Back to Journals » Cancer Management and Research » Volume 12
A Hereditable Mutation of MSH2 Gene Associated with Lynch Syndrome in a Five Generation Chinese Family
Authors Shao WH, Wang CY, Wang LY, Xiao F, Xiao DS, Yang H ![]() , Long XY
, Long XY ![]() , Zhang L, Luo HG, Yin JY
, Zhang L, Luo HG, Yin JY ![]() , Wu W
, Wu W
Received 9 July 2019
Accepted for publication 24 December 2019
Published 27 February 2020 Volume 2020:12 Pages 1469—1482
DOI https://doi.org/10.2147/CMAR.S222572
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Wei-Hua Shao,1– 4,* Cheng-Yu Wang,4,5,* Lei-Yun Wang,1– 4 Fan Xiao,1– 4 De-Sheng Xiao,6 Hao Yang,4,5 Xue-Ying Long,7 Le Zhang,8 Heng-Gui Luo,9 Ji-Ye Yin,1– 4 Wei Wu4,5
1Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha 410078, People’s Republic of China; 2Institute of Clinical Pharmacology, Central South University; Hunan Key Laboratory of Pharmacogenetics, Changsha 410078, People’s Republic of China; 3Engineering Research Center of Applied Technology of Pharmacogenomics, Ministry of Education, Changsha 410078, People’s Republic of China; 4Department of Geratic Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 5National Clinical Research Center for Geriatric Disorders, Changsha, Hunan 410008, People’s Republic of China; 6Department of Pathology, Xiangya Hospital/School of Basic Medicine, Central South University, Changsha 410078, Hunan, People’s Republic of China; 7Department of Radiology, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China; 8Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 9Department of General Surgery, The Central Hospital of Xiangtan City, Xiangtan, Hunan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Wu
Department of Geratic Surgery, Xiangya Hospital, Central South University, Xiangya Road 87, Changsha 410008, Hunan, People’s Republic of China
Tel +86 731 89753053
Email [email protected]
Purpose: In order to clarify which variants of the MMR gene could provide current “healthy” members in affected families a more accurate risk assessment or predictive testing.
Patients and Methods: One family, which meets the criteria according to both Amsterdam I/II and Bethesda guidelines, is reported in this study. The proband and some relatives of the patient have been investigated for whole genome sequencing, microsatellite instability, immunohistochemical MMR protein staining and verified by Sanger sequencing.
Results: A heterozygous insertion of uncertain significance (c.420dup, p.Met141Tyrfs) in MSH2 gene was found in proband (III-16) and part of His relatives. The variant was associated with a lack of expression of MSH2 protein (MMR deficient) and high microsatellite instability analysis (MSI) status in tumor tissues of LS patients. In addition, we found that the variant could affect the expression of MSH2 and the response to chemotherapy drugs in vitro.
Conclusion: We identified an insertion mutation (rs1114167810, c.420dup, p.Met141Tyrfs) in MSH2 in LS using whole genome-wide sequencing (WGS). We further confirmed that this mutation plays an important role in LS patients of this pedigree based on in vivo and vitro study.
Keywords: Lynch syndrome, genetic variation, mismatch repair gene, MSH2, chemotherapy resistance
Introduction
Colorectal cancer is one of the largest public health problems in the world. It is clear that 20% to 30% of colorectal cancers (CRCs) are hereditary, with 5% to 10% related to a known genetic syndrome such as Lynch syndrome (LS).1,2 LS is an autosomal dominant syndrome occurring cancers in colorectum, endometrium, stomach, small bowel, ovarian, ureter, renal pelvis, brain, hepatobiliary tract and sebaceous gland.3–6 Available data show that LS accounts for 1% to 3% of all CRC, and LS patients may suffer from early cancer development and increased risk of extra-colonial manifestations.7–11 The mean age of LS onset is 44–61 years, which is younger than that of sporadic CRC patients.12–15 Besides, colorectal cancer risks are reported to be as high as 75% in LS.16 Furthermore, the average time from onset of a polyp to the onset of carcinoma is much shorter in LS (2–3 years) compared with sporadic colorectal cancer (4–10 years).17,18
The etiology of LS is a deficiency of mismatch repair (MMR) system that responsible for the surveillance and correction of errors in DNA during replication, repair and recombination.19,20 Once pathogenic mutations are found to contribute to MMR function deficiency clearly, mutation carriers can be benefited from genetic counseling. Thus, screening pathogenic mutations in MMR genes is critical for diagnosis, monitoring and management of LS.
MutS homologue 2 (MSH2), mutL homologue (MLH1), mutS homologue 6 (MSH6), post-meiotic segregation increased 2 (PMS2) and epithelial cell adhesion molecule (EPCAM) are found to play important roles in MMR.21–24 Germline mutations in MLH1, MSH2, MSH6, PMS2 and EPCAM genes accounts for approximately 50%, 40%, 7–10%, less than 5% and 3% cases of LS.25–28 LS patients with mutations in these genes have a higher rate of spontaneous somatic mutations in microsatellite sequences that result in microsatellite instability (MSI).29 Both large rearrangements and single nucleotide variant (SNV) in these genes may alter MMR protein function.22,30 Insertion, deletion or nonsense SNVs can cause truncated proteins, while exonic or intronic variants located in splicing sites can result in aberrantly spliced mRNA transcripts.31,32 Since there are many mutations in MMR genes, it is important to identify the potential causative genetic variants of LS.
MSH2 was the first identified LS causative gene.33 It interacts with MSH6 or MSH3 to form the MutSα/β complexes, and they translocate into the nucleus to bind with DNA in order to initiate the repair process.33 Many evidences indicated that MSH2 plays a vital role in the MMR system, and numerous germline mutations of MSH2 are discovered.34 At present, 119 variants of MSH2 have been reported in the Chinese population, including 51 substitutions, 25 frameshifts, 10 nonsenses, 27 deletions, 9 insertions and 4 duplications.35 However, how many variations of them are causative genetic variants of LS remains unknown. In addition, it is unclear if there are unknown mutations that contribute to LS. Anyway, clarifying the pathogenic role of genetic mutations in MSH2 may contribute to diagnose, monitor and manage LS.
