Back to Journals » The Application of Clinical Genetics » Volume 13
A Novel APOC2 Mutation in a Colombian Patient with Recurrent Hypertriglyceridemic Pancreatitis
Authors Pinilla-Monsalve GD, Lores J ![]() , Pachajoa H
, Pachajoa H ![]() , López-Ponce de León JD, López A, Rodríguez-Rojas LX, Nastasi-Catanese JA
, López-Ponce de León JD, López A, Rodríguez-Rojas LX, Nastasi-Catanese JA ![]()
Received 20 December 2019
Accepted for publication 20 February 2020
Published 26 March 2020 Volume 2020:13 Pages 63—69
DOI https://doi.org/10.2147/TACG.S243148
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Gabriel D Pinilla-Monsalve,1,* Juliana Lores,1,2,* Harry Pachajoa,1,2 Juan D López-Ponce de León,1,3 Alejandro López,1,4 Lisa X Rodríguez-Rojas,1,2 José A Nastasi-Catanese1,2
1Faculty of Health Sciences, Universidad Icesi, Cali 760032, Colombia; 2Department of Genetics, Fundación Valle del Lili, Cali 760032, Colombia; 3Department of Cardiology, Fundación Valle del Lili, Cali 760032, Colombia; 4Department of Endocrinology, Fundación Valle del Lili, Cali 760032, Colombia
*These authors contributed equally to this work
Correspondence: José A Nastasi-Catanese Cra. 98 No. 18-49, Cali 760032, Colombia
Tel +57 323 514-7106
Fax +57 2 3319090
Email [email protected]
Abstract: Hypertriglyceridemia is a common disease with only 2% of cases exhibiting monogenic mutations. Familial chylomicronemia syndrome (FCS) is a rare genetic condition associated with recurrent and severe episodes of pancreatitis and is mainly caused by mutations in the LPL gene, with few cases related to abnormal function of apolipoprotein C-II. This is a 50-year-old female with a past medical history of arterial hypertension, miscarriage and recurrent pancreatitis. In the last four years, her triglycerides and lipase concentration reached > 3000 mg/dL and > 700 U/L, respectively. The patient was not responsive to statins, fibrates, or tetrahydrolipstatin. A novel homozygous frameshift mutation on exon 3 of the APOC2 gene was detected, c.133_134delTC. Subsequent Sanger sequencing confirmed that three first-degree relatives were carriers of the same mutation. To the best of our knowledge, we are reporting the first Colombian patient with FCS due to an APOC2 mutation. We propose that this mutation caused recurrent hypertriglyceridemic pancreatitis.
Keywords: hypertriglyceridemia, hyperlipoproteinemia type I, apolipoprotein C-II, pancreatitis
Introduction
Dyslipidemia is one of the most common diseases in the world with a prevalence that reaches almost 70–80% while hypertriglyceridemia (HTG) has a frequency that varies from 10 to 60% according to the studied population.1–4 Among patients with severe HTG, only 2% have monogenic mutations.5 On the other hand, pancreatitis is one of the most common gastrointestinal diagnoses in Europe and the USA but only 10% is attributable to HTG, in which patients have increased blood viscosity leading to pancreatic ischemia.6
Familial chylomicronemia syndrome (FCS), categorized as hyperlipoproteinemia type 1 in the Fredrickson’s classification, is a rare genetic condition affecting around 1 in 1.000.000 patients worldwide.7,8 This disease is characterized by severe forms of HTG or chylomicronemia with onset in infancy or adolescence, recurrent episodes of pancreatitis, hepatosplenomegaly and other manifestations like eruptive xanthomas and lipemia retinalis.9 Particularly, 70% of FCS patients have had six or more pancreatitis events with an incidence of 42 episodes per 1000 person/year.5,10
Deleterious biallelic mutations in five autosomal genes encoding for proteins in the lipolytic pathway have been found responsible for the phenotype: lipoprotein lipase (LPL, MIM: 609708), apolipoprotein A-V (APOA5, MIM: 606368), apolipoprotein C-II (APOC2, MIM: 608083), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1, MIM: 612757) and lipase maturation factor 1 (LMF1, MIM: 611761), with LPL accounting for the vast majority of cases (95%).11,12
The APOC2 gene encodes for apolipoprotein C-II (ApoC-II), a 79 amino acid (9 kDa) molecule and an important cofactor for the activation of LPL, resulting in clearance of chylomicrons from circulation.13,14 FCS due to APOC2 mutations is also known as hyperlipoproteinemia type Ib or APOC2 deficiency (MIM: 207750) and accounts for only 2% of all FCS cases.8 Since the publication by Breckenridge in 1978, 22 mutations in the APOC2 gene have been reported in literature, only one of them in the Latin-American population.14–16 Here, we report a Colombian patient with severe HTG and recurrent pancreatitis who presents a novel mutation in the APOC2 gene.
