient’s sperm exhibited flagellar abnormalities, including coiled, irregular, short, and absent sperm flagella (red arrow: coiled or absent sperm flagella; brackets: short sperm flagella). From the trajectory diagram, the patient’s sperm showed no motile sperm (the yellow trajectories represent immotile sperm, the blue trajectories represent motile sperm, and the red trajectories represent progressive motility sperm). |
Physical examination revealed normal development of secondary sexual characteristics and a normal appearance of the external genitalia. No varicoceles were palpated. The bilateral testicles were about 15mL in size, with normal texture, and no nodules were found in the bilateral epididymide. The vas deferens was palpable. A testicular biopsy was performed and sperm cells were present but exhibited zero motility. Pathological examination of the right testis biopsy revealed a variable reduction in spermatogenic cells at different stages within the seminiferous tubules, accompanied by a corresponding decrease in sperm cells. Chromosomal karyotyping and analysis of the AZF region were performed, and the results were normal.
WES and Sanger Sequencing
WES was conducted on the proband, his younger brother, and parents. WES was conducted to identify a novel homozygous splice site variant, c.1890+5G>C, in the CFAP44 gene, which has not been annotated in the NHLBI Exome Sequencing Project (ESP), 1000 Genomes Project, ExAC Browser, or gnomAD database. The variant was subsequently subjected to Sanger sequencing, confirming that this patient carried the homozygous c.1890+5G>C variant. His parents and younger brother were heterozygous carriers (Figure 2). The semen test results for his father and brother were normal. Based on ACMG criteria (PM2+PM3_supporting+PP3), the c.1890+5G>C variant was classified as having uncertain significance.
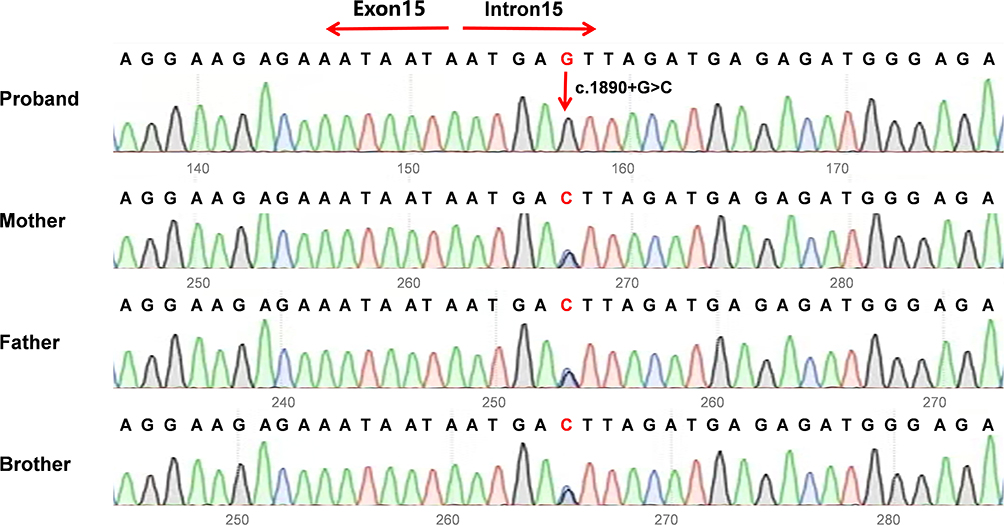 |
Figure 2 The Sanger sequencing results revealed a homozygous intron variant in the proband and heterozygous intron variant in the parents and younger brother (c.1890+5G>C). The red arrow indicates the position of the variant. |
Kinship Analysis
The correlation versus overlap_percent scatterplot (Figure 3a) showed that pairs of samples cluster according to their kinship category. For the core family highlighted in red, the comparison between the child and father samples fell within the “parent-child” region of the plot. This pair showed a correlation of about 0.55 and an overlap_percent of about 75%, consistent with a first-degree parent-offspring relationship, confirming the father-child relationship in that family. The comparison between the child and the mother also fell in an intermediate position between the “mother-child” and “sibling” regions. The comparison between the father and the mother deviated from the typical cluster of unrelated individuals: their correlation (0.50) and overlap_percent (70%) were significantly greater than those of typically unrelated pairs. This result suggested that the father and mother may share some genetic relationships (such as distant relative). This suggested a possible distant familial connection between the father and the mother.
