")
Back to Journals » International Medical Case Reports Journal » Volume 9
A newborn with very rare von Voss-Cherstvoy syndrome: a case report
Authors Sharma D, Gupta B, Shastri S, Sharma P
Received 18 March 2016
Accepted for publication 5 May 2016
Published 20 July 2016 Volume 2016:9 Pages 201—205
DOI https://doi.org/10.2147/IMCRJ.S108746
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ronald Prineas
Deepak Sharma,1 Basudev Gupta,2 Sweta Shastri,3 Pradeep Sharma4
1Department of Pediatrics, Pt. Bhagwat Dayal Sharma Post Graduate Institute of Medical Sciences, Rohtak, 2Department of Pediatrics, Civil Hospital, Palwal, Haryana, 3Department of Pathology, N.K.P. Salve Medical College, Nagpur, Maharashtra, 4Department of Medicine, Mahatma Gandhi Medical College and Research Institute, Jaipur, Rajasthan, India
Introduction: von Voss-Cherstvoy syndrome is a part of a group of syndromes with radial and hematologic abnormalities, and until now approximately ten cases have been reported in the literature. This syndrome is characterized by a triad of radial ray defects, occipital encephalocele, and urogenital abnormalities.
Case presentation: We report a neonate from Indian ethnicity who was diagnosed with von Voss-Cherstvoy syndrome. The neonate had radial ray defect, occipital encephalocele, tetralogy of Fallot, and bilateral agenesis of kidney, ureter, and bladder. The neonate was suspected to have von Voss-Cherstvoy syndrome on the basis of clinical features, which was further confirmed by fibroblast analysis showing somatic mosaicism for del(13q).
Conclusion: von Voss-Cherstvoy syndrome is a very rare syndrome that can be suspected on the basis of typical clinical features and confirmed by fibroblast analysis showing somatic mosaicism for del(13q). This adds a second case of this chromosome anomaly described in this syndrome.
Keywords: von Voss-Cherstvoy syndrome, radial ray defects, occipital encephalocele, urogenital abnormalities, somatic mosaicism for del(13q)
Introduction
von Voss-Cherstvoy syndrome is a very rare syndrome that is characterized by a triad of radial ray defects, occipital encephalocele, and urogenital abnormalities.1 The inheritance of this syndrome is thought to be autosomal recessive in the Online Mendelian Inheritance in Man library as few case reports showed consanguinity and the majority of the case reports were sporadic in nature. The inheritance pattern still remains undetermined, and this syndrome has equal sex predilection.2 We report a case of von Voss-Cherstvoy syndrome that was suspected on the basis of clinical features and confirmed by fibroblast analysis showing somatic mosaicism for del(13q). This adds a second case of this chromosome anomaly described in this syndrome, and the chromosome anomaly provides further evidence of at least some cases of von Voss-Cherstvoy syndrome.
Case presentation
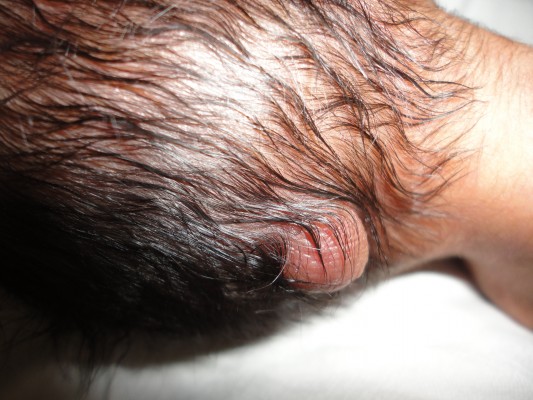
A preterm male child, Indian in origin with a birth weight of 2.4 kg and with an Apgar score of 7, 8, and 8 at 1 minute, 5 minutes, and 10 minutes, respectively, was referred to our hospital at the age of 2 days in view of fracture of femur and malformation. This index case was the first child to a nonconsanguineous couple. The mother had one antenatal scan done, which was suggestive of severe oligohydramnios with an amniotic fluid index of 2.1. The infant was born by a cesarean section with fetal transverse lie with difficult extraction. There was history of energetic traction during delivery. The fracture of femur was thought secondary to energetic traction during the time of birth. The infant was diagnosed antenatally as a case of left subarachnoid cyst with hydrocephalus. There was no history of any drug intake or any substance abuse by the mother. The detailed examination of the infant showed wide-open bulging anterior fontanel, all cranial sutures wide opened, small