Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
A “turn-on” fluorescent microbead sensor for detecting nitric oxide
Received 25 September 2014
Accepted for publication 18 November 2014
Published 19 December 2014 Volume 2015:10(1) Pages 115—123
DOI https://doi.org/10.2147/IJN.S74924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Lan-Hee Yang,1,2 Dong June Ahn,3 Eunhae Koo1
1Advanced Materials Convergence Division, Korea Institute of Ceramic Engineering and Technology, Seoul, Republic of Korea; 2Department of Biomicrosystem Technology, Korea University, Seoul, Republic of Korea; 3Departments of Biomicrosystem Technology, Chemical & Biological Engineering, KU-KIST Graduate School, Korea University, Seoul, Republic of Korea
Abstract: Nitric oxide (NO) is a messenger molecule involved in numerous physical and pathological processes in biological systems. Therefore, the development of a highly sensitive material able to detect NO in vivo is a key step in treating cardiovascular and a number of types of cancer-related diseases, as well as neurological dysfunction. Here we describe the development of a fluorescent probe using microbeads to enhance the fluorescence signal. Microbeads are infused with the fluorophore, dansyl-piperazine (Ds-pip), and quenched when the fluorophore is coordinated with a rhodium (Rh)-complex, ie, Rh2(AcO-)4(Ds-pip). In contrast, they are able to fluoresce when the transition-metal complex is replaced by NO. To confirm the “on/off” mechanism for detecting NO, we investigated the structural molecular properties using the Fritz Haber Institute ab initio molecular simulations (FHI-AIMS) package. According to the binding energy calculation, NO molecules bind more strongly and rapidly with the Rh-core of the Rh-complex than with Ds-pip. This suggests that NO can bond strongly with the Rh-core and replace Ds-pip, even though Ds-pip is already near the Rh-core. However, the recovery process takes longer than the quenching process because the recovery process needs to overcome the energy barrier for formation of the transition state complex, ie, NO-(AcO-)4-(Ds-pip). Further, we confirm that the Rh-complex with the Ds-pip structure has too small an energy gap to give off visible light from the highest unoccupied molecular orbital/lowest unoccupied molecular orbital energy level.
Keywords: nitric oxide, microbead, fluorescence, rhodium complex, ab initio molecular simulation
Introduction
Since nitric oxide (NO) was identified as an endothelium-derived relaxation factor in 1987,1,2 NO has become of interest in many scientific and technological areas. For instance, NO is now known to be a ubiquitous messenger molecule in the cardiovascular, nervous, and immune systems, and is a major factor in tumor progression.3–12 Therefore, detection and manipulation of the NO concentration in biological systems is crucial in treating cardiovascular and many types of cancer-related diseases, as well as neurological dysfunction. However, because NO is highly reactive, it is rapidly and easily converted to other species,6,13 making it very difficult to detect and monitor concentrations in vivo.
Much effort has gone into developing efficient methods for detection of NO, including electrochemicals, electron paramagnetic resonance spectroscopy, chemiluminescence, and fluorescence detection.14–19 Compared with other methods, fluorescence detection can provide highly spatial and temporal data on the distribution of both intracellular and extracellular NO. Lippard et al have been developing reversible, fluorescence-based NO detection adducts using fluorophores coordinated with metal-complex scaffolds.20 However, because the fluorophore adducts do not function as NO sensors in aqueous medium, a silastic polymer-based membrane was used as a NO-permeable membrane. One strategy for enhancing the ability to detect NO in aqueous solution is to use microbead-based probes with a surface layer that serves as a barrier membrane to protect against H2O molecules.
In this paper, we describe fluorescence-based “turn-on/off” microbeads infused with the fluorophore adduct, Rh2(AcO−)4(Ds-pip), consisting of dansyl-piperazine (Ds-pip) and rhodium (II) acetate Rh2(AcO−)4. As shown in Figure 1, microbeads containing Ds-pip lose their fluorescence when the Rh-acetate-complex is coordinated with Ds-pip. However, the fluorescence from Ds-pip is recovered when Ds-pip is separated from Rh-core by NO molecule. As a result, the microbeads give off fluorescent light. Because the microbeads can easily be infused with a high concentration of the fluorophore adduct, they show strong fluorescence at a low NO concentration. The microbeads are spherical and composed of mesoporous silica with a pore size of 20–30 nm. In comparison with nonporous beads, the encoded, porous beads showed 100 times brighter and five times more uniform fluorescence signals.21,22 The surface of the microbead is modified by a layer of octadecyl silane, which serves as a NO-permeable membrane, sequestering the fluorophore adduct from water. In addition, to elicit the “turn-on/off” mechanism related to the associative or dissociative reactions between the dirhodium complex, fluorophore, and NO, we performed ab initio computational calculations using the Fritz Haber Institute ab initio molecular simulations (FHI-AIMS) package to calculate the binding energy and highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) energy levels of practicable molecular structures, in this case [Rh2(AcO−)4(Ds-pip)x(NO)y].
