Back to Journals » Journal of Pain Research » Volume 14
A KCa3.1 Channel Opener, ASP0819, Modulates Nociceptive Signal Processing from Peripheral Nerves in Fibromyalgia-Like Pain in Rats
Authors Takeshita N, Oe T ![]() , Kiso T, Kakimoto S
, Kiso T, Kakimoto S
Received 30 July 2020
Accepted for publication 20 November 2020
Published 12 January 2021 Volume 2021:14 Pages 23—34
DOI https://doi.org/10.2147/JPR.S274563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Nobuaki Takeshita, Tomoya Oe, Tetsuo Kiso, Shuichiro Kakimoto
Drug Discovery Research, Astellas Pharma Inc, Ibaraki, Japan
Correspondence: Nobuaki Takeshita
Drug Discovery Research, Astellas Pharma Inc., 21 Miyukigaoka, Tsukuba, Ibaraki 305-8585, Japan
Tel +81-29-852-1111
Fax +81-29-850-5120
Email [email protected]
Purpose: Although abnormal peripheral and central pain processing has been observed in fibromyalgia (FM) patients, the biomechanics and pathophysiology, surrounding the peripheral mechanism are not well understood. An intermediate conductance channel, KCa 3.1, is expressed in peripheral sensory nerve fibers where it maintains the resting membrane potential and controls nerve firing, making it a plausible target for peripheral therapeutic interventions. ASP0819, a KCa 3.1 channel opener, is an orally available molecular entity and is used in this investigation to elucidate the role of KCa 3.1 in signal processing of pain in FM.
Methods: Human or rat KCa 3.1 channel-expressing cells were used for evaluating the main action of the compound. Effects of the compound on withdrawal behavior by mechanical stimulation were examined in reserpine-induced myalgia (RIM) and vagotomy-induced myalgia (VIM) models of rats. In addition, in vivo electrophysiological analysis was performed to examine the peripheral mechanisms of action of the compound. Other pain models were also examined.
Results: ASP0819 increased the negative membrane potential in a concentration-dependent manner. Oral administration of ASP0819 significantly recovered the decrease in muscle pressure threshold in rat FM models of RIM and VIM. The in vivo electrophysiological experiments showed that Aδ- and C-fibers innervating the leg muscles in the RIM model demonstrated increased spontaneous and mechanically evoked firing compared with normal rats. Intravenous infusion of ASP0819 significantly reduced both the spontaneous activity and mechanically evoked responses in Aδ-fibers in the rat RIM model. ASP0819 significantly reduced the number of abdominal contractions as an indicator of abdominal pain behaviors in the rat visceral extension model and withdrawal responses in the osteoarthritis model, respectively.
Conclusion: These findings suggest that ASP0819 may be a promising analgesic agent with the ability to modulate peripheral pain signal transmission. Its use in the treatment of several pain conditions should be explored, chief amongst these being FM pain.
Keywords: potassium channel, fibromyalgia, pain
Introduction
Fibromyalgia (FM) is a complex syndrome characterized by chronic widespread musculoskeletal pain that often occurs alongside symptoms of depression, fatigue, sleep disturbances, and/or cognitive complaints.1 Pain in FM typically includes deep musculoskeletal pain with tender points in the shoulder, girdle, torso, hips, and extremities. FM may be associated with various somatic symptoms, such as headache and irritable bowel syndrome. The overall prevalence of FM in the general population is approximately 2.0%, with higher prevalence observed in women aged 50 years and older.2
While the pathophysiological mechanism of FM has yet to be established, the dominant theory in FM is that the central processing of nociceptive information is altered.3,4 However, accumulating evidence suggests that there may be an abnormality in both peripheral nerve fibers and central pain processing in patients with FM.5,6 Such studies suggest that peripheral abnormalities in the sensory nerves may contribute to and/or possibly maintain FM symptoms.
Drugs currently used to treat FM provide only modest relief of symptoms at best and cause various side effects, including sedation and dizziness. According to findings from randomized clinical trials, approved medications for the treatment of FM (United States: pregabalin, duloxetine, milnacipran; Japan: pregabalin) show a modest ability to reduce pain in patients with FM. These studies show that treatment with these medications results in approximately 36% to 47% of patients experiencing a reduction in pain of 30%, compared to 28% to 34% of patients experiencing a 30% reduction in pain in response to placebo.7
Calcium-activated potassium channels are expressed in various animal cells and play an important role in the regulation of cellular functions. In response to an increase in intracellular calcium in excitable and non-excitable cells, these receptors open their channels to facilitate the influx of potassium, thereby regulating membrane potentials through induction of after-hyperpolarization. These potassium channels are classified into three categories of conductance channels: large (BK, KCa1.1), small (SK, KCa2.2), and intermediate (IK1, KCa3.1). Among these, expression of the KCa3.1 channel has been observed in lymphocytes, red blood cells, the gastrointestinal tract, peripheral nerves, and dorsal root ganglion, and is thought to be involved in diseases that target those tissues which expressed the KCa3.1 channel.8 However, a study reported that while the KCa3.1 channel is expressed in the sensory nervous system, its expression levels were unchanged in neuropathic and inflammatory pain models.9 Therefore, there is currently no clear evidence of a relationship between the KCa3.1 channel and any pain disorder. Furthermore, there is no report showing that a KCa3.1 channel activator is effective for the treatment of fibromyalgia in animal models.
Here, we examined the in vitro and in vivo profiles of a novel KCa3.1 channel opener (ASP0819; Figure 1) and its effects on analgesia in several animal models for pain.
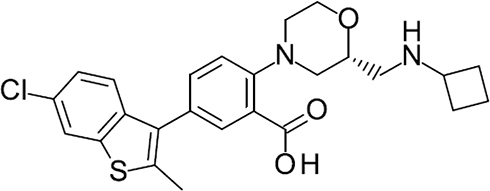 |
Figure 1 Chemical structure of ASP0819 (free base). |
Methods
Animals
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Astellas Pharma Inc. (Ibaraki, Japan), which has been awarded accreditation status by the Association of Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Animals were group-housed under a 12-h light-dark cycle at a constant room temperature (23 ± 2°C) and humidity (55 ± 15%), with free access to standard laboratory chow and water. All efforts were made to minimize animal numbers and suffering. We used male Sprague-Dawley (SD) rats (Japan SLC, Inc., Hamamatsu, Japan or Charles River Laboratories Japan, Yokohama, Japan) for reserpine-induced myalgia (RIM; 7–8 weeks), vagotomy-induced myalgia (VIM; 8 weeks), and monoiodoacetate-induced arthritis (MIA; 6 weeks) models; male Lewis rats for adjuvant-induced arthritis (AIA; 8 weeks); and male Wistar rats (Japan SLC, Inc.) for colorectal distension (CRD; 11–13 weeks). FM-like models, RIM,10 and VIM models11 were generated according to previously published methods.
