Back to Journals » Journal of Inflammation Research » Volume 19
A Functional Shift of IGFBP3 in Osteoarthritis: From Cartilage Protection to Disease Promotion
Authors Chen J ![]() , Liu X, Gu Y, Zhang H
, Liu X, Gu Y, Zhang H ![]() , Yang Y
, Yang Y ![]() , Li Z
, Li Z
Received 3 February 2026
Accepted for publication 22 March 2026
Published 12 April 2026 Volume 2026:19 497779
DOI https://doi.org/10.2147/JIR.S497779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ujjwol Risal
Jian Chen,1,* Xiangzhong Liu,2,* Yanchao Gu,1 Haoyu Zhang,1 Yi Yang,1 Zhanghua Li2
1School of Sports Medicine, Wuhan Sports University, Wuhan, 430079, People’s Republic of China; 2Department of Orthopedics, Wuhan Third Hospital, Wuhan, 430079, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhanghua Li, Department of Orthopedics, Wuhan Third Hospital, 216 Guanshandadao, Hongshan District, Wuhan, 430079, People’s Republic of China, Email [email protected]
Abstract: Osteoarthritis (OA) is a prevalent degenerative joint disease driven by complex interactions among mechanical stress, inflammation, and metabolic dysregulation. Insulin-like growth factor binding protein 3 (IGFBP3) has emerged as a key regulator of cartilage homeostasis, yet its role in OA remains controversial. Accumulating evidence indicates that IGFBP3 exerts both insulin-like growth factor 1 (IGF-1)-dependent and IGF-1-independent effects on chondrocytes, including modulation of proliferation, apoptosis, and nuclear signaling. However, existing studies report contradictory findings, showing both protective and inhibitory actions of IGFBP3, likely reflecting differences in disease stage, cellular microenvironment, and experimental models. Notably, diabetic OA represents a distinct pathological subtype. Emerging data suggest that metabolic conditions reshape IGF signaling and IGFBP3 function, highlighting important differences between primary OA and diabetic OA. IGFBP3 activity is further regulated by endocrine factors, extracellular matrix components, proteases, and epigenetic mechanisms, and mediates multicellular crosstalk among chondrocytes, synovial fibroblasts, macrophages, mesenchymal stem cells, and bone cells. This review integrates these divergent findings and proposes IGFBP3 as a molecular link between metabolic disturbance and joint degeneration, providing a novel framework for subtype-specific therapeutic targeting in OA.
Keywords: osteoarthritis, IGFBP3, chondrocyte, transcript factor, cellular interaction
Introduction
Osteoarthritis (OA) is a complex, multifactorial joint disorder characterized by progressive cartilage degeneration, metabolic imbalance, and chronic low-grade inflammation.1 Beyond mechanical wear, accumulating evidence highlights chondrocyte metabolic reprogramming,2 oxidative stress,3 and disrupted growth factor signaling4 as central drivers of disease progression. Despite extensive investigation into OA-associated genes and pathways, major uncertainties remain regarding how anabolic and catabolic signals are integrated within the joint microenvironment and how systemic endocrine cues intersect with local tissue regulation.
Among regulators of cartilage homeostasis, insulin-like growth factor 1 (IGF1) has emerged as a pivotal modulator. Classically, Insulin-like growth factor binding protein 3 (IGFBP3) recognized as the principal carrier of circulating IGF-1.5 (Figure 1). IGFBP3 also exerts IGF-independent actions through interactions with extracellular matrix (ECM) components, proteases, and nuclear signaling machinery. Emerging evidence from multi-omics profiling of osteoarthritic joints indicates that IGFBP3/IGF-1 system is aberrantly expressed in both cartilage and synovium, positioning it as a key regulator of chondrocyte phenotype, inflammation, and tissue remodeling.6,7 However, context-dependent and sometimes contradictory effects of IGFBP3 underscore unresolved controversies regarding its precise functional role in OA.
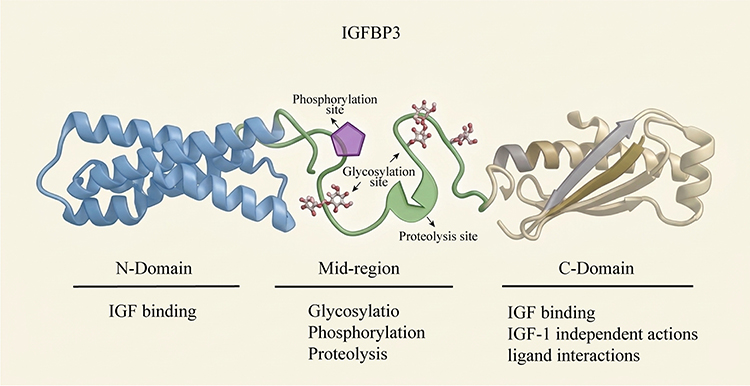 |
Figure 1 IGFBP3 Protein Structure. The IGFBP3 protein comprises three domains: the N-domain, the C-domain, and the middle region. The N-domain contains the IGF-binding site. The C-domain not only includes IGF-binding regions but also provides binding sites for metal ions, heparin, and fibronectin, and plays a crucial role in mediating the IGF-1 independent effects of IGFBP3. The middle region is subject to post-translational modifications, including glycosylation, phosphorylation, and proteolysis. |
Distinct from prior reviews that emphasize IGF signaling or isolated molecular pathways, this review conceptualizes IGFBP3 as an integrative regulatory hub linking gene expression, signaling cascades, epigenetic modulation, and multicellular communication within the joint. We synthesize recent advances in IGFBP3 biology in OA, critically evaluate existing inconsistencies, and identify key knowledge gaps, thereby providing a framework for the rational development of IGFBP3-targeted therapeutic strategies.
