Back to Journals » Infection and Drug Resistance » Volume 11
A genomic infection control study for Staphylococcus aureus in two Ghanaian hospitals
Authors Donkor ES ![]() , Jamrozy D, Mills RO
, Jamrozy D, Mills RO ![]() , Dankwah T, Amoo PK, Egyir B, Badoe EV
, Dankwah T, Amoo PK, Egyir B, Badoe EV ![]() , Twasam J
, Twasam J ![]() , Bentley SD
, Bentley SD
Received 8 March 2018
Accepted for publication 15 May 2018
Published 11 October 2018 Volume 2018:11 Pages 1757—1765
DOI https://doi.org/10.2147/IDR.S167639
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Eric S Donkor,1 Dorota Jamrozy,2 Richael O Mills,3,4 Thomas Dankwah,3 Philip K Amoo,5 Beverly Egyir,6 Ebenezer V Badoe,7 Joana Twasam,8 Stephen D Bentley2
1Department of Medical Microbiology, School of Biomedical and Allied Health Sciences, College of Health Sciences, University of Ghana, Accra, Ghana; 2Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; 3Central Laboratory, Korle-Bu Teaching Hospital, Accra, Ghana; 4Department of Biomedical Sciences, University of Cape Coast, Cape Coast Ghana; 5Public Health Unit, Korle-Bu Teaching Hospital, Accra, Ghana; 6Bacteriology Unit, Noguchi Memorial Institute for Medical Research, University of Ghana, Accra, Ghana; 7Department of Child Health, School of Medicine and Dentistry, University of Ghana, Accra, Ghana; 8Laboratory Unit, Lekma Hospital, Accra, Ghana
Background: Whole genome sequencing analysis (WGSA) provides the best resolution for typing of bacterial isolates and has the potential for identification of transmission pathways. The aim of the study was to apply WGSA to elucidate the possible transmission events involved in two suspected Staphylococcus aureus hospital outbreaks in Ghana and describe genomic features of the S. aureus isolates sampled in the outbreaks.
Methods: The study was carried out at Korle-Bu Teaching Hospital and Lekma Hospital where the suspected outbreaks occurred in 2012 and 2015, respectively. The S. aureus isolates collected from the two hospitals were from three sources including carriage, invasive disease, and the environment. Whole genome sequencing of the S. aureus isolates was performed and the sequence reads were mapped to the S. aureus reference genome of strain USA300_FPR3757. A maximum-likelihood phylogenetic tree was reconstructed. Multilocus sequence typing together with the analysis of antimicrobial resistance and virulence genes were performed by short read mapping using the SRST2.
Results: The S. aureus isolates belonged to diverse sequence types (STs) with ST15 and ST152 most common. All isolates carried the blaZ gene, with low prevalence of tetK and dfrG genes also observed. All isolates were mecA negative. The pvl genes were common and observed in distinct lineages that revealed diverse Sa2int phages. At Korle-Bu Teaching Hospital, the genomics data indicated several transmission events of S. aureus ST15 involving contamination of various surfaces in the pediatric emergency ward where the outbreak occurred.
Conclusion: The pattern of dissemination of the ST15 clone in the emergency ward of Korle-Bu Teaching Hospital highlights a basic problem with disinfection of environmental surfaces at the hospital. Diverse phage population rather than a single highly transmissible phage type likely mediates the high prevalence of pvl genes among the S. aureus isolates.
Keywords: Staphylococcus aureus, sequencing, transmission, outbreak, Ghana
Introduction
Staphylococcus aureus causes a range of serious infections including meningitis, septicemia, pneumonia, endocarditis, and osteomyelitis.1 Methicillin-resistant S. aureus (MRSA) is of particular concern due to its extensive resistance to antibiotics and association with persistent outbreaks in hospital and community settings.2,3In addition to the public health significance of MRSA, methicillin-susceptible S. aureus (MSSA) is commonly implicated in bacteremia and skin and soft tissue infections (SSTI).4 A striking feature of clinical MSSA isolates from Africa is the relatively high prevalence of pvl genes,4–7 a pore-forming toxin that is associated with SSTI and severe necrotizing pneumonia.8 This characteristic of African MSSA isolates is of interest since the highly successful community-associated MRSA clones are also characterized by frequent carriage of pvl genes.9
The field of microbial genomics is advancing at a fast rate with the introduction of next-generation sequencing technologies. Whole genome sequencing analysis (WGSA) is superior to bacterial typing methods as it provides better resolution of bacterial isolates10 and is particularly suitable for highly clonal bacteria such as S. aureus.11 In previous studies, WGSA was applied to S. aureus and used to describe the intercontinental and local transmission of MRSA,12 investigate outbreaks,13,14 predict antimicrobial resistance,15 and type bacterial strains.10 Generally, applications of WGSA to study S. aureus and other microbial pathogens have been carried out to a very limited extent in many countries in Africa, including Ghana.