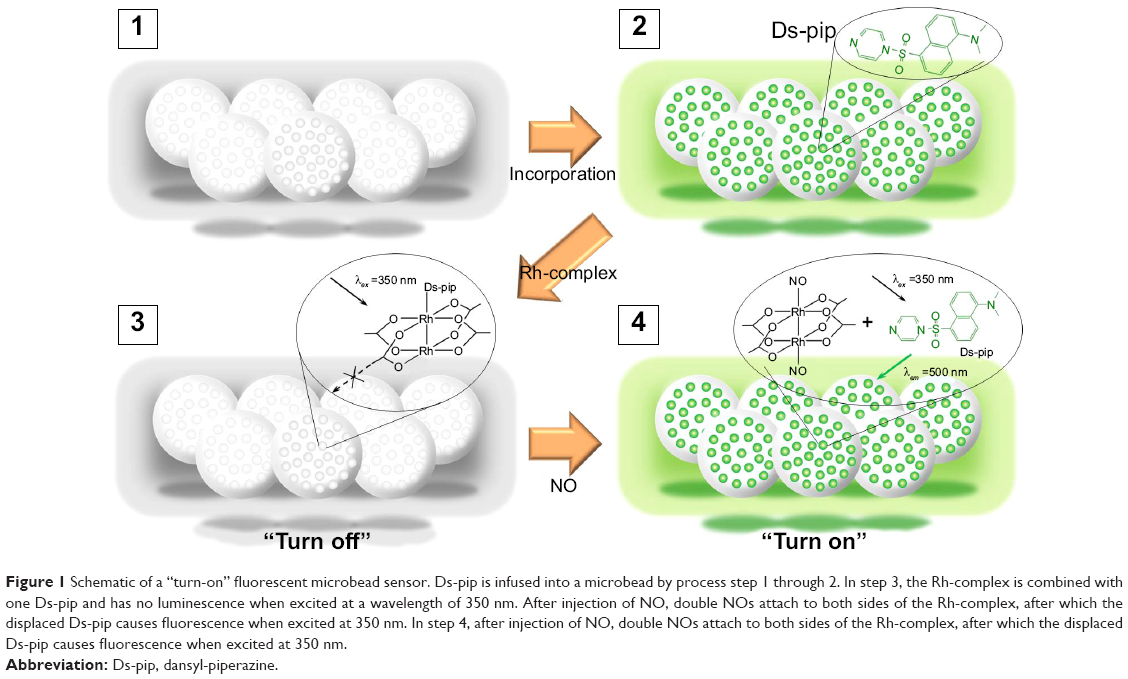 | Figure 1 Schematic of a “turn-on” fluorescent microbead sensor. Ds-pip is infused into a microbead by process step 1 through 2. In step 3, the Rh-complex is combined with one Ds-pip and has no luminescence when excited at a wavelength of 350 nm. After injection of NO, double NOs attach to both sides of the Rh-complex, after which the displaced Ds-pip causes fluorescence when excited at 350 nm. In step 4, after injection of NO, double NOs attach to both sides of the Rh-complex, after which the displaced Ds-pip causes fluorescence when excited at 350 nm. |
Materials and methods
Synthesis of materials
In order to synthesize the microbeads23–25, a triblock copolymer Pluronic P123 (4 g, BASF, Ludwigshafen, Germany) was dissolved in acidic solution (10 mL of HCl and 65 mL of H2O), after which 4 g of 1, 3, 5-trimethylbenzene were added. Dissolved P-123 forms micelles that can be used as a backbone to make structured mesoporous materials. The reaction mixtures were then heated to 37°C–40°C and stirred for 2 hours. Next, 9.2 mL of tetraethoxysilane were put into the reaction mixtures and stirred for 5 minutes. The reaction mixtures were ripened at 40°C for 24 hours in an autoclave. Next, 46 mg of NH4F was added to the reaction mixtures and ripened at 100°C for 24 hours. Finally, to obtain spherical mesoporous silica, the precipitates were calcinated at 550°C for 6 hours in air. Ds-pip was prepared by adding dansyl chloride solution (18.5 mmol, 10 mL) in dichloroethane dropwise to a solution of piperazine (111 nmol, 20 mL) for 5 minutes. After stirring for 15 minutes, the reaction mixture was washed with water and Na2SO4 five times. Finally, the Ds-pip was separated using rotary evaporation.
Incorporation, quenching, and surface derivatization
Microbeads infused with Ds-pip were obtained by adding 2 mL of 100 μM Ds-pip solution in dichloroethane to 0.143 g of mesoporous silica beads dispersed in 20 mL of toluene. After 12 hours of stirring, the mixture was centrifuged to obtain the Ds-pip-infused microbeads. In order to quench the fluorescent microbeads, Rh-acetate-dimer solution in 5 μM, 10 μM, and 20 μM dichloroethane was added to a 10 μM solution of incorporated microbeads. Finally, 0.03 mL of octadecylsilane was added and the surfaces of the microbeads were modified in hexane over 6 hours.
Characterization
The microstructure of the mesoporous microbeads was investigated using a field emission scanning electron microscope (Tecnai G2 F30 S-twin, 300 kV, FEI, Hillsboro, OR, USA) and a transmission electron microscope (JEOL 6300F, JEOL, Japan). The solution incorporating the microbeads was characterized using a fluorescence spectrometer (FluoroMate FS-2, Scinco Co, Seoul, Republic of Korea) and a fluorescence microscope (IX-71, Olympus, Tokyo, Japan).