Drugs
ASP0819 hydrobromide salt was used for in vitro membrane potential assay, in vivo VIM, AIA, and electrophysiological studies. Hydrochloride salt was used for other experiments. Both were synthesized at Astellas Pharma Inc. For in vitro experiments, the compound was dissolved in DMSO. For in vivo withdrawal behavior experiments, ASP0819 was suspended in 0.5% methylcellulose solution to yield free-base concentrations of the indicated doses and was orally administered before behavioral testing. For in vivo electrophysiology experiments, ASP0819 was dissolved in vehicle (10% N,N-dimethylformamide (DMF; Sigma-Aldrich Co. LLC., St. Louis, MO, USA), 10% propylene glycol (PG; Sigma-Aldrich Co. LLC.), and 80% normal saline), and was intravenously (IV) administered. Diclofenac (Sigma-Aldrich Co. LLC.) was dissolved in 0.5% methylcellulose solution and orally administered 4 h before behavioral testing. The drugs were administered to rats at 5 mL·kg−1.
In vitro Experiments
Measurement of Membrane Potential
Membrane potential was measured in human or rat KCa3.1 channel-expressing CHO cells (American Type Culture Collection [ATCC] CRL-9096, Manassas, VA, USA) using a FLIPR® membrane potential assay kit (Molecular Devices, LLC., Sunnyvale, CA, USA) with FMP dye (Blue). It is reported that the fluorescent values correlate the membrane potential (mV) as a function of depolarizing KCl concentrations using whole-cell current clamp.31 CHO cells expressing the human or rat KCa3.1 channel were plated at a density of 2 x 104 cells/well in a 384-well plate. This assay was conducted according to the protocol with the kit. Hank’s Balanced Salt Solution (HBSS) containing 20 mM HEPES [pH 7.5] and 0.02% CHAPS was used and so as to be 0.3% of DMSO concentration in the assay. ASP0819 (final 1–30,000 nM) and NS309 (final 3000 nM; Sigma-Aldrich Co. LLC.)12 were subsequently added. NS309 is the compound that has action of KCa3.1 opener. We used the compound as a control. Membrane potential was measured using a fluorometric imaging plate reader (FLIPR Tetra, Molecular Devices, LLC.), and the maximal increase in fluorescent signal (excitation wavelength: 510–545 nm, emission wavelength: 565–625 nm) was recorded. The response at each concentration of ASP0819 was expressed as a percentage, with the average responses to 3000 nM NS309 and vehicle defined as 100% and 0%, respectively. The half-maximal effective concentration (EC50) value was defined as the concentration of the test compound that produces 50% response of maximum efficacy (Emax; 3 independent experiments in quadruplicate) and was expressed as the geometric mean and 95% confidence interval.
Ion Channel Assays
The activity of ASP0819 in assays for rat small conductance Ca2+-activated K+ channel type 2 (rKCa2.2) and human large conductance Ca2+-activated K+ channel (hKCa1.1) was measured using a FLIPR® membrane potential assay kit (Blue) (Molecular Devices, LLC.) with HEK293 cells (ATCC CRL-1573) stably expressing rKCa2.2 channel and MG63 cells (ATCC CRL-1427) endogenously expressing hKCa1.1, respectively. Cells were plated onto each 384-well plate and incubated with culture medium for approximately 24 h after cell plating, then diluted test compounds and vehicle in HBSS assay buffer containing 20 mM HEPES-NaOH (pH 7.4), 0.01% pluronic acid, and 0.02% CHAPS were added to respective wells. For hKCa1.1 assay, 1 μM ICA17043 (Astellas Pharma Inc.), a KCa3.1 inhibitor, was added into in HBSS assay buffer. The plates were incubated at room temperature for 1 h, and change in relative fluorescence units (RFU) (excitation wavelength: 530 nm, emission wavelength: 590 nm) was recorded as an indicator of change in membrane potential which means channel activity. For rat L-type Ca2+ channel inhibition activity assay, A7r5 cells (ATCC CRL-1444) were plated onto 96-well plate and incubated with culture medium. Two days after culturing, A7r5 cells loaded with Fluo3 AM (Dojindo, Kumamoto, Japan) were used in HBSS buffer containing 20 mM HEPES-NaOH (pH 7.6), 2.5 mM probenecid, 0.04% pluronic acid, and 0.02% CHAPS for 1 h at 37°C and intracellular Ca2+ ([Ca2+]i) response was measured using FLIPR® Calcium assay kit (Molecular Devices, LLC.). Veratridine-stimulated [14C]-guanidine hydrochloride uptake assay was used to evaluate the effect on rat Na+ channel activity. Rat brain primary cells were prepared from rat E19 cortex and incubated with culture medium for 2 days. Reaction mixture solution was constructed with [14C]-guanidine hydrochloride (400 μM, 12kBq/assay), veratridine (200 µM), and diluted test compounds in assay buffer including 135 mM choline chloride, 5 mM KCl, 1 mM MgSO4, 5.5 mM glucose, and 1 mg·mL−1 bovine serum albumin. The radioactivity count was measured with a liquid scintillation counter. The activities for rKCa2.2, rat L-type Ca2+ channel, hKCa1.1, and rat Na+ channel were examined at concentrations up to 10, 10, 100, and 100 µM of ASP0819, respectively.
Off-Target Effects
The inhibitory activity of ASP0819 against radioligand binding to 57 other receptors, ion channels, and transporters, along with 3 enzyme reaction assays, was examined at 10 µM according to the procedures of Sekisui Medical Co., Ltd. (Tokyo, Japan).
The inhibition rate was calculated by subtracting the binding or reaction ratio from 100 (ie, 100 – binding or reaction ratio), where the binding or reaction ratio = [(B – N)/(B0 – N)] x 100 (%), where
B = Bound radioactivity in the presence of the test substance, or radioactivity or fluorescence intensity in the tube for calculation of inhibition rate (individual value);
B0 = Total bound radioactivity in the absence of the test substance, or radioactivity or fluorescence intensity of the tube for calculation of total reaction (mean value); and
N = Non-specific bound radioactivity, or radioactivity or fluorescence intensity of the tube for calculation of non-specific reactions (mean value).
In vivo Efficacy in Animal Models
Measurements of Withdrawal Behavior
The RIM model, which mimics features of FM, including hyperalgesic symptoms, was generated according to a previously reported method.10 Reserpine, a CNS monoamine depleting substance (Nacalai Tesque Inc., Kyoto, Japan) was dissolved in glacial acetic acid and diluted to a final concentration of 0.5% with distilled water. Rats were administered repeated subcutaneous (SC) injections of 1 mg·kg−1 reserpine 3 times (once daily, for 3 consecutive days). Drugs were evaluated using a muscle pressure test on Day 7 after the final injection of reserpine. After the pre-drug muscle pressure test, reserpine-treated rats were allocated to groups based on their muscle pressure threshold, with each group receiving either vehicle or a dose of the drug. The muscle pressure test was conducted again after administration of vehicle or the drug. Vehicle or ASP0819 was orally administered on Day 8, and the muscle pressure threshold was measured 4 h after administration.