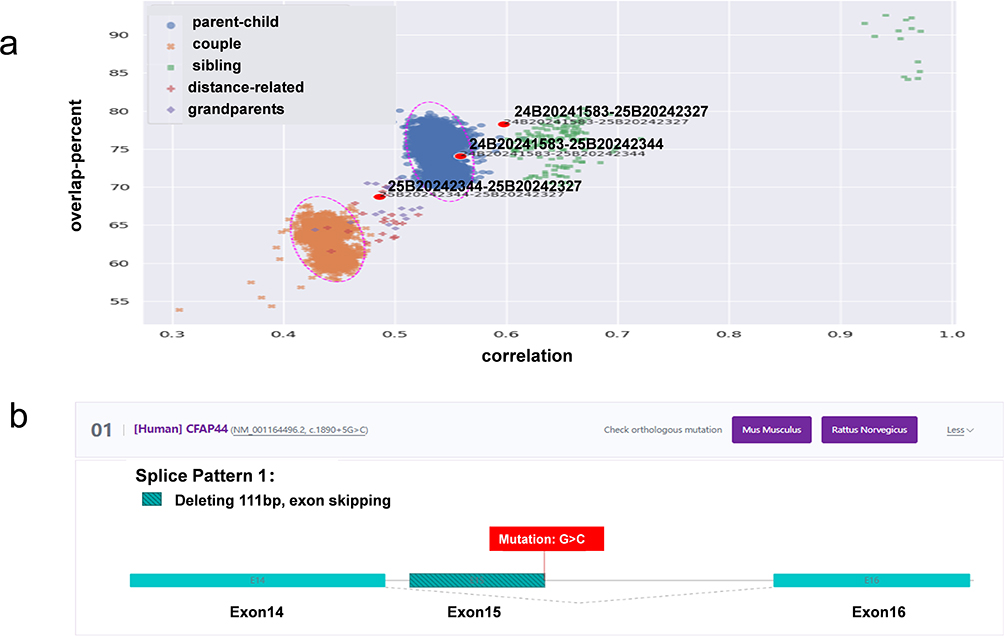 |
Figure 3 (a) kinship analysis. 24B20241583 is proband sample. 25B20242344 is father sample. 25B20242327 is mother sample. “Proband-father” comparison and “proband-mother” comparison were fell in first-degree parent-offspring relationship. “Father-mother” comparison deviated from the typical cluster of unrelated individuals. The purple dotted circles indicate approximate empirical clustering regions derived from the distribution patterns of historical WES samples with known relationships on our platform, and are shown for visualization purposes only rather than as statistically defined boundaries. (b) In silico variant analysis: Splice site prediction through Rare Disease Data Center. |
In silico Variant Analysis
This variant is extremely rare and absent in the ExAC and 1000 Genomes databases. Splice site prediction suggested that this variant may be a disease-causing mutation. With the RDDC splice site prediction program, the c.1890+5G>C variant leads to a 111 bp transcriptional deletion and exon 15 skipping (Figure 3b). The other two prediction programs showed that the donor splice site mutated from G to C resulted to the donor splice site disappearing. Moreover, the mutation tester tool predicted the c.1890+5G>C variant as “pathogenic”.
Minigene Assay
To ascertain the pathogenicity of this intronic variant, we performed an in vitro splicing assay with a minigene vector incorporating the MT and WT c.1890+5G>C CFAP44 genes, which span exons 14–16 (Figure 4a). As suggested by the RT-PCR analysis, cells that contained the WT plasmid generated a 554 bp amplicon. In contrast, those subjected to c.1890+5G>C variant sequence transfection produced a fragment of 443 bp (Figure 4b). According to Sanger sequencing, exon 15 skipping was identified from the mutant fragments, resulting from a 111-bp deletion in the mutated sequence (Figure 4c). The above results revealed that this CFAP44 gene variant generated an abnormality in cDNA splicing (Figure 4d). The mutation tester tool predicted the c.1890+5G>C variant as “pathogenic”.