In this study, we identified an insertion mutation (rs1114167810, c.420dup, p.Met141Tyrfs) in MSH2 which has not been reported in LS using whole genome-wide sequencing (WGS). We further confirmed that this mutation plays an important role in LS patients of this pedigree based on in vivo and vitro study.
Methods and Materials
Patients and Samples
Samples (including 102 whole blood samples and 3 colorectal cancer tissues) of patients and the family, as well as basic information, were collected from Xiangya Hospital, Central South University (Changsha, Hunan, China) from 2015 to 2018. The purpose of this study was informed to all patients or their relatives, and consent forms were signed by them. The study protocol was approved by the Ethics Committee of Xiangya School of Medicine, Central South University.
DNA Extraction
Germline DNA for whole genome sequencing and Sanger sequencing was extracted from the whole blood of the participants using the QIAamp DNA Blood Midi Kit (Qiagen; Valencia, CA, USA). Formalin-fixed paraffin-embedded (FFPE) DNA for microsatellite instability (MSI) analysis was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen; Valencia, CA, USA).
Whole Genome Sequencing
Firstly, DNA was sheared using Covaris focused-ultrasonication (Covaris, MA, USA), and DNA fragments were enriched by 6 cycles of PCR. Then, libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer. Finally, the DNA libraries were sequenced on Illumina Hiseq platform and 150-bp paired-end reads were generated as raw reads.
Data Processing
Raw reads in FastQ format that contained adapter contamination and low-quality/unrecognizable nucleotides were discarded using Trimmomatic (0.36 version). Reads after quality control were aligned to the UCSC human reference genome (GRCh37) using BWA.37 Samtools and Picard were used for sorting, removing PCR duplicates, and building an index for the bam files. Then, base quality recalibration was performed by GATK to generate final BAM files for mutation calling.38 Germline single nucleotide variations (SNVs) and small insertions and deletions (InDels) were called by GATK, the resulting variants were annotated and prioritized by ANNOVAR.39 The pathogenicity of missense variants was evaluated by SIFT, PolyPhen2, and MutationTaster. We focused on pathogenic and expected pathogenic mutations in several genes (including: MLH1, MSH2, MSH6, PMS2, EPCAM, APC, MUTYH, PTEN, STK11, TP53, SMAD4 and BMPR1A) that were defined to be high-risk genes in CRC by the American College of Medical Genetics. Only coding and splice region variants were considered and all variants identified in this study were manually inspected using Integrative Genomics Viewer (IGV version 2.3.86).40 Control-FREEC and Breakdancer were utilized to detect copy number variations (CNVs) and structural variations, respectively,41,42 All these variant results were visualized using Circos.43
Sanger Sequencing
Sanger sequencing was used to validate the candidate variants identified above. PCR primers were designed with Primers5 tools. PCR amplification was carried out in ABI 9700 Thermal Cycler. Sequence data comparisons and analysis were performed by SeqMan (DNASTAR, Madison, Wisconsin, USA). Sequences of primers for identifying these candidate loci are given in Table S1.
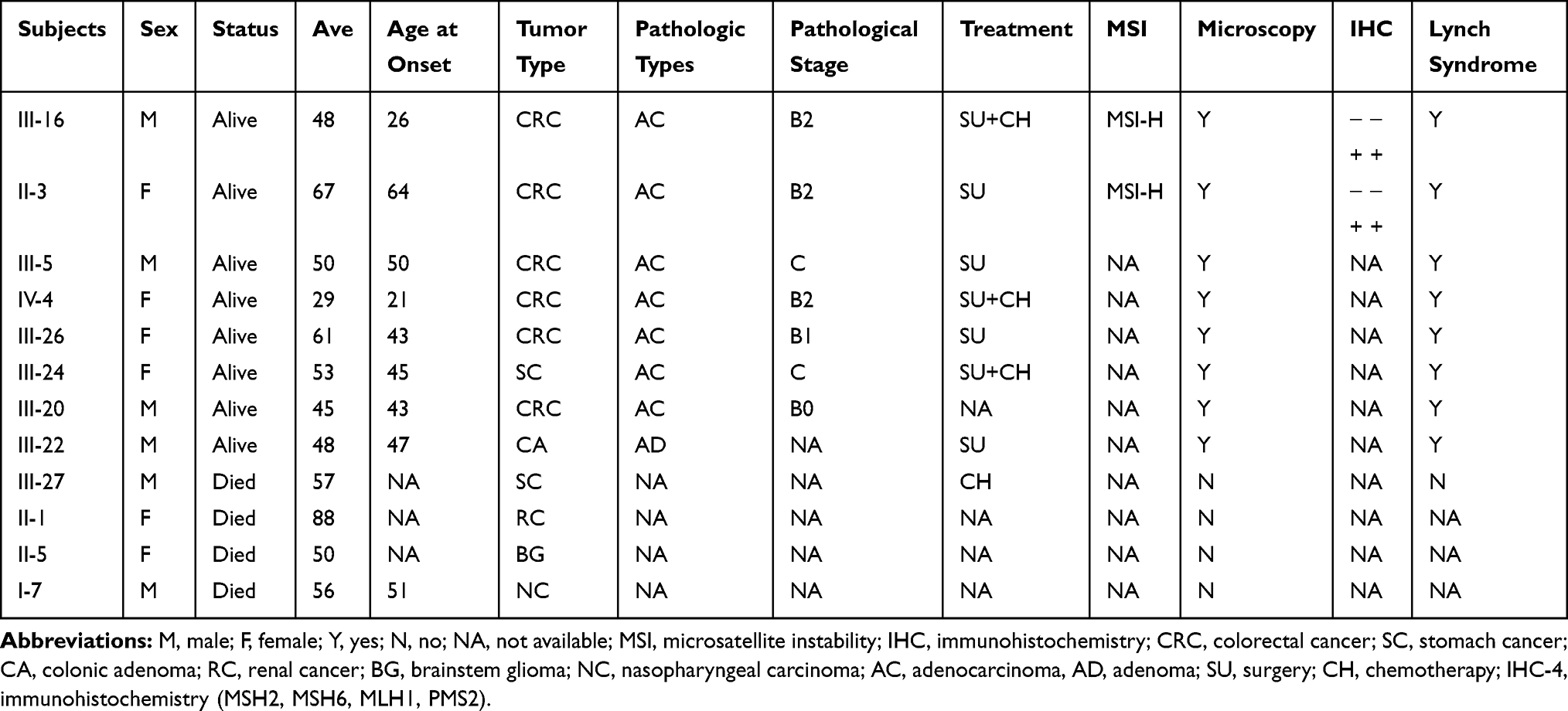 |
Table 1 Clinical Characteristics of Patients in LS Family |
Microsatellite Instability Analysis
MSI was performed for the proband’s mother (II-3) with genomic DNA which was isolated from tumor samples and corresponding blood, respectively. A 5-marker panel including two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D2S123, D5S346 and D17S250) which recommended by the National Cancer Institute Workshop on MSI for Cancer Detection and Familial Predisposition was used.44 Oligonucleotide primers were fluorescently labelled and PCR products were evaluated using 3500DX Genetic Analyzer. Tumors were classified as highly unstable (MSI-H) if at least 40% of the markers showed instability.