Case Report
This is the case of a Colombian 50-year-old non-diabetic and obese female (BMI 31.5 Kg/m2) with a past medical history of severe HTG (onset after her menarche and worsening during pregnancies), arterial hypertension and one miscarriage. She attended the Emergency Department four years ago complaining of several episodes of vomiting, abdominal distension, and epigastric pain after a copious meal. There were no peritoneal irritation signs and laboratory results included high concentrations of lipase (2.691 U/L) and triglycerides (TG) (1.148 mg/dL). She was started on intravenous fluids, antispasmodics, and analgesics with complete resolution of symptoms and was therefore discharged.
The patient was lost on follow-up and apparently replaced allopathic treatment for dyslipidemia (fibrate plus high-potency statin) with homeopathic medications. After three years, she complained again of abdominal epigastric pain irradiated to right lumbar quadrant. Basic blood tests were otherwise normal with the exception of high levels of transaminases (AST 427.1 U/L, ALT 374 U/L). Abdominal contrasted computed tomography and magnetic resonance showed inflammation of the pancreatic head, neck and uncinated process with plastron and signs of portal hypertension (Figure 1). Based on normal serum lipase (43 U/L), these findings were considered as incidental non-acute pancreatitis and treatment was disregarded; on the contrary, a diagnosis of toxic hepatitis caused by homeopathic drugs was suggested.
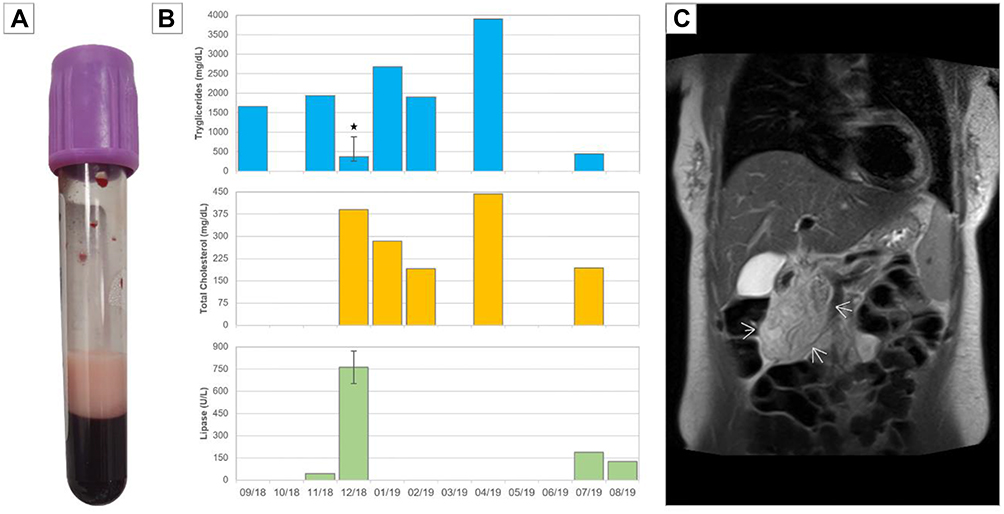 |
Figure 1 Laboratory and imaging findings. (A) Creamy supernatant from drawn blood. (B) Progression of serum concentrations over time for TG, total cholesterol and lipase. Star (*) specifies heparin infusion and error bars represent interquartile range (TG n=6, lipase n=2). (C) Coronal MRI showing acute pancreatitis. Arrows designate inflammatory plastron. |
One month later, nausea, abdominal pain, and distension recurred on epigastrium and right hypochondrium. Laboratories obtained were positive for HTG (2.066 mg/dL) with elevated lipase (982 U/L) in the settings of acute pancreatitis (APACHE II score: 8). Suddenly, she exhibited an altered state of consciousness and hypotension (MBP 40 mmHg) and was hospitalized in the Intensive Care Unit for four days. Considering that the clinical picture was consistent with a very-severe hypertriglyceridemic pancreatitis and that worrisome characteristics were present, the gastroenterology team started her on unfractionated heparin infusion for 3 days (5000 UI/5 hrs) that resulted in a reduction of up to 89.30% (221 mg/dL) and 44.7% (543 U/L) on TG and lipase, respectively. Symptoms resolved completely. A fourth and short hospitalization occurred in the last semester, referring the aforementioned symptoms but lipase concentrations were 443 mg/dL.