SD rats were anesthetized with pentobarbital sodium and the region around the xiphisternum was shaved. A midline incision was made, the stomach and esophagus under the diaphragm were exposed, and the vagus nerves on either side of the esophagus were transected. The muscle and skin around the wound were sutured. A sham operation was performed without transection of the vagus nerves as a control. The analgesic effect of ASP0819 was evaluated in the rat VIM model after single or repeated administration. One week after the operation, the rats were allocated to groups based on both body weight and muscle pressure threshold. Vehicle or ASP0819 was orally administered once or once a day for 14 days starting from 1 week after the sham or vagotomy operation, and the muscle pressure threshold was measured 2, 4, 24, and 48 h after the final administration.
This study was performed by LSI Medience Corporation (Tokyo, Japan). Briefly, arthritis was induced by administering 50 µL/site of Complete Freund’s Adjuvant (0.1% Mycobacterium Tuberculosis Suspension: Sigma-Aldrich Co. LLC.) into the footpad of the right hind paw of rats (Day 1). Grouping was performed using the stratified randomization method based on the mean of three withdrawal threshold values and body weight (Day 2). The withdrawal threshold (mmHg) was measured using the Randall-Selitto method (cut-off value: 500 mmHg) 4 h after administration of vehicle (0.5% methylcellulose solution), ASP0819 (0.3, 1, and 3 mg·kg−1), or the positive control drug (diclofenac: 1 mg·kg−1). Measurement was performed three times per individual hindpaw using an analgesy-meter (TK-201, Unicom Corporation, Ageo, Japan).
Sodium monoiodoacetate (Sigma-Aldrich Co. LLC.) was dissolved in physiological saline. The MIA model was generated as described previously.13 Briefly, rats were anesthetized with isoflurane and given a single intraarticular injection of 1 mg sodium monoiodoacetate or saline (normal) through the infrapatellar ligament of the right knee. Weight distribution was measured using an incapacitance tester (Linton Instrumentation, Norfolk, UK) and used as a behavioral measure of musculoskeletal discomfort.14 Rats were placed in an angled plastic chamber so that each hind paw rested on a separate force plate. The force exerted by each hind paw (measured in grams) was averaged over a 5-s period. The change in hind paw weight distribution was calculated by determining the difference in weight between the left (contralateral control) and right (osteoarthritic) paws. Each data point was the mean of three measurements. If the animal moved during the measurement, the data were excluded and an additional measurement was conducted. Body weight and hind paw weight distribution were measured 13 days after the MIA injection. Weight distribution less than 20 g was excluded. The rats were subsequently assigned to the following four treatment groups to minimize differences in both average hind paw weight distribution and body weight among the groups: (1) MIA-vehicle, (2) MIA-ASP0819 0.1 mg·kg−1, (3) MIA-ASP0819 0.3 mg·kg−1, and (4) MIA-ASP0819 1 mg·kg−1. The normal group was only acclimated to the measurement environment. From 23 to 27 days after the MIA injection, vehicle or ASP0819 was orally administered as a single daily dose. At 23 (single administration) and 27 (repeated administration for 5 days) days after the MIA injection, hind paw weight distribution was measured 2 h after administration. The normal group was orally administered vehicle and hind paw weight distribution was measured in the same manner as the MIA group.
The colorectal distension test was performed in awake and unrestrained rats. A 5-cm long latex balloon was inserted into the colorectal area via the anus under isoflurane anesthesia. The balloon was positioned such that the end was 1 cm proximal to the anus and was secured in place by taping the balloon catheter to the base of the tail. The free end of the balloon catheter was connected to a computerized barostat (DISTENDER SERIES IIR, G&J Electronics Inc., Toronto, Canada). After recovering from the anesthesia, 30-mmHg distension was applied twice for 30 s at a 5-min interval to acclimatize the rats to the balloon distension procedure. ASP0819 (0.1, 0.3, and 1 mg·kg−1) or vehicle was orally administered 1 h before starting the distension procedure, which consisted of distention at 15 to 60 mmHg with stepwise increases of 15 mmHg. The number of abdominal contractions during each distension step was counted as an indicator of abdominal pain.
Muscle Pressure Test
Muscle pressure threshold (MPT) was measured using a previously described method15 in RIM and VIM model. Briefly, the rats were placed inside a sock, and the right hind limb was positioned such that incremental pressure could be applied to the mid-gastrocnemius muscle using a Randall-Selitto apparatus (Muromachi Kikai, Tokyo, Japan). Linearly increasing mechanical force was applied to the gastrocnemius muscle until a hind limb withdrawal response was elicited. The mean withdrawal threshold in 3 trials was defined as the threshold value.
Blinded measurements were performed in behavioral withdrawal tests.
In vivo Electrophysiology Experiments
RIM rats were used for these experiments. The spontaneous and mechanically evoked nerve firing rate in single primary sensory neurons of the dorsal root were measured to examine the mechanism of ASP0819. A laminectomy covering segments L3–L6 was carried out to expose dorsal roots. The dura was carefully removed under a dissecting microscope and an oil pool was formed over the spinal cord dorsal surface by placing the skin on a metal frame. The L5 dorsal root was carefully separated and repeatedly teased to facilitate single fiber recordings (Figure 4A). The neural activity was amplified and monitored using standard electrophysiological recording techniques (NeuroSolution LTD., Coventry, UK) and recorded on to a PC using CED Spike 2 software (Cambridge Electronics Design). Mechanically evoked responses were examined using a series of von Frey hairs (1, 2, 4, 6, and 10 g). Each hair was applied to the receptive field of the gastrocnemius muscle according to a one-sec-on-one-sec-off protocol and repeated 10 times at intervals of 10 min. The conduction velocity (C.V.) criteria for C- and Aδ-fibers were as follows: C-fibers: C.V. < 2 m·s−1; Aδ-fibers: 2 m·s−1 < C.V. <10 m·s−1 (Supplemental Figures 1 and 2). ASP0819 dissolved in the vehicle solution was administered IV via an injection pump at 3 mL·kg−1·h−1 over 60 min. Lidocaine hydrochloride (Sigma-Aldrich Co. LLC.) was dissolved in saline to 3.3 mg·mL−1. The spontaneous and mechanically evoked firing rate were expressed as a percentage of the baseline control (normalized to baseline) and compared before and after vehicle or compound perfusion.