Results and discussion
Figure 1 describes a microbead sensor for detecting NO. Ds-pip is infused into a microbead by process step 1 through 2. In step 3, the Rh-complex coordinates with one Ds-pip, which has no luminescence when excited at a wavelength of 350 nm. After the injection of NO, NO attached to both sides of the Rh-complex, after which Ds-pip separated from Ds-pip-Rh-complex gave off a fluorescence signal when excited at 350 nm. Transmission electron micrographs show spherical microbeads with a pore size of 20–30 nm, as shown in Figure 2. The microbeads were prepared by injecting the Ds-pip solution dispersed in dichloroethane into porous silica beads in toluene. As shown in Figure 3A and B, the microbeads infused with Ds-pip show a significant fluorescence signal in the photoluminescence spectra and in the fluorescence microscopy image. In contrast, there was no fluorescence signal in the supernatant solution (red line). This indicates that most of the Ds-pip entered into the silica beads. Compared with the peak in the fluorescence spectra at about 530 nm in Figure 3A, it should be noted that the peak for the microbeads is about 510 nm in Figures 4A and 5A. In Figure 3A, the red-shift fluorescence emission from about 510 nm to 530 nm may be because of aggregation of the fluorophore in the pore of the mesoporous silica.
 | Figure 2 Electron microscopy images of spherical mesoporous silica beads on (A) field emission scanning electron microscopy and (B) transmission electron microscopy. |
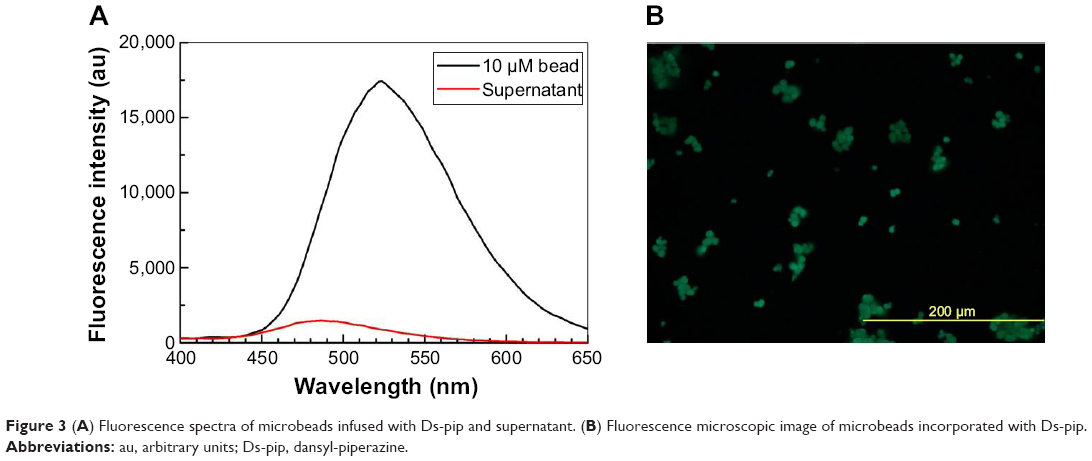 | Figure 3 (A) Fluorescence spectra of microbeads infused with Ds-pip and supernatant. (B) Fluorescence microscopic image of microbeads incorporated with Ds-pip. |
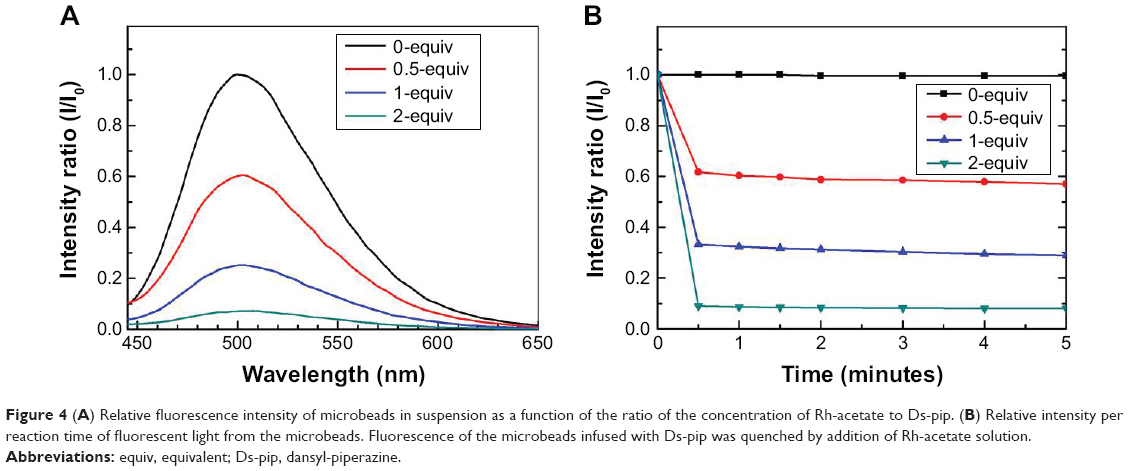 | Figure 4 (A) Relative fluorescence intensity of microbeads in suspension as a function of the ratio of the concentration of Rh-acetate to Ds-pip. (B) Relative intensity per reaction time of fluorescent light from the microbeads. Fluorescence of the microbeads infused with Ds-pip was quenched by addition of Rh-acetate solution. |
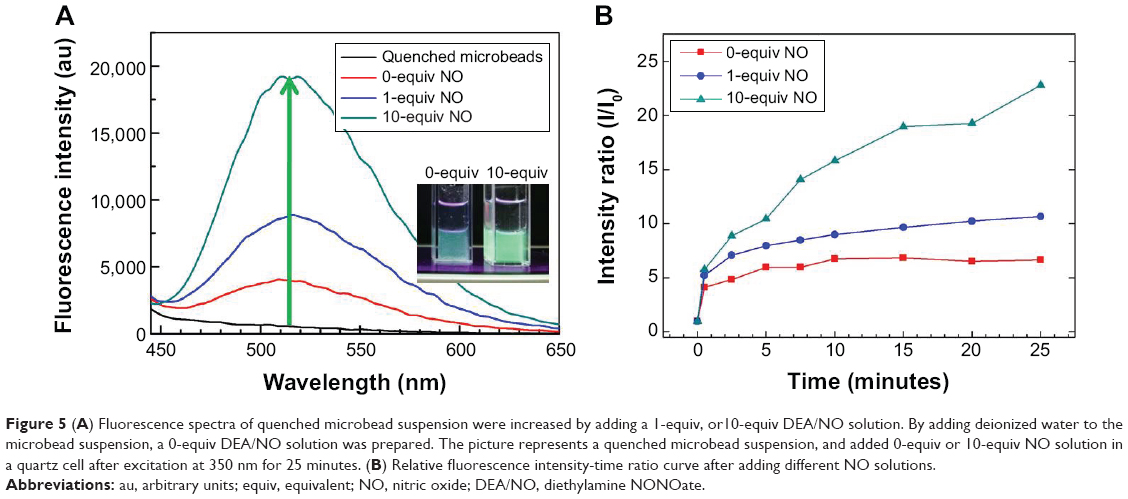 | Figure 5 (A) Fluorescence spectra of quenched microbead suspension were increased by adding a 1-equiv, or10-equiv DEA/NO solution. By adding deionized water to the microbead suspension, a 0-equiv DEA/NO solution was prepared. The picture represents a quenched microbead suspension, and added 0-equiv or 10-equiv NO solution in a quartz cell after excitation at 350 nm for 25 minutes. (B) Relative fluorescence intensity-time ratio curve after adding different NO solutions. |