Over this year, fibrates have been rotated from gemfibrozil to fenofibrate and ciprofibrate due to fluctuations on TG levels (Figure 1). She was also prescribed metformin, orlistat and eicosapentaenoic acid with a mild effect. The ambulatory comprehensive analysis included a transthoracic echocardiogram that only reported a mildly-enlarged left atrium (diameter: 42 mm and volume: 37 mL/m2) and a carotid duplex ultrasound which found no atherosclerosis. Evaluation of cardiovascular risk revealed a low 10-year risk of myocardial infarction or death of 2.3% (adjusted Framingham score for Colombian population), with a borderline ASCVD risk of 5.5%. Taking into account the international recommendations on primary prevention of cardiovascular disease, a coronary scan with calcium score was requested and discarded significant atheromatous disease (5 HU).
Subsequently, a familial chylomicronemia syndrome diagnosis was suspected considering the severity of HTG (1148 mg/dL [IQR 375–2001] with total cholesterol/TG ratio 7.5 [IQR 3.5–9.2]), refractoriness to multiple treatments, and the feature of consanguineous parents (second cousins), which increased the probability of an autosomal recessive disorder. Nonetheless, she did not have a history or exhibited lipemia retinalis or xanthomas.
Further investigation was performed using multigene panel testing via next-generation sequencing. Specifically, blood samples underwent automatized extraction and purification processes for obtaining genomic DNA (QIAsymphony SP®, Qiagen). Library preparation was performed using SureSelect Reagent Kit (Agilent) for Illumina multiplexed paired-end sequencing following manufacturer’s instructions and included 47 genes related to primary/polygenic hypertriglyceridemia and familial combined hyperlipidemia. Enrichment for regions of interest was based on SureSelect Probe Kit (Agilent) that selectively captures codifying regions and flanking intronic sequences from the designated genes. After the generation of clusters, the DNA libraries were sequenced on the Illumina HiSeq 1500 platform. The trimmed reads were mapped to the human reference genome (GRCh38) using the Burrows-Wheeler aligner software.
A novel frameshift pathogenic variant in homozygous state of the APOC2 gene (NM_000483.4) was identified: c.133_134delTC (p.Ser45Glnfs*24) and confirmed by Sanger sequencing. This 2-nucleotide deletion on exon 3 converts the serine codon at position 45 into glutamine, which causes a frameshift resulting in an early stop codon 23 residues later (Figure 2). This variant has not been reported previously in population databases or current medical literature. No other gene variants were identified in this case. The patient’s mother (75 years old), brother (52 years old), and son (23 years old) consented for genetic testing and were found to be heterozygous for this pathogenic variant (Figure 3). They exhibited mildly elevated TG (164 mg/dL, IQR 113–166) and total cholesterol levels (211 mg/dL, IQR 188–213).