Most of what is known about S. aureus emanates from the developed world, partly because that is where most outbreaks are detected, but also because in Africa, the focus of attention tends to be more toward bacterial pathogens with a greater burden of mortality, such as Mycobacterium tuberculosis. Though S. aureus is one of the most common causes of infections reported in hospitals in Ghana,16 there are very limited surveillance data on the pathogen in the country. Recently in Ghana, several hospital-associated outbreaks of S. aureus infection have occurred, placing this pathogen high on the agenda of public health issues. In these outbreaks, extensive infection control investigations were carried out. However, characterization of the associated isolates was limited to basic phenotypic tests, leaving several important epidemiological questions unanswered, which require genomic analysis. To help address some of these concerns, we applied whole genome sequencing to retrospectively investigate carriage, environmental, and clinical isolates of S. aureus that were collected during two suspected hospital outbreaks in Accra, Ghana. The aim of the study was to generally describe genomic features of the S. aureus isolates, and the possible transmission events involved in the suspected outbreaks.
Methods
The study sites
The study sites were two hospitals located in Accra, the capital city of Ghana, namely, Korle-Bu Teaching Hospital (KBTH) and Lekma Hospital (LH). The KBTH is the largest hospital in Ghana and serves as a major referral center in Ghana, while LH is a district hospital. The KBTH has a bed capacity of 1,500 and has 17 departments,17 while LH has a bed capacity of 100 (J Twasam, Lekma Hospital, personal communication, 2017). Both hospitals have an infection control unit, which supervises and coordinates hygienic practices to prevent and control outbreaks. The infection control unit has close links with the bacteriology laboratory of the hospital, where routine identification of bacteria from clinical specimens and susceptibility testing is carried out. Facilities available in the bacteriology laboratory of KBTH and LH permit only limited phenotypic characterization of bacteria, and the most common organisms reported in the laboratory are Escherichia coli, S. aureus, and Pseudomonas aeruginosa.17
The study isolates
The S. aureus isolates used in this study were collected during suspected S. aureus outbreaks at the pediatric wards of KBTH and LH in 2012 and 2015, respectively. The isolates were from three sources: disease cases of patients; nasal carriage of patients, health workers, and hospital visitors; and equipment and surfaces in the hospital wards. Unfortunately, isolates from the initial S. aureus cases that constituted the suspected outbreaks and therefore led to the study had lost viability and therefore could not be sequenced. Overall, there were 17 isolates included in the study. Thirteen of the isolates were from KBTH and were collected mainly from wards in the pediatric unit of the hospital, particularly the emergency ward. The other four isolates were from LH, and these were collected from mothers of babies who were resident in the wards of the hospital. An epidemiological map showing time of sampling, location of patients, and S. aureus isolate source at KBTH, where most of the isolates were collected, is shown in Figure 1.