Figure 3 shows the relative intensity of the fluorescence signal upon adding different amounts of the Rh-complex in dichloroethane solution. Dansyl molecules can be easily coordinated at the axial site of the dirhodium core of the Rh-complex via the nitrogen atoms in piperazine. In the quenching process shown in Figure 4A and B, the relative fluorescence intensities with the control solution (0-equivalent [equiv] Rh-complex, no adding Rh-complex) decrease rapidly from 60% to 10% within a minute when 0.5-equiv, 1-equiv, and 2-equiv Rh-complex is added. H2O molecules in NO solution can bond to rhodium ions more strongly than Ds-pip. This would negatively affect detection of NO, so the surface of the microbeads was modified with octadecylsilane. By the surface derivative reaction of octadecylsilane with long alkyl chains, the surface of mesoporous silica beads with hydroxyl groups can be easily changed from hydrophilic to hydrophobic. The surface polymer-network layer of octadecylsilane serves as a membrane, which can allow NO molecules to pass through and stop H2O molecules.
To investigate the recovery process for detecting NO, deionized water (0-equiv), and 10 μM (1-equiv) and 100 μM (10-equiv) solutions of diethylamine NONOate (DEA/NO, a 2-(N,N-diethylamino)-diazenolate-2-oxide diethylammonium salt) were added to a solution containing microbeads incorporated with 10 μM of Ds-pip-Rh-complex. DEA/NO is a complex of diethylamine and NO, and is used to generate controlled release of NO in solution. The fluorescence intensity increased because of the binding energy of the Rh-complex with NO being stronger than that of the Rh-complex with Ds-pip. Figure 5A shows the fluorescence spectra and an image of the microbeads in solution-upon adding different amounts of DEA/NO solution as much as 0-equiv, 1-equiv, and 10-equiv of Ds-pip. Figure 5B shows the relative fluorescence intensity as a function of the recovery time for the quenched microbeads in solution. When 10-equiv of NO were added, the fluorescence signal of the microbeads reached a maximum in 25 minutes, and was about 23 times stronger than that of the initial fluorescence.
Bases on Figures 4 and 5, it should be noted that the recovery process takes longer than the quenching process. This is because the recovery process needs to overcome the energy barrier for formation of the transition state complex, NO-(AcO−)4-(Ds-pip), as shown in detail in Figure 6. The quenching process is one-step bimolecular reaction between the Rh-complex and Ds-pip, while the recovery process is carried out by two-step reaction. The first step is the reaction in which the quenched (AcO−)4-(Ds-pip) becomes the transition state complex, NO-(AcO−)4-(Ds-pip), by addition of NO. The second step is the reaction in which Ds-pip is separated from the transition state complex. Considering the binding energy, the first step in the recovery process is to overcome the energy barrier for formation of the transition state complex, ie, NO-(AcO−)4-(Ds-pip).