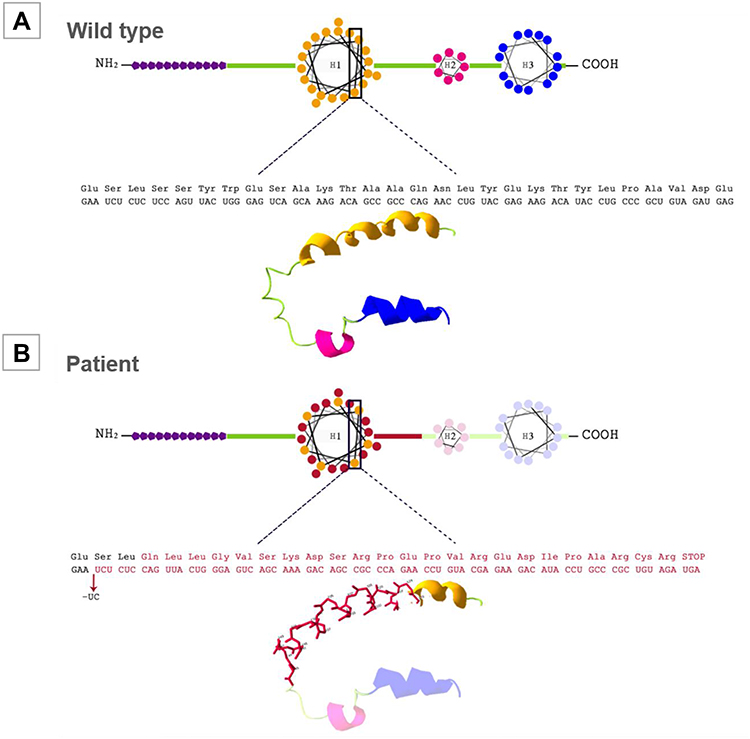 |
Figure 2 APOC2 protein scheme and three-dimensional visualization via Swiss-PDB Viewer. (A) Wild-type APOC2 protein. The purple polygons indicate the signal peptide domain. H1: helix 1; H2: helix 2; H3: helix 3. (B) Suggested structure of mutated APOC2 protein (p.Ser45Glnfs*24). The structure variation due to a frameshift mutation in protein is shown. In red: the aminoacid change; light color: the regions lost due to the premature stop codon. |
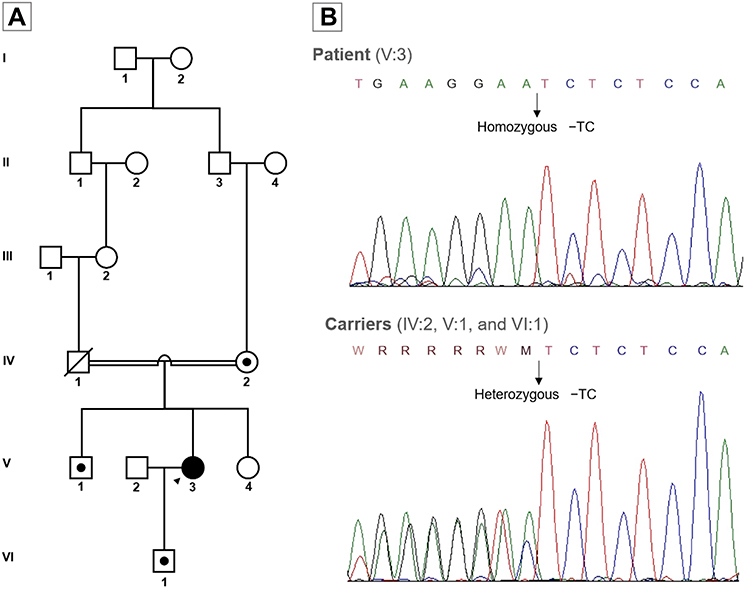 |
Figure 3 Family pedigree and Sanger sequencing. (A) Pedigree demonstrating consanguineous relationship in the patients’ parents and carrier status of the frameshift mutation c.133_134delTC in exon 3 of the APOC2 gene. Arrow: index case; black symbol: familial chylomicronemia syndrome; black dot: heterozygous carriers (B) Sanger sequencing electropherogram of the patient and carriers of the frameshift mutation in the APOC2 gene. |
In the last follow-up patient was overweight (BMI 26.3 Kg/m2; body fat percentage 29.7%) with symptoms compatible with binge-eating disorder. There were no physical complaints or findings.