occipital meningocele (Figure 1), low-set ears, downward slanted eyes, short neck, wide-spaced nipples, cleft palate, anteverted nostrils, bilateral microtia, bilateral absence of thumbs, shortening of both upper limbs with aplasia of both radial deviation and medial deviation of the hands, and single umbilical artery (Figure 2). Examination of the cardiovascular system showed grade 2/6 pansystolic murmur in the left parasternal border. The genital examination showed hydrocele with hypospadias. The baby was evaluated with echocardiography that was suggestive of tetralogy of Fallot, and ultrasound of abdomen and kidney was suggestive of bilateral agenesis of kidney, ureter, and bladder. The ultrasound of the head was suggestive of hydrocephalus with ventricle sizes of 20 mm each and left arachnoid cyst, with agenesis of the corpus callosum. The radiograph of the infant was suggestive of bilateral absence of radius bone, with hypoplastic low-volume lungs (Figure 3). The blood count showed thrombocytopenia with a platelet count of 94,000/mm3. The infant had saturation of 50% with 100% oxygen. The fracture of femur was managed with immobilization of femur as advised by the pediatric orthopedician. The infant expired on the third day after birth as the parents were not willing for aggressive management of the neonate. The karyotype analysis of the lymphocyte showed 46XY pattern. Based on the physical features, the diagnosis of von Voss-Cherstvoy syndrome was kept, and the fibroblast analysis of the skin taken from the left lower limb showed somatic mosaicism for del(13q) that confirmed our diagnosis of von Voss-Cherstvoy syndrome. Written informed consent was obtained from the patient’s legal guardian including publication of images. The ethics committee of Civil Hospital, Palwal does not require ethics approval to be sought for case reports.
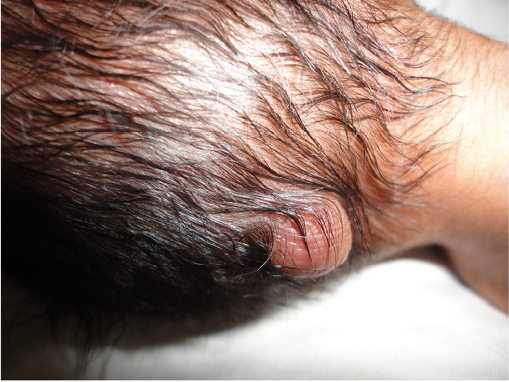 | Figure 1 A photograph showing occipital encephalocele in the present case. |
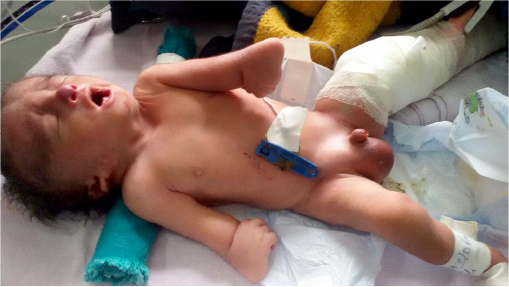 | Figure 2 A photograph showing low-set ears, downward slanted eyes, short neck, wide-spaced nipples, anteverted nostrils, bilateral microtia, bilateral absence of thumbs, and shortening of both upper limbs with aplasia of both radius bone and medial deviation of the hands. |
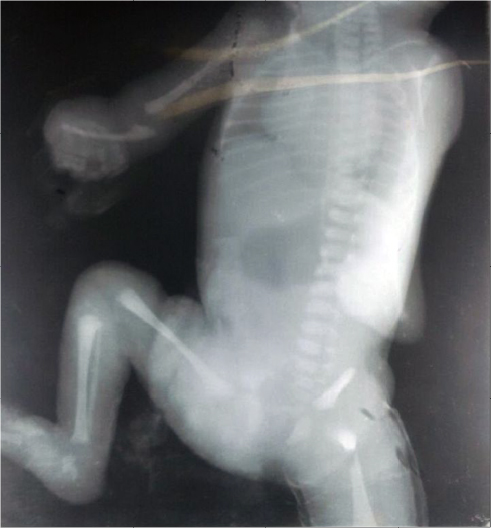 | Figure 3 A radiograph of the infant suggestive of absence of right radius bone. Notes: The left upper limb was rotated and hence the absence of radius could not be seen. In addition, low-volume lung in the index case with five intercostal spaces and fracture of shaft of the femur were noted. |
Discussion
von Voss-Cherstvoy syndrome (Mendelian Inheritance in Man [MIM] 223340) is also known by various other names that include DK phocomelia or phocomelia, thrombocytopenia, encephalocele, and urogenital malformations syndrome. The first case was reported by von Voss et al in 1979, and the syndrome was named after him. They reported a child with phocomelia, meningoencephalocele, and hypoplastic thrombocytopenia, among other findings.1 The second case was reported by Cherstvoy et al,3 and they coined the term DK phocomelia syndrome, which is a misnomer as the limb anomalies are usually radial bone abnormalities. Till date, there are very few case reports in the medical literature that describe this syndrome. The clinical findings and fibroblast analysis of our patient confirmed that our patient had von Voss-Cherstvoy syndrome. The analysis of our patient strengthens the findings of Bamforth and Lin.4 This syndrome is included in the family of syndromes with radial ray and hematologic abnormalities. In all case reports, only the upper limbs are affected with severity ranging from radial agenesis and phocomelia to virtual amelia. This syndrome involves majority of the systems, including the central nervous system, cardiovascular system, respiratory system, hematological system, urogenital system, skeletal system, and gastrointestinal system. The various malformations reported till now have been summarized in Table 1.