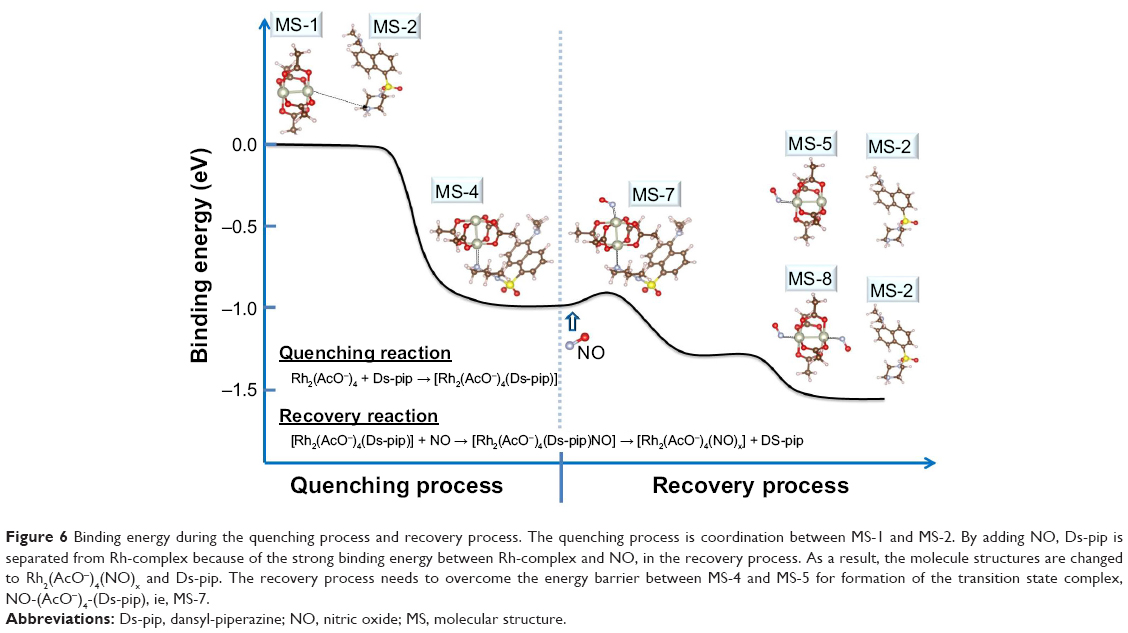 | Figure 6 Binding energy during the quenching process and recovery process. The quenching process is coordination between MS-1 and MS-2. By adding NO, Ds-pip is separated from Rh-complex because of the strong binding energy between Rh-complex and NO, in the recovery process. As a result, the molecule structures are changed to Rh2(AcO−)4(NO)x and Ds-pip. The recovery process needs to overcome the energy barrier between MS-4 and MS-5 for formation of the transition state complex, NO-(AcO−)4-(Ds-pip), ie, MS-7. |
In addition, to separate the microbeads completely from the H2O molecules in DEA/NO solution and to reduce the response time by increasing the direct reaction area, the microbeads were put on glass and separated with a polyethylene membrane, as shown in Figure 7A. When DEA/NO solution was dropped onto the isolator, only NO molecules could pass through the membrane and react with the microbeads. Therefore, the microbeads on the glass turn out to be a bright-green image as “turned on” state by the addition of DEA/NO solution. As a result, the relative intensity increased up to 52% in 10 minutes as shown as Figure 7B. Compared with addition of 0-equiv of NO solution, (Figure 7C), a stronger fluorescence signal is obtained on adding 10-equiv of NO solution to microbeads in 10 μM of Ds-pip-Rh-complex (Figure 7D).
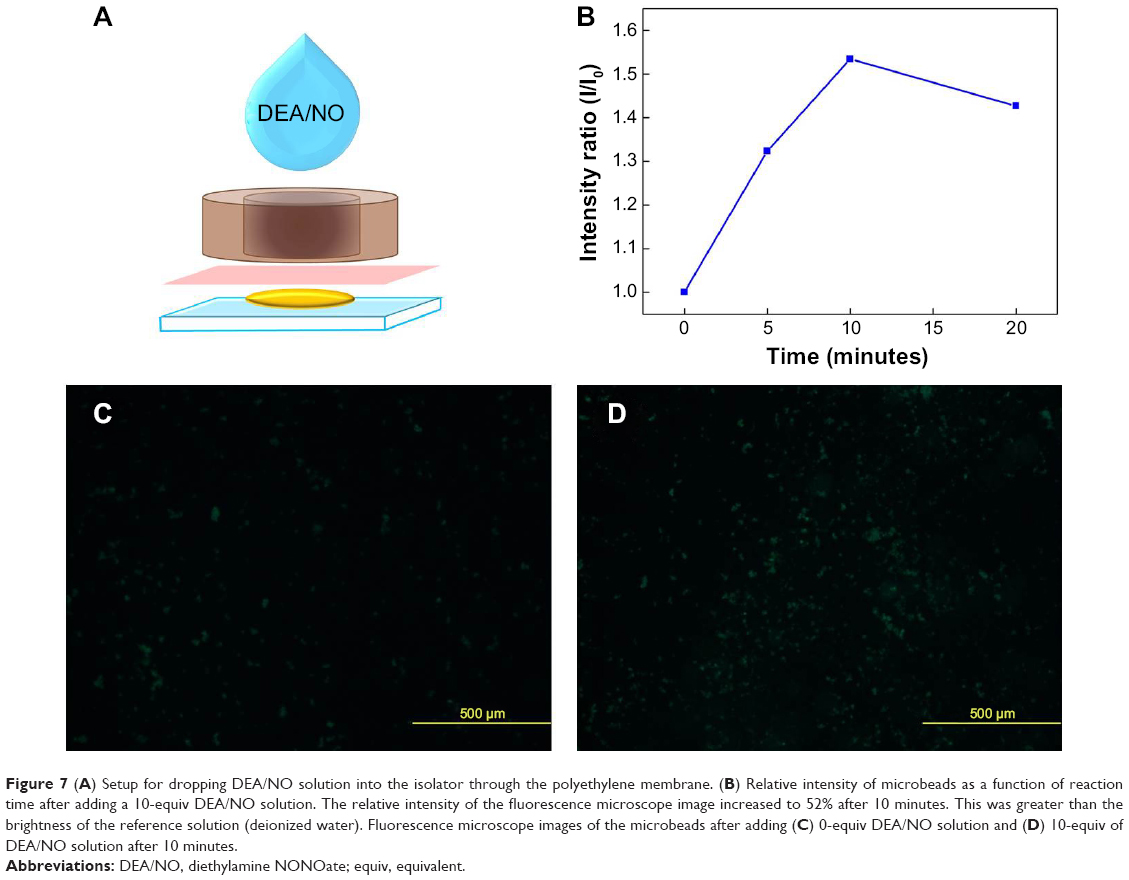 | Figure 7 (A) Setup for dropping DEA/NO solution into the isolator through the polyethylene membrane. (B) Relative intensity of microbeads as a function of reaction time after adding a 10-equiv DEA/NO solution. The relative intensity of the fluorescence microscope image increased to 52% after 10 minutes. This was greater than the brightness of the reference solution (deionized water). Fluorescence microscope images of the microbeads after adding (C) 0-equiv DEA/NO solution and (D) 10-equiv of DEA/NO solution after 10 minutes. |
To confirm the proposed “on/off” mechanism and the experimental results, we investigated the structural molecular properties for various compositions using the FHI-AIMS package, ie, all-electron electronic structure theory with a numeric atom-centered basis function and coordinating different numbers of ligands of Ds-pip and NO with the Rh-complex Rh2(AcO−)4(Ds-pip)x(NO)y. We used density function theory with an all-electron full-potential scheme and “tight” numerical atom-centered orbitals. When Rh-complex molecular structure (MS)-1 bonded with Ds-pip MS-2, a no-fluorescence signal was the “turned-off” state [MS-4]. However, when Ds-pip was knocked out of the Rh-complex upon injection of NO, the intensity of fluorescence emission increased.