Discussion
Including this case, 95 subjects (probands and kindred) have been reported with APOC2 mutations. Among them, 41 homozygous patients were diagnosed at a median age of 27 years (IQR 6–38), and 24 (58.53%) were females. Additionally, 36.58% (n=15) were Europeans, 34.14% (n=14) Americans, and 19.51% (n=8) Asians. Five patients had a history of xanthomata and median body mass index for adults was 22.9 Kg/m2 (IQR 20.8–26.8; n=11); maximum TG concentrations were 2928 mg/dL (IQR 1787–5055; n=40) but, interestingly, levels were <2000 mg/dL in 13 patients. ApoC-II was undetectable in 22 out of 27. Papers by Okubo and Wolska have synthesized the biochemical and genetic characteristics of these patients.14,16,17
Out of 23 mutations, 7 (30.43%) were missense, 6 (26.08%) frameshift, and 5 (21.73%) nonsense. A case initially published in 1986 by Posner et al about a 4 month-old Venezuelan male patient has been the only report in Latin-America with an APOC2 mutation.16,17 In Colombia, there has been another report of a 45-year-old patient with a clinical diagnosis of FCS, but in which the genetic etiology was not identified.12
The APOC2 gene is located in chromosome 19, as part of a cluster named APOE-APOC1-APOC4-APOC2, with a complex transcriptional regulation.14 It gives rise to a 101-amino acid peptide, which undergoes cleavage leaving a mature protein containing 79 residues. ApoC-II is an important component of lipoprotein metabolism, and is expressed in the liver primarily, but also in enterocytes, macrophages and many other cells. It is one of the multiple cofactors that have an effect on the activation/inactivation of LPL. ApoC-II enhances LPL-mediated hydrolysis for energy delivery. Most mutations are located in helix 1 of the protein, as in this case, but this specific mutation creates a premature stop codon which shortens the peptide chain and produces a protein without helix 2 and 3 (Figure 2).
According to the recent review by Chyzhyk and Brown, diagnosis of FCS is suspected in patients with consecutive TG concentrations >885 mg/dL, total cholesterol/TG ratio >5, levels of apolipoprotein B 100 (ApoB-100) <75 mg/dL and total TG/ApoB-100 ratio >8.8.8,14 Creamy supernatant can be observed as well in drawn blood (Figure 1). Furthermore, common treatment such as statins and fibrates are not significantly effective when comparing multifactorial chylomicronemia syndrome with FCS (p=0.002) as these patients are unable to metabolize chylomicrons increasing the suspicion.10 Patients usually complain of multiple episodes of pancreatitis or abdominal pain without an identifiable cause and there is no history of alcoholism, diabetes, hypothyroidism, pregnancy or relevant medicines consumption. Gene sequencing is the gold standard while family carrier status assessment is suggested.8 In this patient, it was curious that HTG apparently started after her menarche (although we have no data); estrogens are known to almost doubled after the first menstrual period and to induce HTG by impairing LPL function.18,19 Indeed, oral contraceptives use has been associated with hypertriglyceridemic pancreatitis in FCS patients.20
Therapeutic alternatives for FCS are scarce and regulation of dietary intake of fat to 30–50 g/day is effective but difficult for patients to apply consistently.8 Based on the available literature, episodes of pancreatitis can be treated with plasmapheresis (especially in those with a worrisome picture), low-calorie parenteral nutrition, and fresh plasma transfusion while others sources claim that insulin (LPL synthesis regulating factor) for diabetic patients and/or heparin infusions are valid alternatives in the acute setting.21–23 Plasmapheresis in HTG-pancreatitis was effective in the study by Gubensek et al for removing TG molecules and consequently reduced its concentrations in 59% compared to 27% in the conservative management group (p<0.001). Non-fatal episodes were associated with higher serum levels of sodium and calcium, shorter in-hospital stay and use of citrate anticoagulation instead of heparin.24
Unfractionated heparin (UFH) infusion enhances the release of LPL attached to the endothelium and therefore reduces plasma TG by converting them into free fatty acids (FFA).23 In the study by Shirakawa’s team, levels of TG were reduced from 74 (49–100) and 108 (73–150) mg/dL to 47 (31–74) and 73 (45–105) mg/dL, at fasting and postprandial states, 15 mins after injecting 50 heparin U/Kg in normal volunteers.25 Unfortunately, anticoagulant treatment does not seem to improve HTG in the long term due to hepatic LPL degradation, induced FFA toxicity, and potential hemorrhages.23 In this patient, UFH infusion for 72 hrs caused a maximal reduction of 1845 mg/dL of TG but levels increased again up to 179% two days later.