 | Table 1 The various findings seen in cases of von Voss-Cherstvoy syndrome Abbreviation: GI, gastrointestinal. |
The close differential diagnoses of von Voss-Cherstvoy syndrome include Fanconi syndrome (MIM 227650),5 Thrombocytopenia Absent Radius syndrome (MIM 274000),6 and Roberts syndrome (MIM 268300).7 The common clinical features of thrombocytopenia and limb defects suggest the possibility of involvement of the homeobox family of genes, as these genes are expressed in both cell lineages. The inheritance pattern has been unknown as all the cases have been sporadic, with no definite inheritance pattern. The somatic mosaicism for del(13)(q12) in the mesoderm lineage has been held responsible for this rare syndrome.4
The children with this syndrome do not die universally immediately after birth because the mortality will depend on the severity of the associated malformation in the case. In the index case, the neonate had bilateral renal agenesis and other system involvement that led to neonatal mortality. The long-term outcome of these patients is elusive because of very few cases reported till now and still there are no conclusive data over the long-term outcome. There have been reports of delayed development and seizures in these patients, but one case report showed transient thrombocytopenia and normal psychomotor development.8 A comparison between our case and the patients reported in the literature is shown in Table 2. A recently published case report described the neurocognitive profile of a young adolescent with von Voss-Cherstvoy syndrome. The 12-year-old male with von Voss-Cherstvoy syndrome underwent comprehensive neuropsychological evaluation. The evaluation showed mild impairment in intellectual functioning, with more significant impairment in adaptive skills and academic achievement. The authors also observed that the neuropsychological profile converged with the neurological findings. There was a distinct pattern of strengths and weaknesses that showed functional compromise of posterior brain regions with relatively well-preserved functioning of more anterior regions. The impairments were more evident in perceptual reasoning, visual perception, and visuomotor integration, whereas normal or near-normal functioning was evident in memory, receptive language, social cognition, attention, and most aspects of executive functioning.12
 | Table 2 Comparison of various manifestations of the von Voss-Cherstvoy syndrome in the different cases reported till now |
In another recently published case report, the authors suggested that the various syndromes that have encephalocele and radial defects in common such as VACTERL association (vertebral anomalies, anal atresia, cardiac defects, tracheoesophageal fistula and/or esophageal atresia, renal and radial anomalies, and limb defects), oculo-auriculo-vertebral spectrum, von Voss-Cherstvoy syndrome, and Edwards syndrome (trisomy 18) should be considered as one entity and proposed the name “Encephalocele–radial, cardiac, gastrointestinal, anal/renal anomalies” or “Froster-Iskenius and Meinecke syndrome”.13 The therapeutic option in our index case could have been going for renal dialysis to take care of the renal system as the neonate had renal agenesis. However, the parents were not willing for aggressive management and hence renal dialysis was not done. A novel aspect in the index case was that we were able to show somatic mosaicism for del(13)(q12) in the fibroblast analysis. Moreover, this is the only second case report that has described similar findings. The findings of our index case add to the hypothesis that somatic mosaicism for del(13)(q12) is responsible for von Voss-Cherstvoy syndrome.