Figure 8 shows the molecular structures with binding energy (Eb) between the MS-1 Rh-complex [Rh2(AcO−)4] and ligands (MS-2 Ds-pip or MS-3 NO). The binding energy was obtained by subtracting the energy of the Rh-complex (ERh) and ligands (Eligand), ie, either Ds-pip (EDs-pip) or NO (ENO), from the total energy (Eb = [Etot − ERh − Eligand]/N). Also, the energy was divided by the number of ligands (N) in order to identify the binding energy for each case when the number of ligands was different.
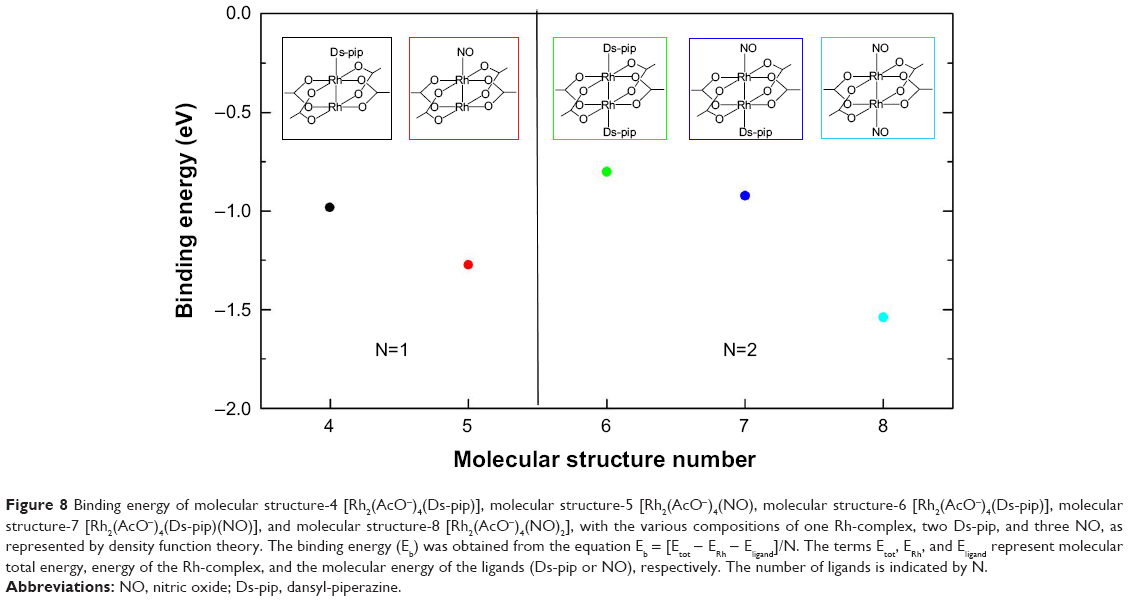 | Figure 8 Binding energy of molecular structure-4 [Rh2(AcO−)4(Ds-pip)], molecular structure-5 [Rh2(AcO−)4(NO), molecular structure-6 [Rh2(AcO−)4(Ds-pip)], molecular structure-7 [Rh2(AcO−)4(Ds-pip)(NO)], and molecular structure-8 [Rh2(AcO−)4(NO)2], with the various compositions of one Rh-complex, two Ds-pip, and three NO, as represented by density function theory. The binding energy (Eb) was obtained from the equation Eb = [Etot − ERh − Eligand]/N. The terms Etot, ERh, and Eligand represent molecular total energy, energy of the Rh-complex, and the molecular energy of the ligands (Ds-pip or NO), respectively. The number of ligands is indicated by N. |
Based on the binding energy, MS-5 [Rh2(AcO−)4(NO)] and MS-8 [Rh2(AcO−)4(Ds-pip)/(NO)2], when bonded with NO, were more stable than the other structures bonded with Ds-pip. In the cases of structures bonding with Ds-pip, MS-4 [Rh2(AcO−)4(Ds-pip)] was more stable than MS-6 [Rh2(AcO−)4(Ds-pip)2] and MS-7 [Rh2(AcO−)4(Ds-pip)/(NO)], although MS-7 was coordinating with NO. Therefore, we would expect that Rh-complexes would assume the shape of MS-4 when combined with Ds-pip. For existing NO, MS-8 would be most stable due to its strong binding energy. It should also be noted that the distance of Rh-N bonding in MS-8 (1.935Å) appears to be shorter than that of MS-6 (2.35Å) due to its weaker binding energy. These are approximate calculation results, using the experimental data, for distance between the Rh ions and nitrogen donor ligands.26,27
Consequently, we could explain the quenching and recovery process by the reaction coordinate versus the binding energy as shown in Figure 6. First, in the quenching process, as MS-1 approaches MS-2, MS-1 and MS-2 are coordinated spontaneously to MS-4 with the dirhodium core of MS-1 via nitrogen atoms in the piperazine of MS-2. However, the recovery process needs to overcome the energy barrier between MS-4 and MS-5 upon adding the NO for formation of the transition state complex, NO-(AcO−)4-(Ds-pip), ie, MS-7. Finally, MS-7 becomes Rh2(AcO−)4(NO)x and Ds-pip spontaneously because of the strong binding energy of Rh-N.