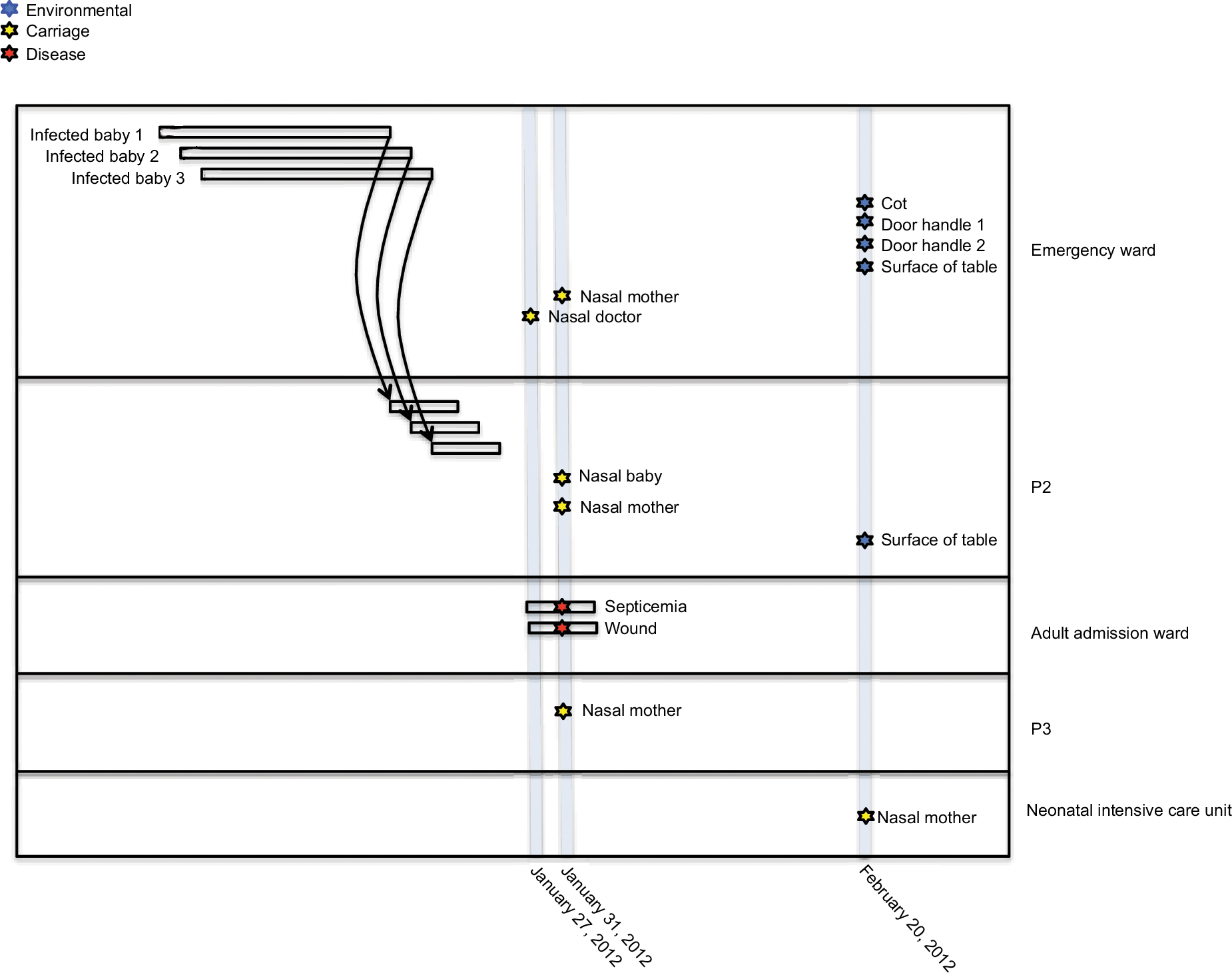 | Figure 1 An epidemiological map showing time of sampling, location of patients and sources of Staphylococcus aureus isolates at Korle-Bu Teaching Hospital. Notes: Three S. aureus cases (infected babies 1, 2, 3) at the emergency ward of Korle-Bu Teaching Hospital led to sampling at this hospital. Thirteen isolates were collected at various wards from a wide range of sources including disease cases, nasal carriage, equipment, and surfaces in the hospital wards. Background epidemiological data of the 13 S. aureus isolates have been reported in Table 1. Unfortunately, isolates from the initial S. aureus cases that led to the study, had lost viability and, therefore, could not be included in the study sample. P2, P3: different pediatric wards at Korle-Bu Teaching Hospital. |
Microbiological analysis
The study isolates were purified on blood agar and mannitol salt agar and confirmed to be S. aureus by the tube coagulase test.18 The isolates were screened for methicillin resistance by cefoxitin disk diffusion test.19
DNA sequencing and analysis
DNA was extracted and prepared for sequencing with the use of a kit (Promega DNA Sample Prep Kit; Epicenter, Madison, WI, USA). Tagged DNA libraries were created according to the Illumina protocol. Whole genome sequencing was performed on the Illumina HiSeq 2000 platform with 100-cycle paired-end runs. Sequence data for all isolates have been submitted to the European Nucleotide Archive (www.ebi.ac.uk/ena), and the accession numbers are provided in Table 1. Annotated assemblies were produced as previously described.20 Briefly, de novo assembly of whole genome sequences was performed using Velvet v1.2,21 with Velvet Optimiser v2.2.5 (http://bioinformatics.net.au/software.velvetoptimiser.shtml). Contigs were scaffolded with SSPACE, and sequence gaps were closed using GapFiller.22,23 The assembled contigs were annotated using Prokka v1.11 and an S. aureus-specific database from RefSeq.24,25
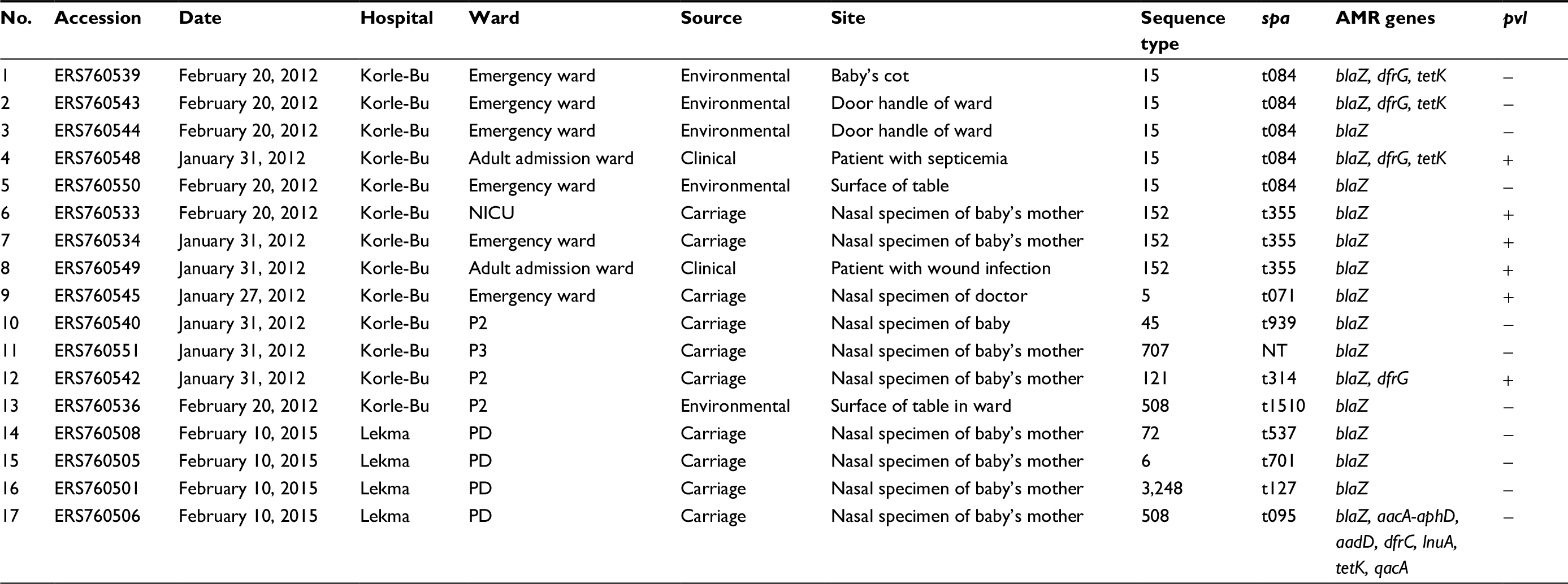 | Table 1 Epidemiological information and genotypes of the analyzed Staphylococcus aureus isolates Note: P2, P3: different pediatric wards at Korle-Bu Teaching Hospital. Abbreviations: AMR, antimicrobial resistance; PD, pediatric ward; NT, non-typeable/unknown; NICU, neonatal intensive care unit. |
To query evolutionary relationships between isolates, single-nucleotide polymorphisms (SNPs) were detected by mapping sequence reads to the S. aureus reference genome of strain USA300_FPR3757,26 using SMALT version 0.7.4 (http://www.sanger.ac.uk/science/tools/smalt-0). Mobile genetic elements (MGEs) were excluded from the whole genome alignment to create the core genome, which was used to reconstruct a maximum-likelihood phylogenetic tree using RAxML version 7.8.6.27
Multilocus sequence typing, together with the analysis of antimicrobial resistance and virulence gene carriage, was performed by short read mapping using the SRST2.28 For spa typing, the spa gene X region was extracted from whole genome assemblies using in silico PCR and previously described primers.29 The spa-type was then determined using an online spa-typer tool (http://spatyper.fortinbras.us/). Genome fragments representing MGEs were extracted from whole genome assemblies as described previously.30 The identified contigs were screened for presence of the pvl gene and antimicrobial resistance genes identified with SRST2. The distribution of the identified pvl-carrying Sa2int phage variants as well as resistance-associated MGEs was performed based on short read mapping with SRST2, further verified by assembly alignment with the use of MUMmer.31