In our case, chronic control of TG concentration has been difficult due to poor response with statins, fibrates, tetrahydrolipstatin and even non-allopathic medicines. Volanesorsen is a novel therapeutic option, not available in Colombia, that induces an antisense-mediated inhibition of hepatic APOC3 mRNA and decreases apolipoprotein C-III (ApoC-III) levels which, among other effects, favors LPL function.8,26 The APPROACH, a randomized clinical trial conducted by Witztum and colleagues in FCS patients treated with volanesorsen, showed a significant reduction of 84% and 77% in ApoC-III and TG at 90 days. Nevertheless, only one patient with APOC2 mutation was enrolled in the trial. The main adverse effects included injection-site reactions, thrombocytopenia, and abdominal pain, an event that seems paradoxical in the context of the disease clinical manifestations.26 Our patient might not benefit from this therapy taking into account the lack of solid evidence for treating HTG caused by APOC2 mutations and current TG levels <500 mg/dL.
Conclusion
We describe a novel mutation in the APOC2 gene resulting in familial chylomicronemia syndrome. To our knowledge, this is the first Colombian patient with molecular confirmation of FCS, and the second in Latin-America. Clinical evidence of recurrent pancreatitis and severe hypertriglyceridemia should raise a suspicion of FCS and prompt genetic testing. Further research is required in terms of prognosis and new therapeutic interventions.
Ethical Approval
The patient provided informed consent for the case details to be published and ethical approval was granted by Fundación Valle del Lili’s Institutional Review Board/Ethical Committee (No. 349/2019).
Acknowledgment
Lorena Díaz, M.Sc. for her collaboration on genetic testing and protein graphing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. González-Juanatey JR, Millán J, Alegría E, Guijarro C, Lozano JV, Vitale GC. Prevalence and characteristics of lipid abnormalities in patients treated with statins in primary and secondary prevention in Spain. DYSIS-Spain Study. Rev Esp Cardiol. 2011;64(4):286–294. doi:10.1016/j.recesp.2010.10.030
2. Galvis-Pérez Y, Barona-Acevedo J, Arias-Cardona A-J. Prevalence of dyslipidemia in a health services provider institution of medellin (Colombia), 2013. CES Medicina. 2016;30(1):3–13. doi:10.21615/cesmedicina.30.1.1
3. Zhang M, Deng Q, Wang L, et al. Prevalence of dyslipidemia and achievement of low-density lipoprotein cholesterol targets in Chinese adults: a nationally representative survey of 163,641 adults. Int J Cardiol. 2018;1(260):196–203.
4. Munguía-Miranda C, Sánchez-Barrera RG, Hernández-Saavedra D, Cruz-López M. Dyslipidemia prevalence and its relationship with insulin resistance on a population of apparently healthy subjects. Salud Pública de México. 2008;50(5):375–382. doi:10.1590/S0036-36342008000500010
5. Garg A, Garg V, Hegele RA, Lewis GF. Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice. Lancet Diabetes Endocrinol. 2019;7(11):880–886. doi:10.1016/S2213-8587(19)30156-1
6. Weiss FU, Laemmerhirt F, Lerch MM. Etiology and risk factors of acute and chronic pancreatitis. Visc Med. 2019;35(2):73–81. doi:10.1159/000499138
7. Hegele RA, Pollex RL. Hypertriglyceridemia: phenomics and genomics. Mol Cell Biochem. 2009;326(1–2):35–43. doi:10.1007/s11010-008-0005-1
8. Chyzhyk V, Brown AS. Familial chylomicronemia syndrome: a rare but devastating autosomal recessive disorder characterized by refractory hypertriglyceridemia and recurrent pancreatitis. Trends Cardiovasc Med. 2019;