In addition, the emission wavelength was calculated using the energy gap indicated by HOMO/LUMO to support the proposed “on-off” mechanism. Figure 9 shows the energy band gap and emission wavelengths from the possible structures. As shown in Figure 9, MS-4 and MS-6 (with Ds-pip) had energy gaps of 1.64 eV (755 nm) and 1.5 eV (814 nm), respectively, and the wavelength of the emitted light was in the infrared region. This explains why Ds-pip with Rh-complexes were in the “turned-off” state, emitting no visible light. However, when NO was added, the molecular structures became MS-5, MS-7, MS-8, and MS-2, which had emission wavelengths of 0 nm, 0 nm, 964 nm, and 483 nm. Therefore, the calculations suggest that the strong binding energy of NO with the Rh-complex causes separation of Ds-pip from the Rh-complex core, and gives off visible green light coming from Ds-pip.
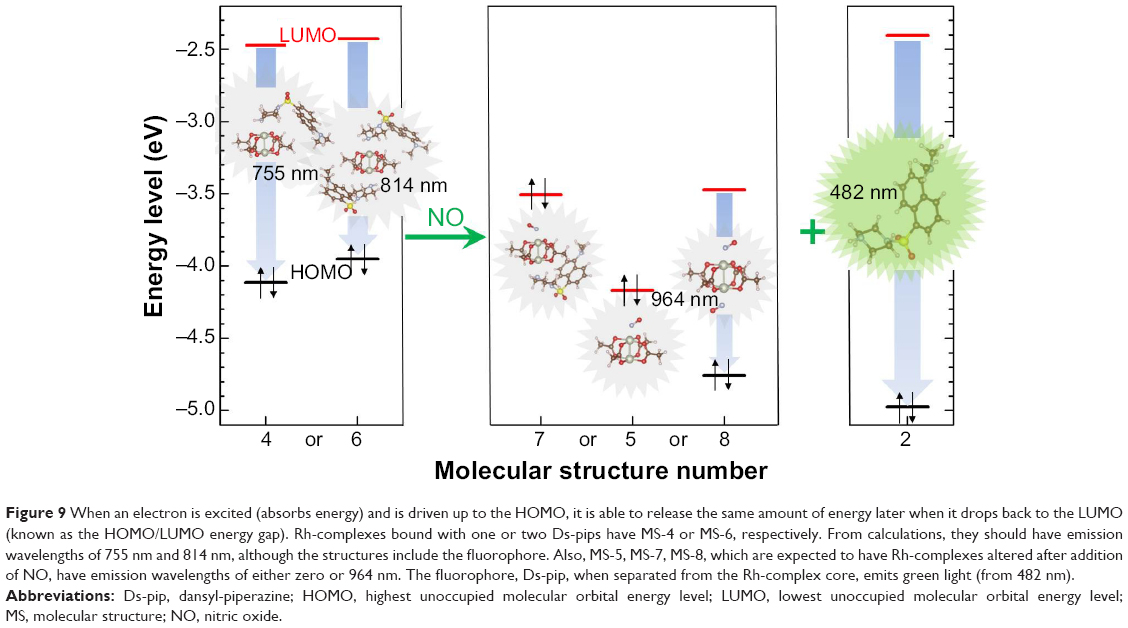 | Figure 9 When an electron is excited (absorbs energy) and is driven up to the HOMO, it is able to release the same amount of energy later when it drops back to the LUMO (known as the HOMO/LUMO energy gap). Rh-complexes bound with one or two Ds-pips have MS-4 or MS-6, respectively. From calculations, they should have emission wavelengths of 755 nm and 814 nm, although the structures include the fluorophore. Also, MS-5, MS-7, MS-8, which are expected to have Rh-complexes altered after addition of NO, have emission wavelengths of either zero or 964 nm. The fluorophore, Ds-pip, when separated from the Rh-complex core, emits green light (from 482 nm). |
Conclusion
In conclusion, we developed a fluorescence “on-off” signal ensemble system for sensing NO, based on a combination of Ds-pip and the Rh-complex, Rh2(AcO−)4(Ds-pip). Emission of fluorescence from Ds-pip is quenched when coordinated with the transition-metal complex, but is restored when the transition-metal complex is replaced by NO. Also, the Ds-pip-Rh-complex combination is infused in mesoporous silica bead, and the surface of the microbead is modified with octadecylsilane, that serves as a NO-permeable membrane. To confirm these experimental results, possible structures for a variety of similar compositions, eg, Rh2(AcO−)4(Ds-pip)x(NO)y, were investigated by means of computational calculations using the FHI-AIMS package. Based on these calculations, we confirm that NO molecules bond with rhodium in the Rh-complex core more strongly than does Ds-pip. In addition, the Rh2(AcO−)4(Ds-pip) molecular structure has too small an energy gap to give off visible light based on the HOMO/LUMO energy level.