Ethical considerations
For this study, the Ethical and Protocol Review Committee of the study hospitals waived ethical approval as it was regarded as part of routine surveillance measures for infection control. Additionally, as the samples used in the study were archived isolates, we could not obtain patients’ consent for use of their clinical data. However, all patients’ data and isolates were de-identified to ensure anonymity.
Results
Description of S. aureus disease cases
A suspected outbreak occurred at the Child Health Department of KBTH in January 2012 with three S. aureus cases. In the first case, S. aureus was isolated from the blood of a 4-month old baby girl (Figure 1; Baby 1) who had been admitted to the emergency ward. She had been at the emergency ward for 5 days and was later transferred to another ward (designated P2), where she died. The second case was reported 4 days after the first case and was isolated from cerebrospinal fluid of a 5-day old baby boy (Figure 1; Baby 2), who had been admitted to the emergency ward. The child was later transferred to the P2 ward. Two days after the second case, S. aureus was isolated from the blood of a 4-month old baby girl (Figure 1; Baby 3). Like the first two children, this baby had been admitted to the emergency ward and was later transferred to the P2 ward.
The Infection Prevention and Control Unit of KBTH requested an investigation into the situation following the death of the baby involved in the first case. It was identified that the babies from which S. aureus were obtained shared the same respiratory equipment. Babies shared the same cot and were connected by tubes (lines of giving set) to one oxygen cylinder. The clinical staff confirmed that they resort to this practice due to inadequate respiratory apparatus.
Following this development, KBTH took steps to disinfect materials (cot and bedding) of the deceased baby. Blood specimens for culture were obtained from babies who were in close proximity and shared tubes with the deceased baby, but the specimens grew no bacteria. A survey on nasal carriage of S. aureus was carried out among babies, their mothers, and health care staff in the unit as well as environmental screening. Nasal carriage of S. aureus and MRSA were 49.7% (88/147) and 4.8% (7/147), respectively. Environmental specimens collected from hospital equipment and surfaces in the wards yielded S. aureus and MRSA prevalence of 27.5% (39/142) and 11.3% (16/142), respectively. During this period, the bacteriology laboratory of KBTH maintained surveillance of S. aureus isolates recovered from all patients who visited the hospital; an average of five clinical isolates of S. aureus are reported in this laboratory daily.
At the LH, there was a suspected outbreak of S. aureus septicemia in the pediatric unit in May 2015. Following this, a survey on nasal carriage of S. aureus was carried out among babies, their mothers, and health care staff at the unit. Nasal carriage of S. aureus and MRSA were 25.2% (26/103) and 4.8% (9.7/103), respectively.