Immunohistochemistry
All tissue specimens were collected via biopsy of paraffin-embedded samples for immunohistochemistry (IHC) analysis in the Pathology Department of Xiangya Hospital or Hunan Provincial Tumor Hospital. Tissue sections (4 um thick) were cut from paraffin-embedded blocks. Antibodies used were: Anti-Human MutL Protein Homolog 1, Clone ES05 (DAKO ref:IR079), Anti-Human Postmeiotic Segregation, Clone EP51 (DAKO ref: IR087), Anti-Human MutS Protein Homolog 6, Clone EP49 (DAKO ref: IR086).
Plasmid Construction
We constructed both wild-type and c.420dup mutant MSH2 expression plasmids in this study. To get the wild-type MSH2 plasmid, full-length MSH2 (NM_000251.2) coding regions were cloned into the pCDNA3.1 vector (Invitrogen, CA, USA) at the multiple cloning site of BamHI and EcoRI. To generate the mutant (c.420dup; p.Met141Tyrfs) MSH2 plasmids, site-directed mutagenesis was carried out. All constructs were directly sequenced to confirm the inserted fragments. Primers used in plasmid construction were also presented in Table S1.
Cell Culture and Transfection
Human colon cancer cell HCT116 cells were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). These cells were cultured in RPMI-1640 media, supplemented with 12% fetal bovine serum (FBS) at 37°C under an atmosphere of 95% air and 5% CO2. The cells for transfection were cultured on six-well plates and grown till 70–90% confluent. Wild-type and c.420dup mutant MSH2 constructs were separately transfected into the cells in comparable amounts using Lipofectamine 3000 (Invitrogen Corporation, Carlsbad, CA, USA).
Western Blot
Twenty-four hours after transfection, the cultured HCT116 cells were harvested and analyzed by Western blotting with an antibody to MSH2. Proteins were extracted using RIPA buffer mixed with protease inhibitors (1:100), phenyl methyl sulfonyl fluoride (PMSF, 1:100) and dithiothreitol (DTT, 1:100) at 4°C for 30-mins incubation. The lysate was centrifuged at 13,000 rpm at 4°C for 15 min. The supernatants were collected and protein concentrations were measured using the BCA method. Forty micrograms of extracts were denatured in SDS loading buffer (TaKaRa), loaded on the gel, separated by 12% SDS-PAGE gel electrophoresis and transferred to PVDF membranes (Millipore, Massachusetts, USA). The membranes were blocked using 5% skim milk and incubated at 4°C overnight with the following primary antibodies: anti-MSH2 antibody (1:1000, CST, China) and anti-β-catenin (1:1000, CST, China), followed by anti-rabbit IgG (CST, China) and anti-mouse IgG (CST, China) at a 1:3000 dilution for 2 hrs at room temperature. Enhanced chemiluminescence (ECL) reagents (Super Signal West Femto Maximum Sensitivity Substrate, Thermo Fisher Scientific, Rockford, USA) were used and the signal was detected with Bio-Rad ChemiDoc XRS. The intensity of them was normalized relative to β-actin bands and was analyzed by ImageJ software. The expression level of MSH2 was calculated as the mean of three independent experiments.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
We extracted total RNA from tissue specimens using Trizol reagent (Takara) according to the manufacturer’s protocol. Nano Drop Spectrophotometer (Shimadzu Biotech, Beijing China) and gel electrophoresis were employed to measure the quality and quantity of extracted RNA. The isolated RNA concentration was calculated and normalized with RNase-free water and then reverse-transcribed into cDNA using PrimeScript™ RT reagent kit with gDNA Eraser (RR047A; Takara, Dalian, China). All cDNA samples were stored at −80°C until use. Light Cycle@480 II (Roche, Basel, Switzerland) was employed to conduct qRT-PCR by using TB GReen™ Premix Ex Taq™ II (Tli RNaseH Plus, Code No: RR820A, Takara Bio Inc.). The qRT-PCR amplification was performed as follows: an initial denaturation at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s, 60°C for 30 s. The 2−ΔΔCt was used to quantify the fold change of MSH2 expression in HCT116 cell. The relative expressions of target genes were calculated after normalization against a reference gene (GAPDH). All primers used for qPCR were listed in Tables.
Cell Viability Analysis
Oxaliplatin, 5-fluorouracil and Irinotecan were purchased from Dalian Meilun Biology Technology Co., Ltd. (Dalian, P.R. China) and was diluted with DMSO (Sigma, St Louis, MO) and stored at the concentration of 100 mM. Cell viability was detected by the MTS approach according to the protocol for Cell Titer 96 Aqueous-One-Solution Cell Proliferation Assay kit (MTS). HCT116 cells transfected with wild and mutation MSH2 were seeded in 96-well plates at a density of 5 × 103 cells in 100-µL medium. Plated cells were incubated with Oxaliplatin, 5-fluorouracil and Irinotecan (CST) for 48 hrs respectively. The medium was then replaced by 100-µL MTS and RPMI-1640 media with a ratio of 1:9. Cell viability was then obtained by measuring the absorbance at a wavelength of 450 nm. All experiments were performed at least three times. The IC50 was calculated and the dose–response curves were depicted using GraphPad Prism 6.0 program.