Conclusion
von Voss-Cherstvoy syndrome is a very rare syndrome and involves the central nervous system, cardiovascular system, hematological system, urogenital system, and respiratory system, with various manifestations. The diagnosis requires high index of suspicion, and confirmation must be done by identification of somatic mosaicism for del(13q) using the fibroblast analysis.
Acknowledgments
My (Dr Deepak Sharma) parents Shri Keshave Dev Sharma and Smt Rajkumari Sharma have been a great source of hard work and sincerity.
Disclosure
The authors report no conflicts of interest in this work.
References
von Voss H, Kramer HH, Gobel U, Kemperdick H. Phokomelie, Meningoenzephalozele und hypoplastische Thrombocytopenie, ein neues Syndrom? Klinische Genetik in der Pädiatrie I. Symposion Kiel, Juli 1978. Stuttgart: George Thieme; 1979:70–74. | ||
Lubinsky MS, Kahler SG, Speer IE, Hoyme HE, Kirillova IA, Lurie IW. Von Voss-Cherstvoy syndrome: a variable perinatally lethal syndrome of multiple congenital anomalies. Am J Med Genet. 1994;52(3):272–278. | ||
Cherstvoy E, Lazjuk G, Lurie I, Ostrovskaya T, Shved I. Syndrome of multiple congenital malformations including phocomelia, thrombocytopenia, encephalocele, and urogenital abnormalities. Lancet. 1980;2(8192):485. | ||
Bamforth JS, Lin CC. DK phocomelia phenotype (von Voss-Cherstvoy syndrome) caused by somatic mosaicism for del(13q). Am J Med Genet. 1997;73(4):408–411. | ||
Chung NG, Kim M. Current insights into inherited bone marrow failure syndromes. Korean J Pediatr. 2014;57(8):337–344. | ||
Greenhalgh KL, Howell RT, Bottani A, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. 2002;39(12):876–881. | ||
Rodríguez JI, Palacios J, Urioste M, Rodriguez-Peralto JL. Tetra-phocomelia with multiple malformations: X-linked amelia, or Roberts syndrome, or DK-phocomelia syndrome? Am J Med Genet. 1992;43(3):630–632. | ||
Brunetti-Pierri N, Mendoza-Londono R, Shah MR, Karaviti L, Lee B. von Voss-Cherstvoy syndrome with transient thrombocytopenia and normal psychomotor development. Am J Med Genet A. 2004;126A(3):299–302. | ||
Bird LM, Newbury RO, Ruiz-Velasco R, Jones MC. Recurrence of diaphragmatic agenesis associated with multiple midline defects: evidence for an autosomal gene regulating the midline. Am J Med Genet. 1994;53(1):33–38. | ||
Urioste M, Paisán L, Martínez-Frías ML. DK-phocomelia syndrome in a child with a long follow-up. Am J Med Genet. 1994;52(3):269–271. | ||
Managoli S, Chaturvedi P, Vilhekar KY, Kumar S. Von Voss-Cherstvoy syndrome. Indian Pediatr. 2002;39(10):967–969. | ||
Antonini TN, Van Horn Kerne V, Axelrad ME, Karaviti LP, Schwartz DD. Neurocognitive profile of a young adolescent with DK phocomelia/von Voss phocomelia/von Voss Cherstvoy syndrome. Am J Med Genet A. 2015;167(7):1632–1636. | ||
Valdez CM, Altmayer SP, Barrow MA, et al. Encephalocele-radial, cardiac, gastrointestinal, anal/renal anomalies: novel evidence for a new condition? Am J Med Genet A. 2014;164A(5):1085–1091. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.