Statistical Analysis
Data are expressed as the mean ± SEM in each group. The significance of differences between two groups was assessed using Student’s t-test, while significance of differences among more than two groups was assessed using Dunnett’s multiple comparisons test with or without Bonferroni correction (GraphPad Prism, GraphPad Software, San Diego, CA, USA). P <0.05 was considered statistically significant. Post hoc tests were only performed if F achieved P <0.05.
Results
Effect of ASP0819 on Membrane Potential in Human and Rat KCa3.1 Receptor-Expressing Cells
The effect of ASP0819 on the membrane potential of human and rat KCa3.1-expressing CHO cells was evaluated using membrane potential-sensitive dye. ASP0819 increased the negative change in membrane potential in a concentration-dependent manner in both cell types. The EC50 and Emax values in human and rat KCa3.1-expressing cells were 102.4 nM and 99.0%, and 392.9 nM and 102.4%, respectively (Table 1).
 |
Table 1 Effect of ASP0819 on Membrane Potential in Human and Rat KCa3.1 Channel-Expressing CHO Cells |
Selectivity of ASP0819
ASP0819 did not show more than 50% activity in the rKCa2.2 assay, hKCa1.1 assay, and L-type Ca2+ channel assay at concentrations up to 10, 100, and 10 µM, respectively (data not shown). Radioligand binding assays and enzyme reaction assays were used to measure the affinity and enzyme reactivity of ASP0819 (Supplemental Table 1–3). ASP0819 (10 µM) did not show more than 50% inhibition of radioligand binding to the 54 proteins inclusive of the various receptors, ion channels, and transporters, or of the enzymatic reactions of the 3 enzymes examined. ASP0819 (10 µM) inhibited radioligand binding to Na+ channel site 2 (rat), the serotonin 5HT4 receptor (guinea pig), and sigma receptor (non-selective) (guinea pig) with inhibition rates of 83.71%, 73.52%, and 57.57%, respectively. However, no more than 50% inhibition was observed at concentrations up to 100 µM for the rat Na+ channel and no meaningful agonistic or antagonistic activity was observed for the mouse 5HT4 receptor at concentrations of up to 10 µM (data not shown).
Effect of ASP0819 on Muscular Withdrawal Responses in RIM Rats
Repeated administration of reserpine (1 mg·kg−1 SC, once daily for 3 consecutive days) significantly decreased the muscle pressure threshold. Oral administration of ASP0819 at doses of 0.1, 0.3, and 1 mg·kg−1 significantly recovered the decrease in muscle pressure threshold (Figure 2). The threshold values (mean ± SEM) were as follows: normal, 129.2 ± 1.1 mmHg; RIM-vehicle, 58.3 ± 1.0 mmHg; RIM-ASP0819 (0.1 mg·kg−1), 83.9 ± 2.7 mmHg; RIM-ASP0819 (0.3 mg·kg−1), 95.2 ± 2.8 mmHg; and RIM-ASP0819 (1 mg·kg−1), 112.1 ± 2.3 mmHg.
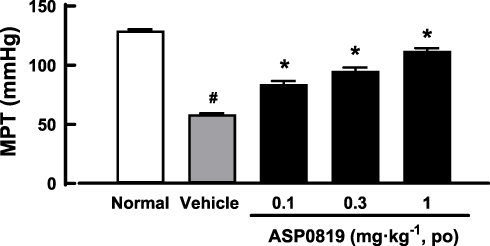 |
Figure 2 Effect of ASP0819 on muscular withdrawal responses in RIM rats. Myalgia was induced by treatment with SC reserpine for 3 days. Vehicle or ASP0819 (0.1, 0.3, and 1 mg·kg-1) was orally administered 8 days later, and the muscle pressure threshold (MPT) was measured 4 h after administration. Data are expressed as the mean ± SEM in each group (n=6). #P<0.05, statistically significant compared to the normal group (Student’s t-test). *P<0.05, statistically significant compared to the vehicle group (Dunnett’s multiple comparisons test). |
Effect of ASP0819 on Muscular Withdrawal Responses in VIM Rats
One week after VIM surgery, there was a decrease in the muscle pressure threshold in all vagotomized rats before grouping. The decrease of threshold in the vagotomy control groups was statistically significant compared to the sham group in the all efficacy evaluation. Four h after the first treatment with ASP0819 (0.1, 0.3, and 1 mg·kg−1), the decreased threshold had significantly recovered (Figure 3A). Similar results were obtained 4 h after the final dose following daily repeated administration across 14 days. At 24 h after the final dose, the analgesic effect of ASP0819 at 0.3 mg·kg−1 was significant, but weak and transient. At 48 h after the final dose, the analgesic effect of ASP0819 at 1 mg·kg−1 had diminished, but the effect remained significant. The maximum efficacy of ASP0819 observed 4 h after administration is shown in Figure 3B.
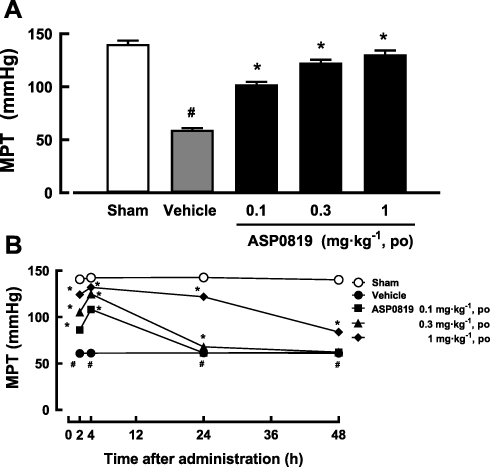 |
Figure 3 Effect of ASP0819 on muscular withdrawal responses in VIM rats (A) 4 h after a single dose and (B) after repeated daily doses across 14 days. (A) The effect of ASP0819 was evaluated in a rat vagotomy model after a single administration. One week after the sham or vagotomy operation, vehicle or ASP0819 (0.1, 0.3, and 1 mg·kg-1) was orally administered, and the muscle pressure threshold (MPT) was measured 4 h after administration. (B) The effect of ASP0819 was evaluated in a rat vagotomy model after repeated administration. Vehicle or ASP0819 (0.1, 0.3, and 1 mg·kg-1) was orally administered once a day for 14 days from one week after the sham or vagotomy operation, and the MPT was measured at 2, 4, 24, and 48 h after the final administration. Data are expressed as the mean ± SEM in each group (n=6). #P<0.05, statistically significant compared to the normal group (Student’s t-test). *P<0.05, statistically significant compared to the control group (Dunnett’s multiple comparisons test). |
Our results demonstrate that treatment with ASP0819 ameliorated hyperalgesia in vagotomized rats and that the drug exhibited similar efficacy after single and repeated administration for 14 days. ASP0819 may therefore exert a continuous analgesic effect against pain in the FM model.