Statistical Analysis
Statistical analysis was performed with SPSS 16.0 (SPSS Inc, Chicago, IL, USA). Results expressed as mean ± SD were analyzed using the Student’s t test. Differences were considered significant when P < 0.05.
Information Collection of MSH2
The information on annotated domains and conservations of MSH2 is collected in Pfam (version 32.0, http://pfam.xfam.org/protein/P43246). The structure prediction of MSH2 is performed by Phyre2 (version V 2.0, http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index).
Results
A 5-Generation LS Pedigree Was Found
The proband (III-16) was a 48-year-old male, whose personal and family history meet Amsterdam II criteria for LS. He was diagnosed with rectal cancer at the age of 28, and then received radical resection of rectal carcinoma and post-operation radiotherapy. At the age of 33, he developed metastatic hepatic cancer and then underwent right hemi-hepatectomy and 5 cycles of chemotherapy. When he was 40, he was diagnosed with rectal cancer and received both surgery and 5 cycles of chemotherapy. He developed colon cancer and subsequently received transverse colonic ostomy and partial small intestine resection when he was 46. Later, he developed sigmoid polyps and rectal polyps at the age of 48 and 49, respectively. The whole process of diagnosis and therapy of this patient is summarized in Figure 1.
 |
Figure 1 Clinical diagnosis and treatment process of the proband (III-16). HE staining of colon tissue from proband showing a colon adenocarcinoma. Immunohistochemical staining results for adenocarcinoma in proband. There is a loss of expression of MSH2 and MSH6 in the neoplastic cells. The tumor cells retain the nuclear expression of MLH1 and PMS2. |
Then, we investigated the five generations of the proband’s family and found proband’s family showed a typical LS pedigree based on Amsterdam II criteria (Figure 2). Among 102 members, five of them (II-3, III-5, III-20, III-26 and IV3) were diagnosed as colorectal cancer. III-24 and III-22 were with stomach cancer and colon adenoma, respectively. Another three family members (II-1, II-5 and II-7) had already died of various cancer. II-7 was diagnosed with nasopharyngeal carcinoma at 50 and died at 56. II-5 was diagnosed with brain stem glioma at the age of 50, and II-1 developed Kidney cancer. Detailed patient family history was collected and summarized in Table 1.
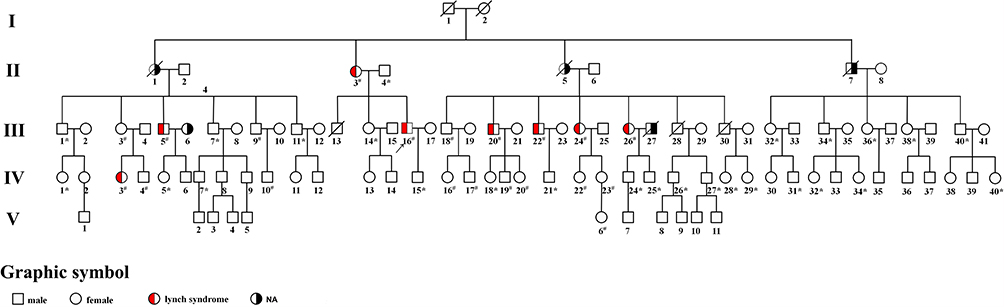 |
Figure 2 Pedigree of the Chinese family with LS. The arrow indicates the proband (III-16). Squares and circles denoted males and females, respectively. Red filled symbols indicate LS members, black filled symbols indicate those members with tumors cannot be diagnosed as Lynch syndrome, and empty symbols indicate unaffected individuals. Sign “#” indicates that family members were tested for mutations and found to carry the mutation in the pedigree; Sign “*” indicates family members who were tested and found not to carry the mutation. |
MSI Was Found in the Pedigree
Since MSI is a symbol of deficiency MMR function, we then performed the MSI test in this pedigree. Tumors from a member who was diagnosed as colorectal cancer (II-3) were collected and used for the MSI test. The 5-marker panel including two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D2S123, D5S346 and D17S250) were used. BAT25 and D2S123 were MSS, while BAT26, D17S250 and D5S346 were MSI (Figure 3A). These results indicated that II-3 was confirmed to MSI-high state because three of five tested loci were found altered (60% MSI).
 |
Figure 3 MSI state of the proband’s mother (II-3). (A) Colon tumor from II-3 showing instability for BAT26, D17S250 and D5S346. (B) Immunohistochemical staining of mismatch repair proteins in II-3 are the same as those in III-16: positive nucleus staining for MLH1 and PMS2, while absent of nuclear staining for MSH2 and MSH6 protein in tumor tissue. |
The Deficiency of MSH2 and MSH6 Were Found in the Pedigree
MSI is often correlated with defect MMR function deficiency, we then detected the expression of MMR proteins (MLH1, MSH2, MSH6 and PMS2) directly to make sure if MMR deficiency exists in this pedigree. Immuno-histochemical staining (× 20) of tumor tissues from two colorectal cancer members (II-3 and III-16) both demonstrate that MSH2 and MSH6 are absent in the tumor cell nuclei while they are presented in the normal internal control (non-tumor cells) (Figure 3B). The II-3 is the mother of III-16 so that they may suffer from a heritable factor. These results are summarized in Table 2.
 |
Table 2 MMR Related Status of Proband and His Mother |
A Frameshift INDEL Mutation in MSH2 Was Found in the Pedigree
To explore the genetic factor for LS, whole-genome sequencing was performed on the proband (III-16). A total of 4023,831 germline SNVs and 852,009 germline InDels were called. All variations, including SNVS, InDels, copy number variations and structural variations are shown in Figure 4A. The distribution of SNVs and Indels in genome regions and the type of mutation in the coding region of exon are showed in Figure 4B. SNVs and InDels which were identified or expected as pathogenic mutations in CRC by the American College of Medical Genetics in several genes including: MLH1, MSH2, MSH6, PMS2, EPCAM, APC, MUTYH, PTEN, STK11, TP53, SMAD4 and BMPR1A. High frequency of variation was found in high-risk genes of Lynch syndrome. And we found that seven mutations which locate in coding and splice region of all these genes (MLH1, MSH2, MSH6, PMS2, EPCAM, MUTYH, PTEN) may relate to the pathogenesis of LS according to their mutant type. Among these mutations, a novel heterozygous insertion (c.420dup, p.Met141Tyrfs) in MSH2 gene [NCBI Reference sequence NM_000251.2] was found in the mutation list. Compared with other SNPs in these genes, this mutation has not been reported in the literature to be associated with colorectal pathogenicity in pedigree, and the frequency of this mutation has not been reported, too. The whole process of screening candidate SNPs for LS is shown in Figure 4C, and the identified genes were listed in Table S1.