9. Cox DW, Breckenridge WC, Little JA. Inheritance of apolipoprotein C-II deficiency with hypertriglyceridemia and pancreatitis. N Engl J Med. 1978;299(26):1421–1424. doi:10.1056/NEJM197812282992601
10. D’Erasmo L, Di Costanzo A, Cassandra F, et al. Spectrum of mutations and long-term clinical outcomes in genetic chylomicronemia syndromes. Arterioscler Thromb Vasc Biol. 2019;39(12):2531–2541. doi:10.1161/ATVBAHA.119.313401
11. Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridemia. J Intern Med. 2012;272(2):185–196. doi:10.1111/j.1365-2796.2012.02516.x
12. Chokshi N, Blumenschein SD, Ahmad Z, Garg A. Genotype-phenotype relationships in patients with type I hyperlipoproteinemia. J Clin Lipidol. 2014;8(3):287–295. doi:10.1016/j.jacl.2014.02.006
13. Brahm AJ, Hegele RA. Chylomicronemia-current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11(6):352–362. doi:10.1038/nrendo.2015.26
14. Wolska A, Dunbar RL, Freeman LA, et al. Apolipoprotein C-II: new findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis. 2017;267:49–60. doi:10.1016/j.atherosclerosis.2017.10.025
15. Breckenridge WC, Little JA, Steiner G, Chow A, Poapst M. Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N Engl J Med. 1978;298(23):1265–1273. doi:10.1056/NEJM197806082982301
16. Xiong WJ, Li WH, Posner I, et al. No severe bottleneck during human evolution: evidence from two apolipoprotein C-II deficiency alleles. Am J Hum Genet. 1991;48(2):383–389.
17. Okubo M, Toromanovic A, Ebara T, Murase T. Apolipoprotein C-II Tuzla: a novel large deletion in APOC2 caused by Alu-Alu homologous recombination in an infant with apolipoprotein C-II deficiency. Clin Chim Acta. 2015;438:148–153. doi:10.1016/j.cca.2014.08.022
18. Widholm O, Kantero RL, Axelson E, Johansson ED, Wide L. Endocrine changes before and after the menarche. I. Urinary excretion of estrogen, FSH and LH, and serum levels of progesterone, FSH and LH. Acta Obstet Gynecol Scand. 1974;53(3):197–208. doi:10.3109/00016347409162157
19. Aljenedil S, Hegele RA, Genest J, Awan Z. Estrogen-associated severe hypertriglyceridemia with pancreatitis. J Clin Lipidol. 2017;11(1):297–300. doi:10.1016/j.jacl.2016.12.006
20. Connelly PW, Maguire GF, Little JA. Apolipoprotein CIISt. Michael. Familial apolipoprotein CII deficiency associated with premature vascular disease. J Clin Invest. 1987;80(6):1597–1606. doi:10.1172/JCI113246
21. Jiang J, Wang Y, Ling Y, Kayoumu A, Liu G, Gao X. A novel APOC2 gene mutation identified in a Chinese patient with severe hypertriglyceridemia and recurrent pancreatitis. Lipids Health Dis. 2016;15:12. doi:10.1186/s12944-015-0171-6
22. De Pretis N, Amodio A, Frulloni L. Hypertriglyceridemic pancreatitis: epidemiology, pathophysiology and clinical management. United Eur Gastroenterol J. 2018;6(5):649–655. doi:10.1177/2050640618755002
23. Gelrud A, Whitcomb D. Hypertriglyceridemia-induced acute pancreatitis. In: UpToDate, Friedman L, editor. Waltham: UpToDate Inc; 2019.
24. Gubensek J, Buturovic-Ponikvar J, Romozi K, Ponikvar R, Rakonczay Z. Factors affecting outcome in acute hypertriglyceridemic pancreatitis treated with plasma exchange: an observational cohort study. PLoS One. 2014;9(7):e102748. doi:10.1371/journal.pone.0102748
25. Shirakawa T, Nakajima K, Shimomura Y, et al. Comparison of the effect of post-heparin and pre-heparin lipoprotein lipase and hepatic triglyceride lipase on remnant lipoprotein metabolism. Clin Chim Acta. 2015;440:193–200. doi:10.1016/j.cca.2014.07.020
26. Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. 2019;381(6):531–542. doi:10.1056/NEJMoa1715944
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.