Effects of ASP0819 on Spontaneous and Mechanically Evoked Activity in Dorsal Root Fibers in a Rat RIM Model
Spontaneous activity and mechanical responses varied greatly from fiber to fiber. In Aδ-fibers, the spontaneous firing rate ranged from 213 spikes·min−1 to 1332 spikes·min−1 before dosing, and the total of mechanically evoked responses to 5 von Frey hairs ranged from 342 spikes/10 trials to 1895 spikes/10 trials. In C-fibers, the spontaneous firing rate ranged from 84.7 spikes·min−1 to 1198 spikes·min−1, and mechanically evoked responses ranged from 343 spikes/10 trials to 2343 spikes/10 trials. Therefore, the spiking rate was expressed as a percentage of the baseline control (normalized to baseline) for standardization before comparing between before and after vehicle or compound perfusion. Example recordings were shown in RIM rats (Supplemental Figure 3A and 3B).
ASP0819 at 0.3 mg·kg−1·h−1 and 1 mg·kg−1·h−1 IV significantly reduced spontaneous activity in Aδ-fibers 30 and 60 min after administration (Figure 4B). The effect of ASP0819 at 0.1, 0.3, and 1 mg·kg−1·h−1 IV on spontaneous activity in C-fibers was comparable to that of vehicle. But the precise effect of the compound on spontaneous activity in C-fibers made it difficult to determine due to time-dependent decrease in the response of vehicle (data not shown). ASP0819 at 0.3 mg·kg−1·h−1 and 1 mg·kg−1·h−1 IV significantly reduced mechanically evoked responses in Aδ-fibers (Figure 4C) and mechanically evoked responses in C-fibers (data not shown). The typical recordings were shown in Supplemental Figure 3A and 3B. Lidocaine at 10 mg·kg−1·h−1 significantly inhibited spontaneous activity and mechanically evoked responses over time. Finally, naive rats had fewer Aδ- and C-fibers with spontaneous activity. Vehicle had no effect on spontaneous activity or mechanically evoked responses in naive rats. These results suggest that ASP0819 significantly blocked both spontaneous and mechanically evoked activity in Aδ-fibers and mechanically evoked responses in C-fibers in RIM models.
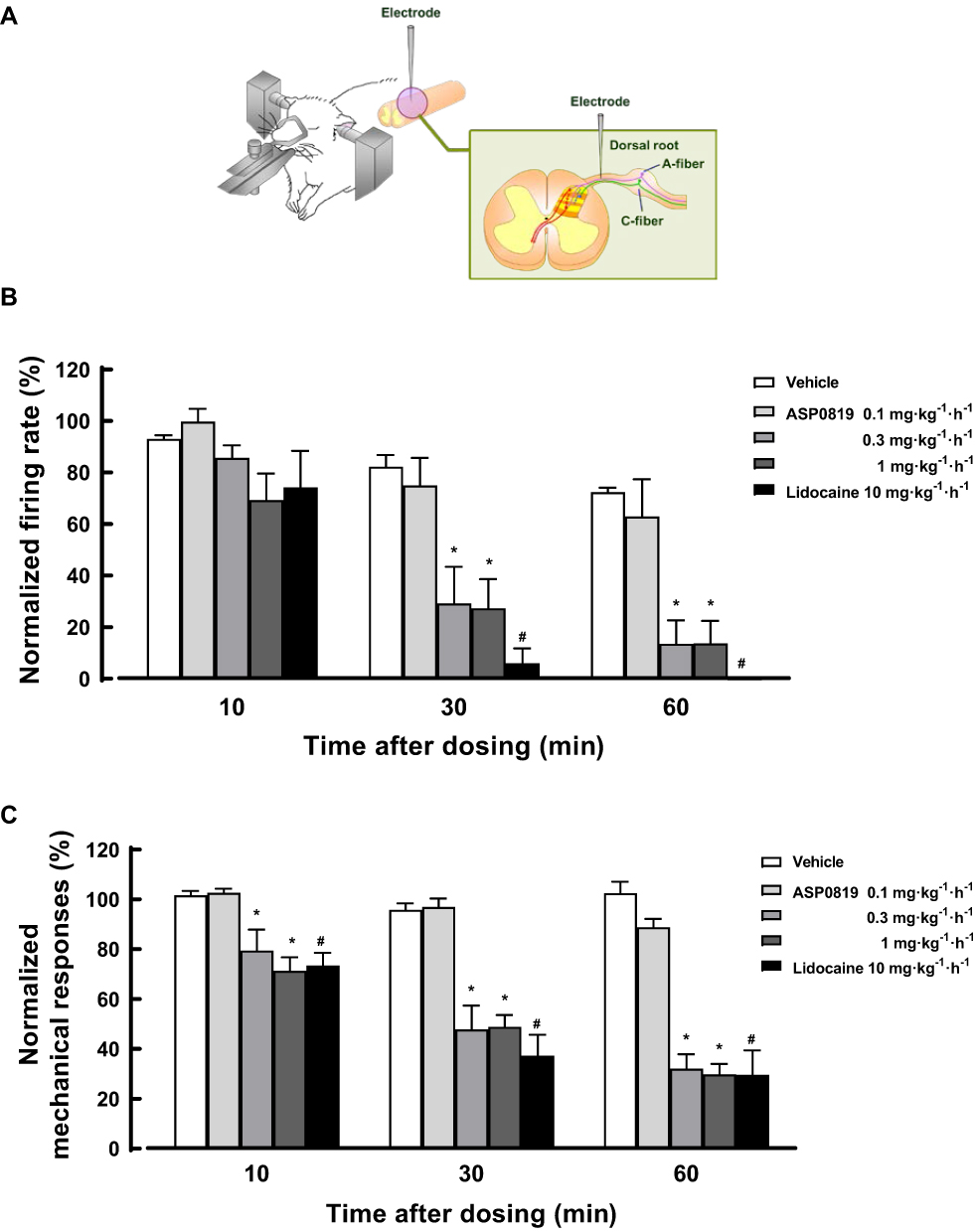 |
Figure 4 In vivo electrophysiology in a rat RIM model. Spontaneous and mechanically-evoked nerve firing in single primary sensory neurons of the dorsal root were recorded in urethane-anesthetized RIM rats (A). Percent changes in spontaneous and mechanically-evoked nerve firing rate in Aδ-fiber are shown in (B and C), respectively. Data are expressed as the mean ± SEM (n=8–10) in each group. #P<0.05, statistically significant compared to the vehicle-treated group at same time point (Student’s t-test). *P<0.05, statistically significant compared to the vehicle-treated group at same time point (Dunnett’s multiple comparisons test). |
Effect of ASP0819 on Withdrawal Threshold in AIA Model Rats
The effects of oral administration of ASP0819 on withdrawal threshold were evaluated in a rat AIA model (Figure 5). Using the Randall-Selitto’s method, the withdrawal threshold in the control group was 51.2 mmHg. The withdrawal threshold (67.6 mmHg) in the 1 mg·kg−1 diclofenac group was significantly higher than that in the control group. The withdrawal threshold in the 0.3, 1, and 3 mg·kg−1 ASP0819 groups 4 h after administration was 52.3, 58.1, and 63.5 mmHg, respectively. The withdrawal thresholds in the 1 and 3 mg·kg−1 ASP0819 groups were significantly higher than that in the control group.