 |
Figure 4 Whole genome sequencing results of the proband (III-16). (A) The landscape of all types of variations in this patient. The columns in the first circle indicated the frequency of single nucleotide polymorphism, while red, yellow, green and blue indicated alt allele “A”, “G”, “C” and “T” mutations, respectively. The second circle indicated the frequency of InDels, while red and green indicated insertion and deletion, respectively. The third circle indicated the CNV of this patient, while red and green indicated increased copy number and decreased copy number, respectively. The copy number ranges from 0 to 161. The inner-circle referred to the SV of this patient. (B) The distribution of SNVs and InDels in genome regions and the type of mutations in the coding region of the exon. The color of blue and yellow indicated SNVs, and the color of green and red-brown indicated InDels. (C) The whole process of screening candidate SNPs for LS. |
Next, we found that this mutation can result in a frameshift and introduce a stop codon (stop in exon 3) in 10 amino acids downstream. So that this mutation was predicted to produce a truncated MSH2 protein as Figure 5A presented, and the structure of full-length MSH2 which predicted by Phyre2 is showed in Figure 5B. Besides, we found that this mutation is highly conserved across several species (Figure 5C). These results suggest that this mutation may play an important role in the function of the MSH2 protein.
 |
Figure 5 The truncation mutation c.420dup in MSH2. Information of this WGS-identified mutation is presented in this figure. (A) The sequence and amino acid change caused by this mutation is indicated in red color. This mutation can cause a large part of domains losing and all annotated domains of MSH2 are marked in different colors. (B) The structure of MSH2 which predicted by Phyre2 is showed. Different colors indicated different domains as presented in (A). (C) The conservation of MSH2 across species is presented. Different colors in the right panel indicated different domains as presented in (A). |
The Frameshift Mutation Was Found in All Cancer Diagnosed Members of This Pedigree
Sanger sequencing, as the golden standard of variation identifying, was performed on this patient (III-16) to validate some of these variants. As Figure 6A and B showed, our result can be validated by Sanger sequencing. To make sure if this mutation is heritable or not, Sanger sequencing was performed on all other members with tumors (II-3, III-5, III-18, III-20, III-22, III-24 and IV3) in the pedigree. Surprisingly, all these members carry this insertion in MSH2 (Figure 2 and Table S1). In other words, this mutation is heritable. Their young offspring also have a great chance to carry this variation. We also performed Sanger sequencing of other mutations on these members. Sequencing results of other mutations were provided in Table S1. Some family members also carry other mutations including MLH1(rs1800734), EPCAM (rs1126497) and MUTYH (rs3219468). The MLH1 variant (rs1800734), which has been reported to be associated with the risk of several cancers including CRC, glioblastoma, endometrial cancer, and lung cancer.45–48 But II-7 was diagnosed with nasopharyngeal carcinoma. However, no literature has reported that patients with nasopharyngeal carcinoma have been diagnosed with Lynch syndrome until now. And His healthy descendants (III-36 and III-40) also carry this mutation. Moreover, homozygous mutation of this SNP was also found carried by two healthy members (III-20 and IV-5), indicated that this mutation is also present in the general population. In addition, unlike the absence of MSH2, our immunohistochemistry (IHC) experiments revealed that the MLH1 protein is still expressed in the tumor tissues. This result indicated that the MSH2 is severely affected by the truncated mutation rs1114167810, while MLH1 is still expressed in this patient. Indeed, the frequency of this mutation in the general population is also as high as 0.24. These evidences indicate that this mutation does not play a major role in the pathogenesis of this family’s LS. Similarly, the mutation frequencies of both EPCAM (rs1126497) and MUTYH (rs3219468) are also quite high, which indicated that these two mutations are unlikely to cause LS. These evidences indicated that rs1114167810 in MSH2 might be the leading cause of this family’s Lynch syndrome.
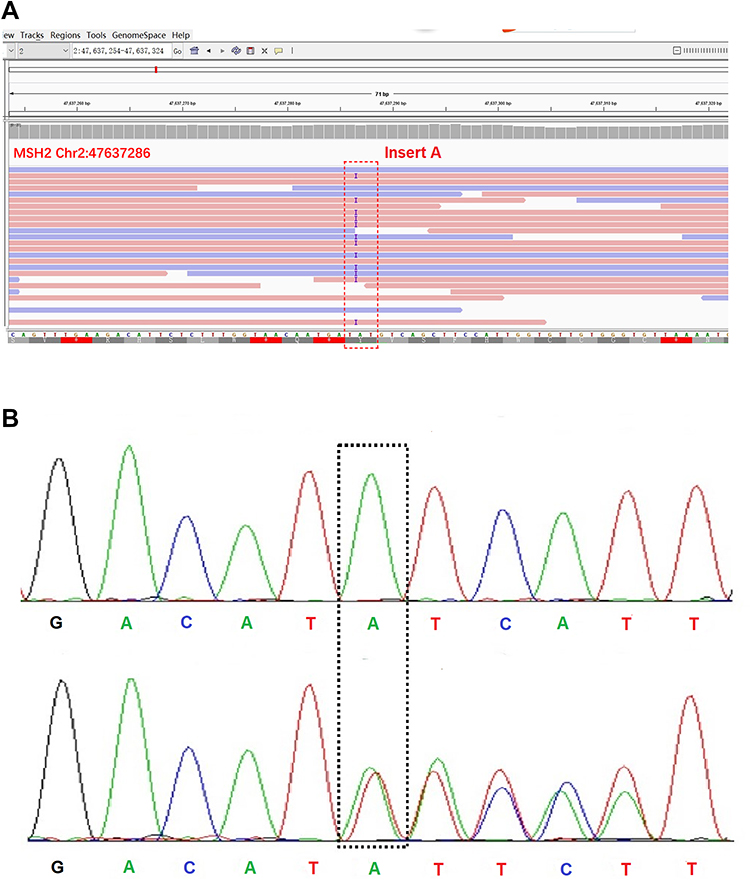 |
Figure 6 Identification of this truncation mutation c.420dup in MSH2. (A) Visualization of MSH2 sequencing reads containing c.420dup mutation with the Integrative Genomics Viewer (IGV), the red box represents the insertion of the A base. (B) Confirmation of c.420dup mutation in the proband and His family members by Sanger sequencing. |
The Mutated MSH2 Causes a Decreased Protein Level and an Increased Sensitivity to Anti-Cancer Drugs in vitro
In order to clarify the effect of the mutation on expression and function of MSH2, His-tagged wild-type or mutant MSH2 constructs were transiently expressed in HCT116 cells. Western blot analysis was carried out using an anti-MSH2 antibody after 48 h. In transfected cells, a 100-kDa band was detected, which agreed with the size of the full-length MSH2 protein in control. The mutant MSH2 showed an obvious decrease in the level of MSH2 compared with the wild-type protein, since the expression levels of the control and mutant samples were comparable (Figure 7A). MSH2 exhibited a markedly lower expression in HCT116 cells with mutant MSH2 constructs than that with wild-type MSH2 constructs (Figure 7B).