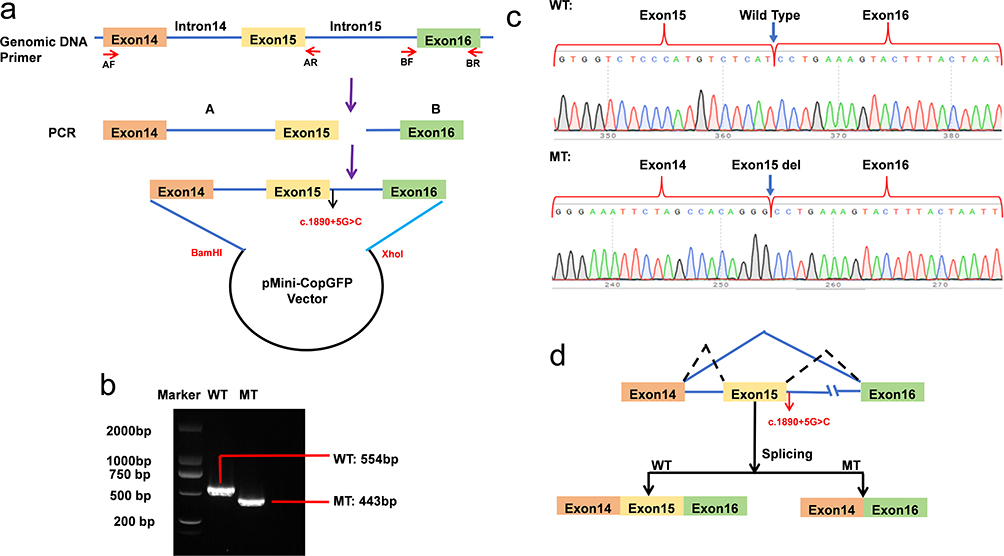 |
Figure 4 Minigene assay: (a) Schematic diagram of plasmid assembly. Red arrows mark the DNA fragments used for minigene assembly and granulization. Black arrow indicates the location of mutation sites. Purple arrow indicates the workflow of plasmid assembly. AF: upstream of fragment A; AR: downstream of fragment A; BF: upstream of fragment B; BR: downstream of fragment B. (b) Electrophoretogram of RT‒PCR products: Lane 1: mutant plasmid (MT) 443bp; Lane 2: wild-type plasmid (WT): 554 bp; Lane 3: marker. (c) Sanger sequencing of RT‒PCR products: variant c.1890+5G>C resulted in a 115 bp deletion caused by exon 15 skipping. Blue arrows indicate the region altered by c.1890+5G>C mutation in RT-PCR products. (d) Schematic representation of the splicing events associated with this CFAP44 variant. The black dashed lines indicate the ligation of exon 14, 15 and 16 in the wild-type transcript sequence. The blue solid lines represent the linkage of exon 14 and 16 in the mutant transcript sequence, with exon 15 skipped. WT: wild type; MT mutant type. |
RT-PCR Assay in vivo
To confirm in vivo splicing alteration of CFAP44, RT-PCR was conducted using blood samples collected from the family members. The proband with c.1890+5G>C homozygous variant displayed a 310 bp fragment, whereas the parents and brother presented heterozygous variants with 421-bp and 310-bp bands. The negative control samples, which consisted of healthy individuals, presented a 421 bp fragment (Figure 5a). The PCR fragments were subjected to sequencing analysis. Sequencing analysis revealed only exon 15 skipping in this homozygous proband. In parents heterozygous for the deletion, exon 15 showed overlapping peak patterns because it contained both a normal allele and an exon 15-skipping allele. A sketch map showing variant splicing events was illustrated in Figure 5b. Pathogenicity was subsequently re-evaluated following ACMG guidelines, and the variant was confirmed as pathogenic based on PVS1+PM2+PM3_Supporting. And following is the explanation of criteria: PVS1: Minigene assay confirmed that this variant induces aberrant splicing, resulting in skipping of exon 15 and a 111 bp deletion, which ultimately impairs protein function. PM2: This variant is not recorded in the 1000 Genomes, ExAC, and gnomAD databases. PM3_supporting: This variant was detected in homozygosity in a patient suffering from oligospermia.