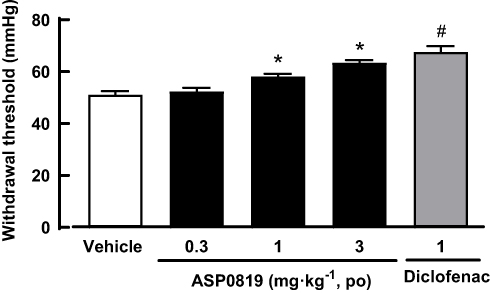 |
Figure 5 Effect of ASP0819 on withdrawal threshold in a rat AIA model. Arthritis was induced by administering complete Freund’s adjuvant into the footpad of the right hind paw of rats. The withdrawal threshold (mmHg) was measured using the Randall-Selitto method 4 h after administration of vehicle, ASP0819 (0.3, 1, and 3 mg·kg-1), or the positive control drug (diclofenac: 1 mg·kg-1). Data are expressed as the mean ± SEM in each group (n = 8). #P<0.05, statistically significant compared to the vehicle-treated group (Student’s t-test). *P<0.05, statistically significant compared to the vehicle-treated group (Dunnett’s multiple comparisons test). |
Effects of ASP0819 on Weight Distribution in MIA Model Rats
A weight-bearing deficit was detected in MIA-treated rats. In the chronic phase of the MIA model, single oral administration of ASP0819 at 0.3 and 1 mg·kg−1 significantly improved the weight-bearing deficit (Figure 6A). Repeated oral administration of ASP0819 at 0.1–1 mg·kg−1 also significantly improved the weight-bearing deficit (Figure 6B).
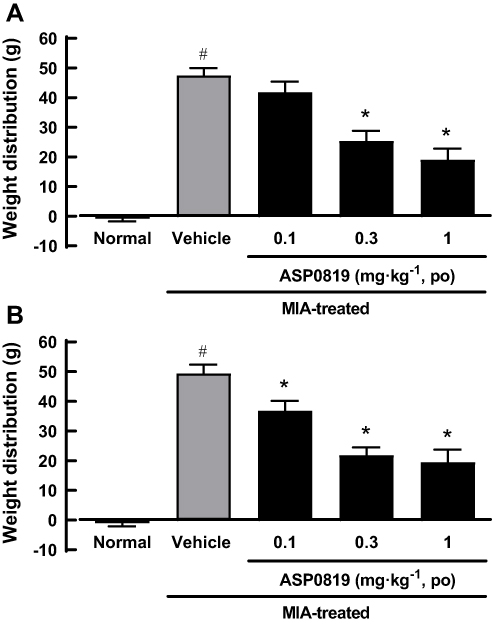 |
Figure 6 Effect of oral administration of ASP0819 on weight distribution in MIA model rats. Rats were given a single intraarticular injection of 1 mg sodium monoiodoacetate or saline (normal) through the infrapatellar ligament of the right knee. From 23 to 27 days after the injection, vehicle or ASP0819 was orally administered as a single daily dose. At 23 (single administration; (A) and 27 (repeated administration for 5 days; (B) days, hind paw weight distribution was measured 2 h after administration using an incapacitance tester. The data represent the mean ± SEM weight distribution (n = 8). #P<0.05, statistically significant compared to the normal group (Student’s t-test). *P<0.05, statistically significant compared to the vehicle-treated group (Dunnett’s multiple comparisons test). |
Effects of ASP0819 on Visceromotor Response Induced by CRD in Rats
The number of abdominal contractions increased with stepwise incremental increases in balloon distension. ASP0819 at 0.3 and 1 mg·kg−1 significantly reduced the number of abdominal contractions induced by colorectal distension (Figure 7).
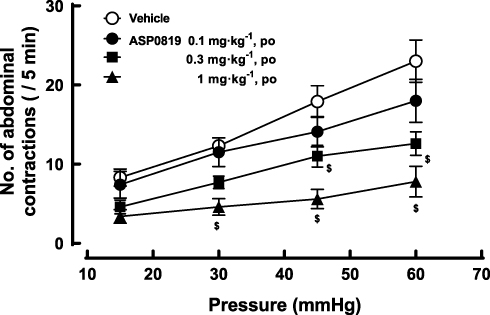 |
Figure 7 Effect of ASP0819 on visceromotor response induced by colorectal distension in rats. One hour after vehicle or ASP0819 (0.1–1 mg·kg-1) was orally administered, the number of abdominal contractions induced by balloon distension from 15 to 60 mmHg at 15-mmHg steps was counted. Each distension period was 5 min and was performed at 5-minute intervals. Data are expressed as the mean ± SEM in each group (n=10). $P<0.05, statistically significant compared to the control group (Dunnett’s multiple comparisons tests with Bonferroni correction). |
Discussion
ASP0819 is a novel KCa3.1 opener with high potency, selectivity, and oral activity that induces analgesia in the animal models of FM, OA, and visceral pain. These findings suggest that the activation of KCa3.1 can ameliorate various types of pain in FM, OA patients, and diseases with visceral pain.
In in vitro studies, ASP0819 activated the KCa3.1 channel (EC50 of about 100 nM) but not the KCa2.2 and KCa1.1 channels at 10 μM. In addition, ASP0819 at 10 μM had no appreciable affinity for 54 other receptors, channels, and transporters, and no appreciable inhibitory effect on 3 enzymes. Additionally, it had no functional activity in several receptor-expressing cells (data not shown). These results indicate that ASP0819 is highly potent, with more than 100 times the selectivity for the KCa3.1 channel than other receptors or channels tested.
In this study, we found that ASP0819 had efficacy in a rat RIM model. These results were of great interest given we previously reported that the rat RIM model mimics the features of FM, including hyperalgesic symptoms.10 For this reason, it was developed and advanced as an animal model of FM. Administration of reserpine once a day for 3 days in this model decreases levels of 5-HT, norepinephrine (NE), and dopamine (DA) in the brain and spinal cord, inducing muscle pain that persists for 1 week or longer in rats.16 The decreased levels of biogenic amines in RIM rats are similar to that observed following dysfunction of biogenic amine-mediated pain control in FM patients.17,18 Additionally, it is interesting from the aspect of predictive validity that RIM rats exhibit sensitivity not only to activators of the biogenic amine system (eg, duloxetine) but also to other anti-FM drugs (eg, pregabalin).16,19 Because of these findings, we further concluded that the RIM model was useful for evaluating agents which modulate the central nervous system.