Acknowledgments
This study was supported by a grant from the Fundamental R&D Program for Core Technology of Materials and the R&D Program for Technology of World Premier Materials funded by the Ministry of Trade, Industry and Energy, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. | ||
Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. | ||
Furchgott RF. Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Angew Chem Int Ed. 1999; 38:1870–1880. | ||
Ignarro LJ. Nitric oxide: a unique endogenous signaling molecule in vascular biology. Angew Chem Int Ed. 1999;38:1882–1892. | ||
Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998; 19:711–721. | ||
Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. | ||
Kerwin JF Jr, Lancaster JR Jr, Feldman PL. Nitric oxide: a new paradigm for second messengers. Med Chem. 1995;38:4343–4362. | ||
Feldman PL, Griffith OW, Stuehr DJ. The surprising life of nitric oxide. J Chem Eng News. 1993;71:26–38. | ||
Bredt DS, Snyder SH. Annu Rev Biochem. Nitric oxide: a physiologic messenger molecule. 1994;63:175–195. | ||
Murad F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Angew Chem Int Ed. 1999;38:1856–1868. | ||
Butler AR, Williams DLH. The physiological role of nitric oxide. Chem Soc Rev. 1993;22:233–241. | ||
Rubbo H, Darley-Usmar V, Freeman BA. Nitric oxide regulation of tissue free radical injury. Chem Res Toxicol. 1996;9:809–820. | ||
Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. | ||
Nagano T, Yoshimura T. Bioimaging of nitric oxide. Chem Rev. 2002; 102:1235–1270. | ||
Bedioui F, Villeneuve N. Electrochemical nitric oxide sensors for biological samples: principle, selected examples and application. Electroanalysis. 2003;15:5–18. | ||
Fujii S, Yoshimura T. A new trend in iron-dithiocarbamate complexes: as an endogenous NO trapping agent. Coord Chem Rev. 2000;198:89–99. | ||
Brien JF, McLaughlin BE, Nakatsu K, Marks GS. Chemiluminescence headspace-gas analysis for determination of nitric oxide formation in biological systems. Methods Enzymol. 1996;268:83–92. | ||
Franz KJ, Singh N, Lippard SJ. Metal-based NO sensing by selective ligand dissociation. Angew Chem Int Ed. 2000;39:2120–2122. | ||
Katayama Y, Soh N, Maeda M. Strategies and development of molecular probes for nitrogen monoxide monitoring. Bull Chem Soc Jpn. 2002; 75:1681–1691. | ||
Hilderbrand SA, Lim MH, Lippard SJ. Dirhodium tetracarboxylate scaffolds as reversible fluorescence-based nitric oxide sensors. J Am Chem Soc. 2004;126:4972–4978. | ||
Gao X, Nie S. Quantum dot-encoded mesoporous beads with high brightness and uniformity: rapid readout using flow cytometry. Anal Chem. 2004;76:2406–2410. | ||
Sathe TR, Agrawal A, Nie S. Mesoporous silica beads embedded with semiconductor quantum dots and iron oxide nanocrystals: dual-function microcarriers for optical encoding and magnetic separation. Anal Chem. 2006;78:5627–5632. | ||
Lee D-S, Lee J-C, Koo E-H. CdSe/Zns-QD incorporated microbeads for ultra-sensitive chemo-sensor applications. J Korean Phys Soc. 2010;57:1111–1114. | ||
Yan J, Estevez C, Smith JE, et al. Dye-doped nanoparticles for bioanalysis. Nano Today. 2007;2:44–50. | ||
Zhao X, Tapec-Dytioco R, Tan W. Ultrasensitive DNA detection using highly fluorescent bioconjugated nanoparticles. J Am Chem Soc. 2003;125:11474. | ||
Koh YB, Christoph GG. Metal-metal bonding in dirhodium tetracarboxylates. Structure of the bis(pyridine) adduct of tetra-.mu.-acetato-dirhodium(II). Inorganic Chemistry. 1978;17:9. | ||
Cotton FA, Walton RA. Multiple Bonds Between Metal Atoms. 2nd ed, Oxford, UK: Oxford University Press; 1993. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.