Genomic investigations
Multilocus sequence and spa typing
We sequenced 17 S. aureus isolates from a variety of sources contemporary to the infections at the two hospitals (Table 1). Multilocus sequence typing revealed a total of eleven distinct sequence types (STs) (Table 1). The highest relative prevalence was observed for ST15, identified only among the isolates from KBTH. The ST15 isolates were isolated from various environmental surfaces of the pediatric emergency ward and also from the blood of a patient on admission to an adult ward (Table 1). The second most common lineage was ST152, also identified at KBTH only. Isolates belonging to ST152 were isolated from anterior nares of two mothers of babies in different pediatric wards (NICU, P2) and also from the wound of a patient on admission to an adult surgical ward (Table 1). Other lineages detected at KBTH included ST5, ST45, ST121, ST508, and ST707. ST45 and its single locus variant ST508 were isolated from anterior nares of a baby and the surface of a table in the pediatric P2 ward. The other STs were isolated from anterior nares of hospital staff or mothers of babies in different pediatric wards, though each was represented by a single isolate only. The isolates of S. aureus from LH yielded diverse and unrelated STs (ST6, ST72, ST508, and ST3248, the latter a single locus variant of ST1).
Isolates represented eleven distinct spa types, which included a single isolate with novel spa type (Table 1). Isolates that belonged to the same ST also revealed identical spa types, except for ST508. As such, all ST15 isolates were spa type t084 whereas all ST152 isolates belonged to spa type t355.
Antimicrobial resistance genes
Analysis of antimicrobial resistance gene carriage revealed that all isolates were mecA negative (Table 1). We screened for presence of other resistance-associated genes, which revealed that all isolates contained the penicillin resistance gene blaZ. Less common were the tetracycline resistance gene tetK and the trimethoprim resistance determinant dfrG. Both genes were observed mostly in ST15 isolates. A single isolate from LH representing ST508 displayed a unique composition of multiple antimicrobial resistance genes, which in addition to the tetK gene also included the aminoglycoside resistance determinants aacA-aphD and addD, the trimethoprim resistance gene dfrC, the lincosamide resistance gene lnuA, and the antiseptic resistance gene qacA. We interrogated the genetic environment of the identified resistance genes to check for association with MGEs. Carriage of the blaZ gene was associated with four distinct MGEs, although the majority of isolates carried a plasmid that closely resembled the 21 kb pSaa6159, identified previously in S. aureus ST93 (accession: CP002115). Less prevalent was the Tn552-like element found in three isolates, with a single isolate carrying a plasmid that matched part of the pLUH02 element from S. aureus (accession: FR714929). In most of the tetK positive isolates, the gene was carried on a plasmid resembling the previously reported SAP095B plasmid (accession: GQ900445). In a single isolate representing ST508, the tetK gene was carried on a plasmid fragment, which also contained the aacA-aphD gene and that shared a high level of sequence identity with a large, multiple resistance plasmid pT33G-1 derived from Staphylococcus lugdunensis (accession: KU882683). This isolate revealed a second plasmid fragment, which also matched the pT33G-1 and contained addD, lnuA, and qacA genes. Finally, the dfrG gene was associated with a chromosomally integrated insertion sequence as described previously,32 which was nearly identical to the dfrG accessory element found in MRSA ST772 strain DAR4145.33,34
pvl gene
Genes encoding pvl were detected in six isolates representing four distinct STs (Table 1). Carriage of the pvl genes was associated with the acquisition of Sa2int-type phages, and three distinct Sa2int phages were identified. All of the ST152 isolates carried a very closely related phage element of 46.6 kb in size, which showed high sequence identity (99%) with Sa2int found in S. aureus ST152 reference genome BB155 (accession: LN854556). In addition, it shared 91% of its sequence with the Sa2int derived from the MRSA USA300_FPR3757 strain.26 The single pvl-positive ST15 isolate carried a distinct 42.7 kb Sa2int phage that shared only 17% of its sequence with the ST152 pvl phage and was most closely related to a phage found in MRSA ST239 strain XN108.35 Also different was the Sa2int found in the pvl positive ST5 isolate, which was most closely related to the phiSa119 (84% sequence coverage), found in MRSA ST772.36 Finally, the single pvl positive S. aureus ST121 isolate contained a short, truncated pvl-associated phage sequence, which matched most closely (99% sequence identity) Sa2int carried by MRSA ST80 reference genomes such as GR2 and 11819–97.37,38
Phylogenetic analysis
A phylogenetic tree based on the core-genome alignment was reconstructed to analyze evolutionary relationships between the isolates (Figure 2). The ST152 isolates revealed high divergence; the three isolates were non-clonal showing pairwise SNP distances of 77, 94, and 95. The ST15 isolates from KBTH formed two clusters. One cluster of the ST15 isolates from KBTH comprised three isolates from a baby’s cot in the emergency ward, door handle of the emergency ward, and a patient with septicemia (Table 1: isolates 1, 2, and 4). Isolates from the door handle (isolate 2) and baby’s cot (isolate 1) in the emergency ward were isolated on the same day and had a pairwise SNP distance of 1, suggesting a possible transmission. The clinical isolate from the septicemia patient (isolate 4) was isolated a few weeks before the isolation of isolates 1 and 2; the SNP distance between isolate 4 and the other two isolates was 97 for isolate 1 and 95 for isolate 2. The other cluster of ST15 isolates from KBTH comprised two isolates, which were isolated from the door handle of the emergency ward and the surface of a table in the ward (Table 1: isolates 3 and 5). The two isolates were isolated the same day and had a pairwise SNP distance of 0 suggesting a hand contamination problem.