Next, we examined the effect of ASP0819 on withdrawal behavior in a rat VIM model. Vagotomized rats, another animal model that mimics the hyperalgesic symptoms of FM20 with constructing dysfunction of the vagus nerve, show muscle pain, sensitization of primary afferent nociceptive neurons, and involvement of dysfunction of a type of potassium channel from the aspect of face validity.21 In addition, from the aspect of predictive validity, VIM rats exhibit sensitivity not only to activators of the biogenic amine system (eg, amitriptyline) but also to other anti-FM drugs (eg, pregabalin).11 Efficacy of ASP0819 on withdrawal behavior in both rat RIM and VIM models was demonstrated in our experiments, suggesting peripheral modulation with drugs may be promising for the treatment of FM pain.
The mechanism of action was further examined using an in vivo electrophysiological method in a rat RIM model. The spontaneous and mechanically evoked peripheral nerve firing rate at the dorsal root was significantly increased in the RIM rats compared with the control rats, while ASP0819 significantly reversed this increased firing rate in the RIM rats. We also examined the plasma concentrations of ASP0819 in normal rats with the same experimental protocol of the in vivo electrophysiology study. After IV infusion of ASP0819 at 0.3 mg·kg−1·h−1, the mean plasma concentrations at 10, 30, and 60 min were 89.1, 714, and 2080 ng·mL−1, respectively. This indicates that ASP0819 showed efficacy at reasonable concentrations. In addition, ASP0819 showed low penetration into the brain (plasma/brain ratio was less than 1%). The pharmacologic effects of ASP0819 suggest that it has therapeutic potential as an analgesic agent by facilitating recovery of normal nerve firing through peripheral mechanisms.
As seen in this and previous studies, opening of the KCa3.1 channel results in a period of reduced excitability after each action potential, which is called after-hyperpolarization. The physiological role of KCa3.1 is thought to be regulation of cellular excitability, which suggests that it is a potential therapeutic target for various diseases associated with abnormal nerve excitation.8 For example, activation of KCa3.1 appears to not only have an analgesic effect on visceral hypersensitivity22 but also to be involved in the analgesic action of peroxisome proliferator-activated receptor (PPAR) agonists.23 The channel is present in Aδ-fibers and C-fibers of dorsal root ganglion and on axons.24 Therefore, KCa3.1 has a role in immunological/inflammatory responses and abnormal nerve excitation. Behavioral analyses of KCa3.1-deficient (KCa3.1−/-) mice in various pain models point to a specific inhibitory role for KCa3.1 in the pain processing of noxious chemical stimuli.25 Palmitoylethanolamide (PEA) is a known PPARα agonist and has been launched as a dietary supplement in Europe. The compound has analgesic effects against various types of pain in humans. In particular, the analgesic effects of PEA are reportedly significantly greater than those of ibuprofen in temporomandibular joint osteoarthritis and synovitis pain.26 These acute analgesic effects are thought to occur through the KCa3.1 channel.23 In contrast, clotrimazole is a known antifungal agent with inhibitory action against the KCa3.1 channel. Clotrimazole induces nociceptive behavior in mice.27 In clinical settings, common side effects of 1% clotrimazole cream include reddening, itching, irritation, and burning pain.28 Taken together, these reports suggest that KCa3.1 may be a novel target for the treatment of pain-inducing disorders associated with neuronal hyperactivity.
We expected ASP0819 would have efficacy in other pain models based on the mechanism of action. Efficacy of NSAIDs for OA pain is thought to have limitations. One of the explanations is that the symptoms of pain in OA patients are mediated not only by inflammatory mechanisms but also by non-inflammatory one which is considered to be less responsive to NSAIDs treatment. Single oral administration of diclofenac and other NSAIDs had no effect on pain in our experimental MIA condition,29,30 indicating that the pain would reflect non-inflammatory pain in the animal model. Meanwhile, ASP0819 has potent analgesic effects on inflammatory (AIA model) and non-inflammatory (MIA model) pain. These results suggest that ASP0819 is expected to have superior effects over NSAIDs in OA pain. Most FM patients also have visceral pain. We also found ASP0819 had efficacy in pain with CRD using these models.
A possible mechanism of action of KCa3.1 openers would be considered. The peripheral nerve firing rate may increase as a result of a decrease in activity in the peripheral descending inhibitory system in FM. The compound ASP0819 reverses the increase in nerve firing rate. ASP0819 is a peripherally acting KCa3.1 channel opener that targets peripheral sensory nerves and is currently being developed as a treatment for FM. ASP0819 is predicted to cause hyperpolarization of primary sensory afferent nerves and subsequent decreased firing. This pharmacological action of ASP0819 should reduce peripheral nerve hyperexcitability, thereby normalizing the frequency of action potentials and reducing the pain experienced by FM patients.
Conclusions
In conclusion, ASP0819 works on the peripheral KCa3.1 channels found on peripheral afferent nerves and may offer a new approach for the treatment of pain in FM, OA patients, and diseases with visceral pain. It may further avoid central adverse effects compared to pregabalin, duloxetine, and opioids — current treatments for these diseases. ASP0819 may also represent a non-opioid treatment option for these patients.
Abbreviations
AAALAC, Association of Assessment and Accreditation of Laboratory Animal Care; AIA, adjuvant-induced arthritis; CRD, colorectal distension; DA, dopamine; DMF, dimethylformamide; EC50, half-maximal effective concentration: Emax, maximum efficacy; FM, Fibromyalgia; HBSS, Hank’s Balanced Salt Solution; MIA, monoiodoacetate-induced arthritis; MPT, muscle pressure threshold; NE, norepinephrine; NSAID, nonsteroidal anti-inflammatory drug; OA, osteoarthritis; PEA, palmitoylethanolamide; PPAR, peroxisome proliferator-activated receptor; RFU, relative fluorescence units; RIM, reserpine-induced myalgia; SD, Sprague-Dawley; VIM, vagotomy-induced myalgia.
Acknowledgments
This research was funded by Astellas Pharma Inc. (Tokyo, Japan). The authors would like to acknowledge Dr. Yasuaki Shimizu for his valuable comments and continued encouragement, and Dr. Ryuichi Takezawa and Dr. Satoshi Kitamura for their expert technical assistance. The authors would also like to acknowledge Astellas Research Technologies Co., Ltd. and Analysis & Pharmacokinetics Research Laboratories, Astellas Pharma Inc. for their contributions to the pharmacokinetics study. Editorial and writing assistance, under the guidance of the authors, was provided by Elizabeth Hermans, PhD of OPEN Health Medical Communications (Chicago, IL) and funded by the study sponsor.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors are employees of Astellas Pharma Inc, during the conduct of the study. Tomoya Oe has patents issued: JP4853844, US8795632, and EP2327299. The authors report no other potential conflicts of interest for this work.