 |
Figure 7 Effect of this truncation mutation c.420dup in MSH2 on the expression of MSH2 expression and cellular response to chemotherapy drugs. (A) The MSH2 expression level in transfected colorectal cancer cells (HCT116). (B) Expression level of MSH2 mRNA in HCT116 cells transfected with wild-type and c.420dup mutant MSH2 constructs. The data are shown the mean of 2–ΔΔCt. (C) Dose–response analyses of 5-fluorouracil effect on inhibition of viable cell numbers (cell viability) were carried out in HCT116 cells which were transfected with wild-type and c.420dup mutant MSH2 constructs. The curve consisted of three independent experiments. (D) Dose–response analyses of Oxaliplatin effect on inhibition of viable cell numbers (cell viability) were carried out in HCT116 cells which were transfected with wild-type and c.420dup mutant MSH2 constructs. The curve consisted of three independent experiments. (E) Dose–response analyses of Irinotecan effect on inhibition of viable cell numbers (cell viability) were carried out in HCT116 cells which were transfected with wild-type and c.420dup mutant MSH2 constructs. The curve consisted of three independent experiments. |
Many studies suggested that MMR genes played important roles in chemotherapy.49–52 Is this mutation important for the prediction of chemotherapy? Considering that the proband’s chemotherapy regimen is oxaliplatin and 5-fu, and irinotecan is a commonly used chemotherapy drug for colorectal cancer, we use these three drugs for cell viability experiments. Two transfected cells were incubated with different concentrations of 5-FU, Oxaliplatin and Irinotecan. In mutant MSH2 transfected HCT116, the IC50 value for 5-fluorouracil is lower than that in HCT116 cells transfected with wild-type MSH2 constructs, although the p-value indicates an insignificant result (17.62 vs 20.49, P>0.05). The IC50 for Oxaliplatin and Irinotecan in HCT116 transfected with mutant MSH2 constructs is significantly lower than wild-type MSH2 constructs.(Oxaliplatin, 10.85 vs 15.19, P<0.05; Irinotecan, 29.88 vs 41.84, P<0.05). (Figure 7C–E). These results indicated that this mutation could decrease the expression of MSH2 and increase the sensitivity of tumor cells to the anti-cancer drug.
Discussion
Germline deleterious defects in MMR genes, mainly MLH1, MSH2 MSH6 and PMS2 were found to be related with LS.21,22 Froggatt et al first independently reported the mutation of MMR genes is associated with Lynch syndrome.53 Nearly all LS and 10%~20% of sporadic colorectal cancer occur gene mutation of MMR gene.54 MMR genes are associated with several cellular functions including repair the DNA mismatch error, DNA double-strand breaks, DNA destabilization and apoptosis. In other words, the MMR proteins are very significant in maintaining both DNA stability and cell-cycle regulation.1,19,20 Immunohistochemical analysis of MMR protein status in the tumor and MSI status can provide useful clues to identify which MMR gene is involved in tumor pathogenesis: indeed, the lack of expression of one of the proteins or the demonstration of MSI-High status might also be relevant in suggesting a correlation with a pathogenic mutation. In this way, genetic testing of MMR genes can be applied to the diagnosis of LS. So far, a large number of pathogenic MMR gene mutations have been reported to be associated with LS in different countries and ethnic groups, in particular in MLH1 and MSH2.
In the present study, we describe a Chinese pedigree suffering from LS. A novel MSH2 mutation (rs1114167810, c.420dup, p.Met141Tyrfs) was identified in a 48-year-old LS patient and his family history fulfilled Amsterdam II criteria. We found some candidate gene mutations including rs1114167810 through whole genome-wide sequencing at first. Using Sanger sequencing, we verified that this insertion in MSH2 is carried by this patient. Sanger sequencing results showed that this mutation was found in all LS diagnosed family members, who underwent genetic tests. However, for non-Lynch syndrome member (II-7), we did not detect this mutation in his offspring. This means that the family of II7 did not inherit this pathogenic mutation, and thus did not cause the occurrence of Lynch syndrome. This result was perfectly explained the phenomenon in Figure 2. On the contrary, we found both Lynch syndrome and non-Lynch syndrome families carry MLH1 mutations (rs1800734). On the other hand, our IHC experiments only revealed the absence of MSH2 and MSH6 protein expression rather than PSM2 and MLH1 in the tumor tissues. In conclusion, we believe that the decrease of MSH2 plays a major role in the occurrence of Lynch syndrome in this family, while this truncated mutation rs1114167810 is the main causer. As for other found mutations with high frequency, we speculated that these variants did not play a major role in the development of LS. We also found that some members in this family carried three candidate variants in tumor-related genes: PTEN (rs71022512), PTEN (rs5786797) and PMS2 (rs374762935). But these tandem repeat mutations may be caused by the insertion of a mismatch repair gene. This assumption was not carried in this study and required further verification.