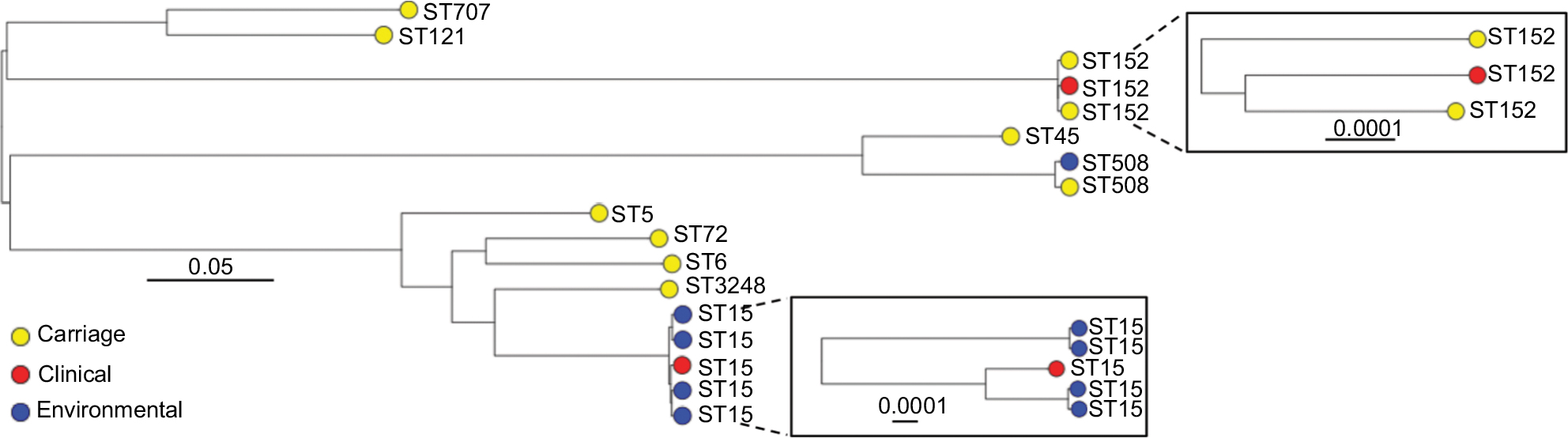 | Figure 2 Mid-point rooted maximum likelihood phylogenetic tree of all Staphylococcus aureus isolates analyzed in this study. Notes: The tree was reconstructed based on core-genome alignment, generated after mapping short reads to the S. aureus USA300_FPR3757 reference genome. The tips are labeled with the isolate’s sequence type (ST) and color coded to show the sample source. Values above scale bars describe number of nucleotide substitutions per site. |
Discussion
This study is the first in Ghana and one of the few in sub-Saharan Africa to employ whole genome sequencing to study S. aureus isolates. The isolates were collected during suspected S. aureus outbreaks at a teaching and a district hospital in Ghana. We describe our investigation of the outbreaks as indirect, as the initial disease S. aureus isolates that led to the investigations were not available for sequencing. At KBTH, the data showed that there was circulation of an S. aureus ST15 clone in the pediatric emergency ward (location of the outbreak) during the period of the outbreak. The data showed that there were two pathways of transmission of the ST15 clone in the pediatric emergency ward of KBTH, both of which could be linked to the door handle of the ward. This concurs with several studies that have shown that door handles could be an important source of S. aureus contamination in health care settings.39,40 Within our data, it is difficult to identify the source of the S. aureus ST15 clone in the pediatric emergency ward of KBTH. However, since S. aureus ST15 represents lineage commonly carried in the anterior nares,41,42 it may have been transferred from this source to the door handle of the emergency ward through contamination of the hand of a health care worker or visitor to the ward. S. aureus is known to survive for as long as 7 months on dry surfaces.43 This survival feature of the organism, coupled with overcrowding at the pediatric emergency ward of KBTH and the fact that equipment is shared among patients, could facilitate its rapid transmission among patients. The ST15 clone of S. aureus appears to be widely disseminated at KBTH as it was isolated from patients in adult wards, and the genomic data showed that these isolates were unrelated to the transmission events at the pediatric ward. Generally, ST15 appears to be one of the common clones of S. aureus in Ghana, and it has been isolated from both diseased and asymptomatic carriers.44 The pattern of dissemination of the ST15 clone in the emergency ward does provide us with some insights about transmission events in this ward, and highlights a basic problem with disinfection of environmental surfaces at this health care facility. Generally, S. aureus and other bacterial pathogens, such as Clostridium difficile and P. aeruginosa, are common contaminants of environmental surfaces and may be resistant to sanitation procedures due to biofilm formation.39,40,45,46 The situation at LH is unlikely to be a true outbreak, as S. aureus strains from this hospital were all of unrelated STs. The S. aureus septicemia situation at LH thus appears to be an isolated one and not related to any transmission events at the hospital. Since the S. aureus outbreak at KBTH in 2012, there has been a heightened awareness of this pathogen in Ghana, and isolated invasive cases like the one in LH have attracted attention and been misinterpreted as outbreaks.