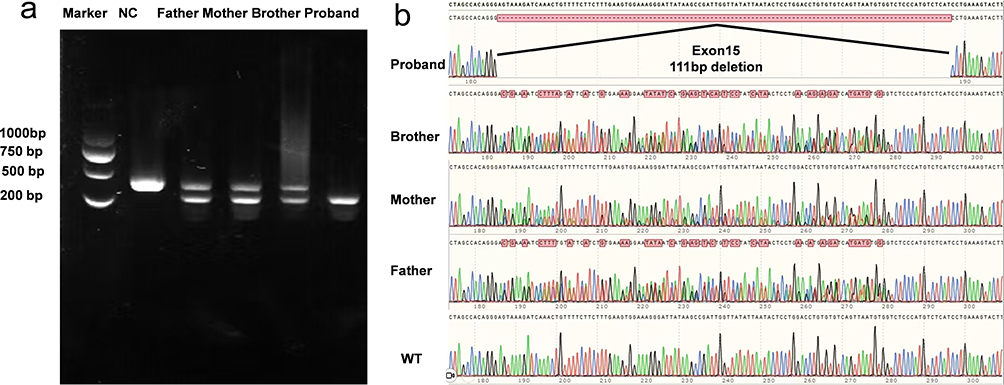 |
Figure 5 In vivo RT-PCR validation of CFAP44 exon 15 deletion in the proband. (a) Nucleic acid electrophoresis results: Lane 1: DNA ladder (Marker). Lane 2: The negative control exhibited a 421 bp fragment without variation. Lane 3, Lane 4 and Lane5: The parents and brother exhibited heterozygous variants with two bands at 421 bp and 310 bp. Lane 6: The proband exhibited a 310 bp fragment with the homozygous variant. (b) Sanger sequencing chromatogram alignment, confirming that the variant c.1890+5 G>C resulted in a 111-bp deletion causing exon 15 skipping. |
Literature Review
After searching the PubMed, Embase, and CNKI databases, only three reports on CFAP44 gene variants were retrieved [5–7]. We identified 10 variants in 12 people from 11 families, including five point variants (c.1769T>A, c.3263G>A, c.1718C>A, c.3175C>T, and c.1387G>A), three deletion variants 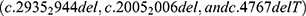 , one duplication variant (c.2818dupG), and one splicing variant
, one duplication variant (c.2818dupG), and one splicing variant 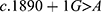 . Except for c.1769T>A, c.3263G>A, and c.1718C>A, the other seven variants might induce premature termination of translation of CFAP44.
. Except for c.1769T>A, c.3263G>A, and c.1718C>A, the other seven variants might induce premature termination of translation of CFAP44.
These patients exhibited severe asthenozoospermia. Almost no spermatozoa with normal morphology were observed (normal proportion 0–0.5%), and the sperm flagella were bent, coiled, irregular, short, or absent. Moreover, the percentage of mobile spermatozoa ranged from 0% to 1.2%. Detailed information on the clinical phenotypes of these patients is summarized in Table 1.
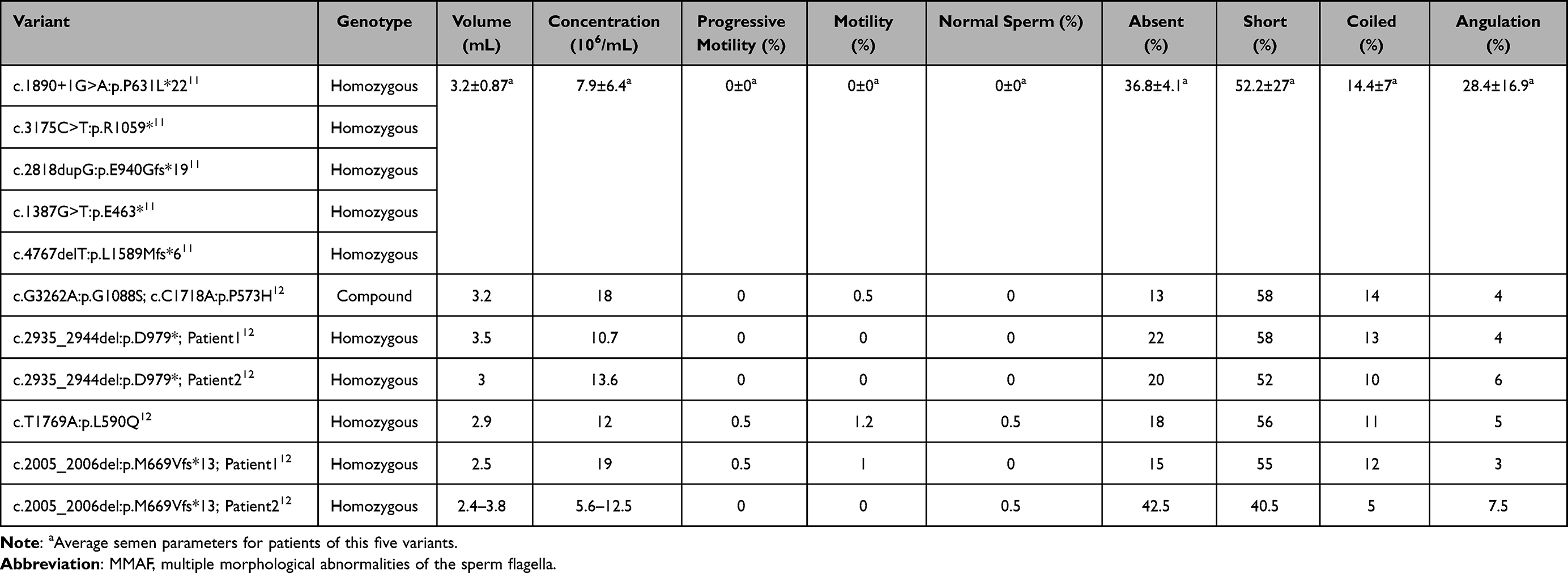 |
Table 1 Summary of the Phenotypes of CFAP44 Variants Related MMAF in Previous Studies |
Discussion
This study represents the fourth reported case worldwide. We detected one new deep-intronic variant, c.1890+5G>C, within the CFAP44 gene of an infertile individual. This variant is associated with type 20 spermatogenic failure, which is characterized by abnormal morphologies: bending of the sperm flagellum, coiling, irregularity, shortness, and/or absence. The results of in silico analysis, minigene assays and in vivo RT-PCR demonstrated that the variant was pathogenic, highlighting that CFAP44 is important for MMAF pathogenesis.