References
1. Bradley LA. Pathophysiologic mechanisms of fibromyalgia and its related disorders. J Clin Psychiatry. 2008;69(Suppl 2):6–13.
2. Wolfe F, Ross K, Anderson J, Russell IJ, Hebert L. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38(1):19–28. doi:10.1002/art.1780380104
3. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2–S15. doi:10.1016/j.pain.2010.09.030
4. Staud R. Brain imaging in fibromyalgia syndrome. Clin Exp Rheumatol. 2011;29(6 Suppl 69):S109–S117.
5. Serra J, Collado A, Solà R, et al. Hyperexcitable C nociceptors in fibromyalgia. Ann Neurol. 2014;75(2):196–208. doi:10.1002/ana.24065
6. Staud R, Smitherman ML. Peripheral and central sensitization in fibromyalgia: pathogenic role. Current Pain Headache Report. 2002;6(4):259–266. doi:10.1007/s11916-002-0046-1
7. Häuser W, Walitt B, Fitzcharles MA, Sommer C. Review of pharmacological therapies in fibromyalgia syndrome. Arthritis Res Ther. 2014;16(1):201. doi:10.1186/ar4441
8. Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14(13):1437–1457. doi:10.2174/092986707780831186
9. Mongan LC, Hill MJ, Chen MX, et al. The distribution of small and intermediate conductance calcium-activated potassium channels in the rat sensory nervous system. Neuroscience. 2005;131(1):161–175. doi:10.1016/j.neuroscience.2004.09.062
10. Nagakura Y, Oe T, Aoki T, Matsuoka N. Biogenic amine depletion causes chronic muscular pain and tactile allodynia accompanied by depression: A putative animal model of fibromyalgia. Pain. 2009;146(1–2):26–33. doi:10.1016/j.pain.2009.05.024
11. Furuta S, Shimizu T, Narita M, et al. Subdiaphragmatic vagotomy promotes nociceptive sensitivity of deep tissue in rats. Neuroscience. 2009;164(3):1252–1262. doi:10.1016/j.neuroscience.2009.09.021
12. Christophersen P, Wulff H. Pharmacological gating modulation of small- and intermediate-conductance Ca2+-activated K+ channels (KCa2.x and KCa 3.1). Channels. 2015;9(6):336–343. doi:10.1080/19336950.2015.1071748
13. Guingamp C, Gegout-Pottie P, Philippe L, Terlain B, Netter P, Gillet P. Mono-iodoacetate-induced experimental osteoarthritis: a dose-response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997;40(9):1670–1679. doi:10.1002/art.1780400917
14. Bove SE, Calcaterra S, Brooker RM, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003;11(11):821–830. doi:10.1016/S1063-4584(03)00163-8
15. Schäfers M, Sorkinb LS, Sommer C. Intramuscular injection of tumor necrosis factor-alpha induces muscle hyperalgesia in rats. Pain. 2003;104(3):579–588. doi:10.1016/S0304-3959(03)00115-5
16. Oe T, Tsukamoto M, Nagakura Y. Reserpine causes biphasic nociceptive sensitivity alteration in conjunction with brain biogenic amine tones in rats. Neuroscience. 2010;169(4):1860–1871. doi:10.1016/j.neuroscience.2010.06.061
17. Russell IJ, Vaeroy H, Javors M, Nyberg F. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35(5):550–556. doi:10.1002/art.1780350509
18. Wood PB. Role of central dopamine in pain and analgesia. Expert Review Neurotherapy. 2008;8(5):781–797. doi:10.1586/14737175.8.5.781
19. Murai N, Fushiki H, Honda S, et al. Relationship between serotonin transporter occupancies and analgesic effects of AS1069562, the (+)-isomer of indeloxazine, and duloxetine in reserpine-induced myalgia rats. Neuroscience. 2015;289:262–269. doi:10.1016/j.neuroscience.2014.12.065
20. Khasar SG, Miao FJP, Jänig W, Levine JD. Modulation of bradykinin-induced mechanical hyperalgesia in the rat by activity in abdominal vagal afferents. European J Neurosci. 1998;10(2):435–444. doi:10.1046/j.1460-9568.1998.00030.x
21. Furuta S, Watanabe L, Doi S, et al. Subdiaphragmatic vagotomy increases the sensitivity of lumbar Aδ primary afferent neurons along with voltage-dependent potassium channels in rats. Synapse. 2012;66(2):95–105.
22. McHugh D, Head V, Panesar MS, Tigani B, Fox A, Lilley E. Effect of small and intermediate conductance potassium channel modulators on visceral hypersensitivity and function. Gastroenterology. 2008;134(Suppl 1):A–544. doi:10.1016/S0016-5085(08)62540-8
23. LoVerme J, Russo R, La Rana G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-α. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi:10.1124/jpet.106.111385
24. Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 2014;37(3):146–158. doi:10.1016/j.tins.2013.12.002
25. Lu R, Flauaus C, Kennel L, et al. KCa3.1 channels modulate the processing of noxious chemical stimuli in mice. Neuropharmacology. 2017;125:386–395. doi:10.1016/j.neuropharm.2017.08.021
26. Marini I, Bartolucci ML, Bortolotti F, Gatto MR, Bonetti GA. PEA was significantly higher than those of ibuprofen in temporomandibular joint. J Orofac Pain. 2012;26(2):99–104.
27. Meseguer V, Karashima Y, Talavera K, et al. Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J Neurosci. 2008;28(3):576–586. doi:10.1523/JNEUROSCI.4772-07.2008
28. Binet O, Soto-Melo J, Delgadillo J, Videla S, Izquierdo I. Flutrimazole 1% dermal cream in the treatment of dermatomycoses: a randomized, multicentre, double-blind, comparative clinical trial with 1% clotrimazole cream. Flutrimazole study group. Mycoses. 1994;37(11–12):455–459. doi:10.1111/j.1439-0507.1994.tb00400.x
29. Yoshimi E, Kumakura F, Hatori C, et al. Antinociceptive effects of AS1892802, a novel Rho kinase inhibitor, in rat models of inflammatory and noninflammatory arthritis. J Pharmacol Exp Ther. 2010;334(3):955–963. doi:10.1124/jpet.110.167924
30. Nagase H, Kumakura S, Shimada K. Establishment of a novel objective and quantitative method to assess pain-related behavior in monosodium iodoacetate-induced osteoarthritis in rat knee. J Pharmacol Toxicol Methods. 2012;65(1):29–36. doi:10.1016/j.vascn.2011.10.002
31. Baxter DF, Kirk M, Garcia AF, et al. A novel membrane-sensitive fluorescent dye improves cell-based assays for ion channels. J Biomol Screen. 2002;7(1):79–85. doi:10.1177/108705710200700110
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.