Like this study, a number of studies have reported that S. aureus isolates from West and Central Africa show a relatively high prevalence of pvl genes.4–7 Based on genomic analysis in the current study, we can now report that high frequency of these genes is likely mediated by diverse phage population rather than a single highly transmissible phage type. Furthermore, the full sequence of each identified Sa2int variant revealed close similarity with pvl-carrying phages identified in globally disseminated MRSA lineages, providing a link between the pvl positive MSSA populations from Africa and the pandemic MRSA clones. An S. aureus lineage that is known to be associated with pvl is ST152,47,48 which is also evident from our data as all the ST152 isolates were pvl positive. As previously observed,49 the ST152 isolates analyzed in this work revealed high divergence.
Analysis of genetic context behind carriage of the identified resistance genes revealed that the majority was associated with MGEs that shared high sequence identity with previously described plasmids, transposons, or other elements. It is important to note the uniquely wide range of resistance genes harbored by the single ST508 strain, which was identified to be MSSA. In a previous community nasal carriage study in Ghana, the only two MRSA strains isolated were ST508.5 ST508 is a single locus variant of ST45, which was also identified among our isolates and represents a lineage that includes the Berlin epidemic clone.50 S. aureus ST508 thus appears to be an interesting and important carriage clone in Ghana, and further studies are needed to elucidate its epidemiological significance.
The main limitation of the study is the limited number of isolates used in the investigations. This resulted from loss of isolates during their storage while funding was being sought for the genomic investigations. Despite this limitation, we have carried out extensive genomic analyses on the isolates, adding to the few studies that have undertaken whole genome analysis of S. aureus isolates in Africa.
Conclusion
The S. aureus isolates studied belonged to diverse STs with ST15 and ST152 being relatively more common. There was a high prevalence of pvl genes among the isolates, which is likely mediated by diverse phage population rather than a single, highly transmissible phage type. Though we were not able to identify the specific strain of S. aureus implicated in the outbreak at the pediatric emergency ward of KBTH, we are confident that its transmission is similar to that of the ST15 clone, which we have elucidated through whole genome sequencing. Contamination of various surfaces in the emergency ward by strains of ST15 highlights the need for more rigorous disinfection of environmental surfaces at KBTH. This investigation also highlights the need for proper storage of bacterial isolates in laboratories in the developing world where facilities for molecular investigations are limited.
Acknowledgments
The study was funded through a grant from the Alborada Research Fund and Wellcome Trust Sanger Institute core funding (Wellcome Trust grant 098051). The authors acknowledge the contributions of the technical staff of the Department of Microbiology, Korle-Bu Teaching Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
Todar K. Todar’s Online Textbook of Bacteriology. Madison, WI: University of Wisconsin–Madison; 2006. | ||
Hall GS. MRSA: lab detection, epidemiology, and infection control. Microbiol Front. 2003;3:1–6. | ||
Chambers HF. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin Microbiol Rev. 1997;10(4):781–791. | ||
David MZ, Boyle-Vavra S, Zychowski DL, Daum RS. Methicillin-susceptible Staphylococcus aureus as a predominantly healthcare-associated pathogen: a possible reversal of roles? PLoS One. 2011;6(4):e18217. | ||
Egyir B, Guardabassi L, Esson J, et al. Insights into nasal carriage of Staphylococcus aureus in an urban and a rural community in Ghana. PLoS One. 2014;9(4):e96119. | ||
Breurec S, Fall C, Pouillot R, et al. Epidemiology of methicillin-susceptible Staphylococcus aureus lineages in five major African towns: high prevalence of Panton-Valentine leukocidin genes. Clin Microbiol Infect. 2011;17(4):633–639. | ||
Schaumburg F, Köck R, Friedrich AW, et al. Population structure of Staphylococcus aureus from remote African Babongo Pygmies. PLoS Negl Trop Dis. 2011;5(5):e1150. | ||
Lina G, Piémont Y, Godail-Gamot F, et al. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999;29(5):1128–1132. | ||
Vandenesch F, Naimi T, Enright MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis. 2003;9(8):978–984. | ||
Salipante SJ, Sengupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol. 2015;53(4):1072–1079. | ||
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–1530. | ||
Harris SR, Feil EJ, Holden MT, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–474. | ||
Harris SR, Cartwright EJ, Török ME, et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis. 2013;13(2):130–136. | ||
Kong Z, Zhao P, Liu H, et al. Whole-genome sequencing for the investigation of a hospital outbreak of MRSA in China. PLoS One. 2016;11(3):e0149844. | ||
Gordon NC, Price JR, Cole K, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol. 2014;52(4):1182–1191. | ||
Newman MJ, Frimpong E, Donkor ES, Opintan JA, Asamoah-Adu A. Resistance to antimicrobial drugs in Ghana. Infect Drug Resist. 2011;4:215–220. | ||
Annual Report of the Korle-Bu Teaching Hospital. 2010. Accra, Ghana: Korle-Bu Teaching Hospital. | ||