The proband had a history of infertility for more than four years after marriage. His sperm cells were 100% immobile and had completely abnormal morphology, including short, coiled, irregular, and absent sperm flagella. Additionally, no motile sperm cells were found through testicular sperm extraction. Considering these findings, the patient’s clinical manifestations were consistent with MMAF.
WES of the family members identified a homozygous intronic variant in CFAP44 c.1890+5G>C in the proband, which was verified through Sanger sequencing. Usually, the regions adjacent to the splice sites, especially the ±1 or ±2 positions within introns, are important for gene splicing. They directly guide intron recognition and excision by the spliceosome throughout RNA splicing.13 Variants in these important positions may substantially affect RNA splicing, disrupting gene expression and causing inherited diseases. However, empirically predicting the pathogenicity of deep-intronic region variants is difficult.
In silico analysis, minigene assays, and in vivo RT-PCR were performed to assess variant pathogenicity. In vivo and in vitro results demonstrated that this deep-intronic variant caused exon 15 skipping, ultimately resulting in impaired gene function. Our results revealed this deep- intronic variant, +5 position in the intron, was pathogenic according to ACMG guidelines. By integrating genetic tests and representative clinical presentations, the proband was diagnosed with CFAP44-related MMAF.
CFAP44-related MMAF was extremely rare and the proband had an autosomal recessive homozygous variant in the CFAP44 gene. Cases from three previously reported studies also involved mainly homozygous variants. These rare autosomal recessive genetic diseases are often associated with consanguineous unions between parents.14 Therefore, we assessed the parents’ consanguinity using WES data in PLINK. The results indicated the possibility of a distant familial relationship between the proband’s parents. Thus consanguineous marriage should be avoided for decreasing genetic autosomal recessive disease risk in offspring.
After the diagnosis was confirmed, we provided the patient with treatment advice. Intracytoplasmic sperm injection (ICSI) assists in obtaining genetic offspring from MMAF cases. MMAF cohorts report promising results after ICSI, and many couples deliver healthy babies.11,12,15 Sha reported similar rates of transferable embryos, implantation, clinical pregnancy, and miscarriage between the CFAP44+ group and the control group.12 But due to the small scale reported in existing literature, the clinical outcome following ICSI for this particular genotype remains to be observed. At last, the proband has decided to accept ICSI-assisted pregnancy.