Although we did not experimentally confirm that this variant causes a truncated protein, we predicted that this variant would lead to the premature termination of codon production. The IHC result and our functional experiment also showed that the MSH2 (c.420dup, p.Met141Tyrfs) variant can decrease the expression of proteins and mRNA. We also investigated the effect of this variant on the response of colorectal cancer cells to clinically used chemotherapy drugs in this study. We found that this variant causes tumor cells (HCT116) to be sensitive to oxaliplatin and irinotecan, but has no effect on 5-fu sensitivity, which should be further validated in a clinical study. This is basically consistent with what has been previously described for Lynch syndrome-related 11 colorectal cancer patients by Maccaroni et al, although this mutation is not referred in 12 this study, perhaps because of its extremely low allele frequency. In addition, other literatures also supported that MMR deficient was associated with poor benefit from 5-FU treatment in CRC.51,55–57 In contrast, MMR deficiency sensitives CRC cells to oxaliplatin.58–61 Oxaliplatin’s specific 1,2-diaminocyclohexane (DACH) ligand prevents the MMR complex from binding to its DNA adduct, resulting in loss of repair capacity and subsequent apoptosis of tumor cells.58 But the clinical results about the relationship between MMR status and CPT-11 were inconclusive, Charara et al demonstrated that high microsatellite instability with loss of mismatch repair protein is predictive of an improved response to neoadjuvant treatment with 5-FU, CPT-11 and radiation therapy,62 while some evidences indicating that MMR status was not associated with the response to CPT-11 in CRC treatment.36 All in all, this means that this mutation may also serve as a target for a patient to predict chemotherapy response, which requires subsequent studies in the clinic to further confirm.
Altogether, our experimental evidence supports a pathogenic role for the MSH2 c.420dup mutation, reinforcing the importance of the variants in offering genetic counseling management, and surveillance in LS families.
Ethical Statement
This study is conducted in accordance with the declaration of Helsinki and is approved by the ethics committee of Xiangya Hospital, the trial number is 2018121120.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81873581, 81571151, 81641039), the Science and Technology Major Project of the Ministry of Science and Technology of Hunan Province, China (2017SK1032), Scientific Research Projects of Health Commission of Hunan Province (C2019176), the National Science and Technology Basic Resources Survey Project of China (2018FY100900) and Hunan Provincial Natural Science Foundation (2016JJ2164).
Disclosure
The authors declare no conflict of interest in this work.
References
1. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterol. 2010;138(6):2044–2058. doi:10.1053/j.gastro.2010.01.054
2. Liu Q, Tan YQ. Advances in identification of susceptibility gene defects of hereditary colorectal cancer. J Cancer. 2019;10(3):643–653. doi:10.7150/jca.28542
3. Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81(2):214–218. doi:10.1002/(ISSN)1097-0215
4. Weissman SM, Burt R, Church J, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J Genet Couns. 2012;21(4):484–493. doi:10.1007/s10897-011-9465-7
5. de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer. 2005;4(3):233–237. doi:10.1007/s10689-004-5811-3
6. Haraldsdottir S, Hampel H, Wei L, et al. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16(7):553–557. doi:10.1038/gim.2013.193
7. Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338(21):1481–1487. doi:10.1056/NEJM199805213382101
8. Samowitz WS, Curtin K, Lin HH, et al. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterol. 2001;121(4):830–838. doi:10.1053/gast.2001.27996
9. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterol. 1999;116(6):1453–1456. doi:10.1016/S0016-5085(99)70510-X
10. Canard G, Lefevre JH, Colas C, et al. Screening for Lynch syndrome in colorectal cancer: are we doing enough? Ann Surg Oncol. 2012;19(3):809–816. doi:10.1245/s10434-011-2014-7
11. Win AK, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol. 2013;20(6):1829–1836. doi:10.1245/s10434-012-2858-5
12. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851–1860. doi:10.1056/NEJMoa043146
13. Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. doi:10.1200/JCO.2008.17.5950
14. Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterol. 2005;129(2):415–421. doi:10.1016/j.gastro.2005.05.011
15. Sinicrope FA. Lynch Syndrome-Associated Colorectal Cancer. N Engl J Med. 2018;379(8):764–773. doi:10.1056/NEJMcp1714533
16. Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer. 2013;12(2):229–240. doi:10.1007/s10689-013-9615-1
17. Edelstein DL, Axilbund J, Baxter M, et al. Rapid development of colorectal neoplasia in patients with Lynch syndrome. Clin Gastroenterol Hepatol. 2011;9(4):340–343. doi:10.1016/j.cgh.2010.10.033
18. Winawer S, Fletcher R, Rex D, et al. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterol. 2003;124(2):544–560. doi:10.1053/gast.2003.50044
19. Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038.