Bannerman TL. Staphylococcus, Micrococcus and other catalase-positive cocci that grow aerobically. In: Murray PR, Baron EJ, Jorgensen JH, Pfaller MA, YolkenRH, editors. Manual of Clinical Microbiology, 8th ed, vol 1. Washington DC: American Society for Microbiology; 2003. | ||
CLSI. Performance Standards for Antimicrobial Susceptibility Testing. CLSI Approved Standard M100-S23. Wayne, PA: Clinical and Laboratory Standards Institute; 2013. | ||
Page AJ, de Silva N, Hunt M, et al. Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb Genom. 2016;2(8):e000083. | ||
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. | ||
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–579. | ||
Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13(6):R56. | ||
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. | ||
Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–135. | ||
Diep BA, Gill SR, Chang RF, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–739. | ||
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. | ||
Inouye M, Dashnow H, Raven LA, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6(11):90. | ||
Kahl BC, Mellmann A, Deiwick S, Peters G, Harmsen D. Variation of the polymorphic region X of the protein A gene during persistent airway infection of cystic fibrosis patients reflects two independent mechanisms of genetic change in Staphylococcus aureus. J Clin Microbiol. 2005;43(1):502–505. | ||
Harris SR, Robinson C, Steward KF, et al. Genome specialization and decay of the strangles pathogen, Streptococcus equi, is driven by persistent infection. Genome Res. 2015;25(9):1360–1371. | ||
Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. | ||
Jensen SO, Lyon BR. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009;4(5):565–582. | ||
Steinig EJ, Andersson P, Harris SR, et al. Single-molecule sequencing reveals the molecular basis of multidrug-resistance in ST772 methicillin-resistant Staphylococcus aureus. BMC Genomics. 2015;16:388. | ||
Manoharan A, Zhang L, Poojary A, Bhandarkar L, Koppikar G, Robinson DA. An outbreak of post-partum breast abscesses in Mumbai, India caused by ST22-MRSA-IV: genetic characteristics and epidemiological implications. Epidemiol Infect. 2012;140(10):1809–1812. | ||
Zhang X, Hu Q, Yuan W, et al. First report of a sequence type 239 vancomycin-intermediate Staphylococcus aureus isolate in Mainland China. Diagn Microbiol Infect Dis. 2013;77(1):64–68. | ||
Sanchini A, del Grosso M, Villa L, et al. Typing of Panton-Valentine leukocidin-encoding phages carried by methicillin-susceptible and methicillin-resistant Staphylococcus aureus from Italy. Clin Microbiol Infect. 2014;20(11):O840–O846. | ||
Sabat AJ, Pournaras S, Akkerboom V, Tsakris A, Grundmann H, Friedrich AW. Whole-genome analysis of an oxacillin-susceptible CC80 mecA-positive Staphylococcus aureus clinical isolate: insights into the mechanisms of cryptic methicillin resistance. J Antimicrob Chemother. 2015;70(11):2956–2964. | ||
Stegger M, Price LB, Larsen AR, et al. Genome sequence of Staphylococcus aureus strain 11819-97, an ST80-IV European community-acquired methicillin-resistant isolate. J Bacteriol. 2012;194(6):1625–1626. | ||
Oie S, Hosokawa I, Kamiya A. Contamination of room door handles by methicillin-sensitive/methicillin-resistant Staphylococcus aureus. J Hosp Infect. 2002;51(2):140–143. | ||
Visalachy S, Palraj KK, Kopula SS, Sekar U. Carriage of multidrug resistant bacteria on frequently contacted surfaces and hands of health care workers. J Clin Diagn Res. 2016;10(5):18–20. | ||
Schaumburg F, Ngoa UA, Kösters K, et al. Virulence factors and genotypes of Staphylococcus aureus from infection and carriage in Gabon. Clin Microbiol Infect. 2011;17(10):1507–1513. | ||
Schaumburg F, Alabi AS, Peters G, Becker K. New epidemiology of Staphylococcus aureus infection in Africa. Clin Microbiol Infect. 2014;20(7):589–596. | ||
Kramer A, Schwebke I, Kampf G. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect Dis. 2006;6:130. | ||
Egyir B, Guardabassi L, Sørum M, et al. Molecular epidemiology and antimicrobial susceptibility of clinical Staphylococcus aureus from healthcare institutions in Ghana. PLoS One. 2014;9(2):e89716. | ||
Abdallah M, Benoliel C, Drider D, Dhulster P, Chihib NE. Biofilm formation and persistence on abiotic surfaces in the context of food and medical environments. Arch Microbiol. 2014;196(7):453–472. | ||
Hammond EN, Donkor ES, Brown CA. Biofilm formation of Clostridium difficile and susceptibility to Manuka honey. BMC Complement Altern Med. 2014;14:329. | ||
Ruimy R, Maiga A, Armand-Lefevre L, et al. The carriage population of Staphylococcus aureus from Mali is composed of a combination of pandemic clones and the divergent Panton-Valentine leukocidin-positive genotype ST152. J Bacteriol. 2008;190(11):3962–3968. | ||
Monecke S, Berger-Bächi B, Coombs G, et al. Comparative genomics and DNA array-based genotyping of pandemic Staphylococcus aureus strains encoding Panton-Valentine leukocidin. Clin Microbiol Infect. 2007;13(3):236–249. | ||
Ruimy R, Maiga A, Armand-Lefevre L, et al. The carriage population of Staphylococcus aureus from Mali is composed of a combination of pandemic clones and the divergent Panton-Valentine leukocidin-positive genotype ST152. J Bacteriol. 2008;190(11):3962–3968. | ||
Wannet WJ, Spalburg E, Heck ME, et al. Emergence of virulent methicillin-resistant Staphylococcus aureus strains carrying Panton-Valentine leucocidin genes in the Netherlands. J Clin Microbiol. 2005;43(7):3341–3345. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.