Conclusion
To summarize, our identified intronic variant (NM_001164496.1:c.1890+5G>C) probably results in a truncated CFAP44 protein resulting from exon 15 skipping, leading to abnormal assembly of flagella. Our findings broaden the spectrum of genetic variants associated with CFAP44-related spermatogenic failure, which presents as MMAF. These endeavors provide deeper insights into the effect of CFAP44 on spermatogenesis. However, the exact underlying mechanism needs to be elucidated.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author Jie Zhang upon reasonable request.
Ethics Approval Statement
This study was approved by the Ethical Review Board of the First People’s Hospital of Yunnan Province (number: 2025-GN023) and conducted in accordance with the World Medical Association Declaration of Helsinki. Consent was provided by the family after the members were adequately informed of this study.
Informed Consent
Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by Doctoral Fund Project of The First People`s Hospital of Yunnan Province (Grant No. KHBS-2022-025); National Natural Science Foundation of China (Grant No.82160319); Ten Thousand People Planning Commission of Yunnan Province (Grant No. XDYC – MY – 2022-0094, YNWR – MY – 2020 – 066); Young and Middle-Aged Academic Leaders Planning Commission of Yunnan Province (Grant No. 202105AC160034); Basic Research Project-Key Projects of Yunnan Province (Grant No. 202301AS070007); Basic Research Projects of Yunnan Province (Grant No. 202501AY070001-021); and High-end Medical Talents of Yunnan Provincial Health Commission (Grant No. L-2024016).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Inhorn MC, Patrizio P. Infertility around the globe: new thinking on gender, reproductive technologies and global movements in the 21st century. Hum Reprod Update. 2015;21(4):411–10. doi:10.1093/humupd/dmv016
2. Zhou Y, Yu S, Zhang W. The molecular basis of multiple morphological abnormalities of sperm flagella and its impact on clinical practice. Genes. 2024;15(10):10. doi:10.3390/genes15101315
3. Khelifa MB, Coutton C, Zouari R, et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2013;94(1):95–104. doi:10.1016/j.ajhg.2013.11.017
4. Martinez G, Barbotin AL, Cazin C, et al. New mutations in DNHD1 cause multiple morphological abnormalities of the sperm flagella. Int J Mol Sci. 2023;24(3):2559. doi:10.3390/ijms24032559
5. Coutton C, Vargas AS, Amiri-Yekta A, et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat Commun. 2018;9(1):686. doi:10.1038/s41467-017-02792-7
6. Sha YW, Wang X, Xu X, et al. Novel mutations in CFAP44 and CFAP43 cause multiple morphological abnormalities of the sperm flagella (MMAF). Reprod Sci. 2017;26(1):26–34. doi:10.1177/1933719117749756
7. Tang S, Wang X, Li W, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2017;100(6):854–864. doi:10.1016/j.ajhg.2017.04.012
8. Available from: https://rddc.tsinghua-gd.org/.
9. Available from: https://www.fruitfly.org/seq_tools/splice.html.
10. Available from: https://services.healthtech.dtu.dk/services/NetGene2-2.42.
11. Wambergue C, Zouari R, Fourati Ben Mustapha S, et al. Patients with multiple morphological abnormalities of the sperm flagella due to DNAH1 mutations have a good prognosis following intracytoplasmic sperm injection. Hum Reprod. 2016;31(6):1164–1172. doi:10.1093/humrep/dew083
12. Sha YW, Wang X, Su ZY, et al. Patients with multiple morphological abnormalities of the sperm flagella harbouring CFAP44 or CFAP43 mutations have a good pregnancy outcome following intracytoplasmic sperm injection. Andrologia. 2018;51(1):e13151. doi:10.1111/and.13151
13. Witten JT, Ule J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011;27(3):89–97. doi:10.1016/j.tig.2010.12.001
14. Anwar S, Taslem Mourosi J, Arafat Y, Hosen MJ. Genetic and reproductive consequences of consanguineous marriage in Bangladesh. PLoS One. 2020;15(11):e0241610. doi:10.1371/journal.pone.0241610
15. Ferreux L, Bourdon M, Chargui A, et al. Genetic diagnosis, sperm phenotype and ICSI outcome in case of severe asthenozoospermia with multiple morphological abnormalities of the flagellum. Hum Reprod. 2021;36(11):2848–2860. doi:10.1093/humrep/deab200
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.