20. Casey G, Lindor NM, Papadopoulos N, et al. Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. JAMA. 2005;293(7):799–809. doi:10.1001/jama.293.7.799
21. Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4(3):227–232. doi:10.1007/s10689-004-7993-0
22. Duraturo F, Liccardo R, Cavallo A, De Rosa M, Rossi GB, Izzo P. Multivariate analysis as a method for evaluating the pathogenicity of novel genetic MLH1 variants in patients with colorectal cancer and microsatellite instability. Int J Mol Med. 2015;36(2):511–517. doi:10.3892/ijmm.2015.2255
23. Duraturo F, Liccardo R, Cavallo A, De Rosa M, Grosso M, Izzo P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. Int J Cancer. 2011;129(7):1643–1650. doi:10.1002/ijc.v129.7
24. Duraturo F, Liccardo R, Izzo P. Coexistence of MLH3 germline variants in colon cancer patients belonging to families with Lynch syndrome-associated brain tumors. J Neuro oncol. 2016;129(3):577–578. doi:10.1007/s11060-016-2203-0
25. Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition – update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4–5):269–276. doi:10.1155/2004/305058
26. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30(2):197–203. doi:10.1002/humu.20942
27. Kuiper RP, Vissers LE, Venkatachalam R, et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat. 2011;32(4):407–414. doi:10.1002/humu.21446
28. Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3ʹ exons of TACSTD1. Nat Genet. 2009;41(1):112–117. doi:10.1038/ng.283
29. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi:10.1093/jnci/djh034
30. Duraturo F, Cavallo A, Liccardo R, et al. Contribution of large genomic rearrangements in Italian Lynch syndrome patients: characterization of a novel alu-mediated deletion. Biomed Res Int. 2013;2013:219897. doi:10.1155/2013/219897
31. van der Klift HM, Jansen AM, van der Steenstraten N, et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med. 2015;3(4):327–345. doi:10.1002/mgg3.145
32. Liccardo R, De Rosa M, Izzo P, Duraturo F. Novel MSH2 splice-site mutation in a young patient with Lynch syndrome. Mol Med Rep. 2018;17(5):6942–6946. doi:10.3892/mmr.2018.8752
33. Fishel R, Wilson T. MutS homologs in mammalian cells. Curr Opin Genet Dev. 1997;7(1):105–113. doi:10.1016/S0959-437X(97)80117-7
34. Woods MO, Williams P, Careen A, et al. A new variant database for mismatch repair genes associated with Lynch syndrome. Hum Mutat. 2007;28(7):669–673. doi:10.1002/(ISSN)1098-1004
35. Bin Wu, Wuyang Ji, Shengran Liang, et al. A novel heterozygous germline deletion in MSH2 gene in a five generation Chinese family with Lynch syndrome. Oncotarget. 2017;8(33):55194–55203. doi:10.18632/oncotarget.19234
36. Braun MS, Richman SD, Quirke P, et al. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol. 2008;26(16):2690–2698. doi:10.1200/JCO.2007.15.5580
37. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi:10.1093/bioinformatics/btp698
38. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi:10.1038/ng.806
39. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
40. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi:10.1038/nbt.1754
41. Boeva V, Zinovyev A, Bleakley K, et al. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics. 2011;27(2):268–269. doi:10.1093/bioinformatics/btq635
42. Chen K, Wallis JW, McLellan MD, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–681. doi:10.1038/nmeth.1363
43. Naquin D, D’Aubenton-Carafa Y, Thermes C, Silvain M. CIRCUS: a package for Circos display of structural genome variations from paired-end and mate-pair sequencing data. BMC Bioinformatics. 2014;15:198. doi:10.1186/1471-2105-15-198
44. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257.
45. Miyakura Y, Tahara M, Lefor AT, Yasuda Y, Sugano K. Haplotype defined by the MLH1-93G/A polymorphism is associated with MLH1 promoter hypermethylation in sporadic colorectal cancers. BMC Res Notes. 2014;7:835. doi:10.1186/1756-0500-7-835
46. Niu L, Li S, Liang H, Li H. The hMLH1-93G>A polymorphism and risk of ovarian cancer in the Chinese population. PLoS One. 2015;10(8):e135822. doi:10.1371/journal.pone.0135822
47. Lo YL, Hsiao CF, Jou YS, et al. Polymorphisms of MLH1 and MSH2 genes and the risk of lung cancer among never smokers. Lung Cancer. 2011;72(3):280–286. doi:10.1016/j.lungcan.2010.10.009
48. Rodriguez-Hernandez I, Perdomo S, Santos-Briz A, et al. Analysis of DNA repair gene polymorphisms in glioblastoma. Gene. 2014;536(1):79–83. doi:10.1016/j.gene.2013.11.077
49. Liu JY, Qian CY, Gao YF, Chen J, Zhou HH, Yin JY. Association between DNA mismatch repair gene polymorphisms and platinum-based chemotherapy toxicity in non-small cell lung cancer patients. Chin J Cancer. 2017;36(1):12. doi:10.1186/s40880-016-0175-2
50. Ma J, Zhang Y, Shen H, et al. Association between mismatch repair gene and irinotecan-based chemotherapy in metastatic colon cancer. Tumour Biol. 2015;36(12):9599–9609. doi:10.1007/s13277-015-3723-5
51. Zhang CM, Lv JF, Gong L, et al. Role of deficient mismatch repair in the personalized management of colorectal cancer. Int J Environ Res Public Health. 2016;13:9. doi:10.3390/ijerph13090892
52. Du P, Zhao H, Peng R, et al. LncRNA-XIST interacts with miR-29c to modulate the chemoresistance of glioma cell to TMZ through DNA mismatch repair pathway. Biosci Rep. 2017;37:5. doi:10.1042/BSR20170696
53. Froggatt NJ, Koch J, Davies R, et al. Genetic linkage analysis in hereditary non-polyposis colon cancer syndrome. J Med Genet. 1995;32(5):352–357. doi:10.1136/jmg.32.5.352
54. Pu C, Ren W, Sun Z, et al. Human mutL homolog 1 expression characteristic and prognostic effect on patients with sporadic colorectal cancer. Int J Clin Exp Med. 2015;8(10):19652–19661.
55. Bras-Goncalves RA, Pocard M, Formento JL, et al. Synergistic efficacy of 3n-butyrate and 5-fluorouracil in human colorectal cancer xenografts via modulation of DNA synthesis. Gastroenterol. 2001;120(4):874–888. doi:10.1053/gast.2001.22440
56. Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterol. 1999;117(1):123–131. doi:10.1016/S0016-5085(99)70558-5
57. Tokunaga E, Oda S, Fukushima M, Maehara Y, Sugimachi K. Differential growth inhibition by 5-fluorouracil in human colorectal carcinoma cell lines. Eur J Cancer. 2000;36(15):1998–2006. doi:10.1016/S0959-8049(00)00200-8
58. Raymond E, Chaney SG, Taamma A, Cvitkovic E. Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol. 1998;9(10):1053–1071. doi:10.1023/A:1008213732429
59. Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. doi:10.1056/NEJMoa032709
60. Des GuetzG, Schischmanoff O, Nicolas P, Perret GY, Morere JF, Uzzan B. Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer. 2009;45(10):1890–1896. doi:10.1016/j.ejca.2009.04.018
61. Aebi S, Fink D, Gordon R, et al. Resistance to cytotoxic drugs in DNA mismatch repair-deficient cells. Clin Cancer Res. 1997;3(10):1763–1767.
62. Charara M, Edmonston TB, Burkholder S, et al. Microsatellite status and cell cycle associated markers in rectal cancer patients undergoing a combined regimen of 5-FU and CPT-11 chemotherapy and radiotherapy. Anticancer Res. 2004;24(5B):3161–3167.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.