Methods
A comprehensive literature search was conducted in PubMed, Web of Science, and Scopus to identify studies published from 1990 to 2026. The search strategy combined MeSH terms and keywords including “IGFBP3,” “insulin-like growth factor binding protein 3,” “osteoarthritis,” “chondrocytes,” “cartilage metabolism,” and “IGF signaling.” English-language original articles and reviews focusing on IGFBP3-related gene expression, signaling pathways, and regulatory mechanisms in cartilage or joint tissues were included. Studies not relevant to OA or joint biology were excluded after title and abstract screening. Additional relevant publications were identified through manual screening of reference lists from key articles.
Changes in IGFBP3 Expression in OA
The maintenance of IGFBP3 and IGF expression within specific ranges is crucial for ensuring the turnover of normal chondrocytes and the ECM in cartilage. However, following injury, aberrant changes occur in their expression levels. Studies involving cultured cartilage tissues from OA patients have demonstrated a significant upregulation in IGFBP3 secretion,8,9 while IGF expression remains either unchanged or shows only slight elevation.10 This notable disparity in expression dynamics between IGF and IGFBP3 disrupts their coordinated regulation, thereby affecting cellular viability and synthetic metabolism, playing a pivotal role in disease progression. However, whether these changes signify dysregulation of expression mechanisms in response to cellular damage or beneficial regulatory responses for self-repair remains unclear. Furthermore, genome-wide association studies have identified two single nucleotide polymorphisms upstream of the IGFBP3 gene, which are strongly associated with the incidence of hip OA.6 This indicates that genetic variations at the IGFBP3 gene level influence OA onset. Moreover, both downregulation and overexpression of IGFBP3 have been linked to chondrocyte hypertrophy and metabolic alterations.6 These findings collectively underscore the detrimental impact of abnormal IGFBP3 expression changes on the maintenance of the chondrocyte phenotype.
During various developmental stages of cartilage, the expression of IGFBP3 exhibits dynamic changes. In the early stages of chondrocyte differentiation within articular cartilage, IGFBP3 expression is notably upregulated, followed by subsequent downregulation post-hypertrophy.11,12 In murine models, IGFBP3 expression is relatively low in one-week-old mice but markedly increases by three weeks of age.12 These fluctuations likely reflect the organism’s adaptive responses to developmental demands during distinct time frames. Similarly, following OA, IGFBP3 expression demonstrates diverse patterns across different phases. For instance, in murine temporomandibular joint arthritis, IGFBP3 displays a biphasic expression pattern, characterized by initial upregulation followed by subsequent downregulation.13 Notably, this expression pattern correlates with the progression of OA.14 These observed variations in IGFBP3 expression likely stem from differing pathological mechanisms during distinct phases of OA. Furthermore, the sensitivity of IGFBP3 as an indicator underscores its potential utility in signaling changes during various stages subsequent to OA.
IGFBP3 exhibits regional expression variations within cartilage, holding significant implications for understanding pathological states. Within the epiphyseal growth plate of fetal sheep bones, IGFBP3 expression is confined solely to the proliferative and hypertrophic zones, with no detection in the differentiation zone.15 Moreover, its expression is relatively diminished in the hypertrophic zone,16 suggesting a potentially heightened requirement for IGFBP3 expression in regions characterized by active cell proliferation. In articular cartilage, IGFBP3 predominantly localizes to the superficial layer,17 likely reflecting the region’s heightened metabolic activity. However, in OA, IGFBP3 expression patterns diverge. While there is a notable increase in IGFBP3 expression within the middle and deep layers of cartilage, its expression declines in the superficial layer.17,18 Nevertheless, conflicting findings emerge, with some studies reporting elevated IGFBP3 expression across all layers of OA cartilage, correlating with disease severity.19 The complex pathological mechanisms of OA may contribute to these disparities, given that different cartilage layers undergo distinct pathological processes. Upon stimulation with growth factors, IGFBP3 expression demonstrates heterogeneous trends across various layers of OA cartilage: the middle region exhibits a swifter and more pronounced increase, followed by a gradual increase in the deep layer, while the superficial layer displays the slowest increase.18 These observations underscore a significant association between IGFBP3 expression dynamics and the pathological status of cartilage cells.
Moreover, IGFBP3 expression is influenced by mechanical stress. Orthopedic corrective devices have been shown to reduce IGFBP3 expression in the condylar cartilage of rat mandibles under mechanical stress, thereby facilitating cell growth and differentiation.20 After exercise, an elevation in IGFBP3 and IGF-1 levels is observed in the synovial fluid of ponies.21 Proper mechanical stress may enhance cellular metabolism and proliferation by regulating the coordinated expression of IGF/IGFBP3. Conversely, non-physiological mechanical stress disrupts this coordination. Adverse tensile stress hinders the expression of IGF-1 and IGFBP3 in temporomandibular joint disc chondrocytes,22 and heightened matrix hardness affects cell differentiation by upregulating IGFBP3 expression.23 Stress is an essential factor in stimulating cartilage growth and development, as well as a significant contributor to OA occurrence. IGFBP3 plays a pivotal role in cellular responses to mechanical stimuli. IGFBP3 represents a crucial mechanism for regulating the proliferation and differentiation of cartilage cells and serves as a pivotal indicator of functional disruption following cellular damage.
The Mechanism of IGFBP3 Effects
IGF-1 Dependent Effect
A common mechanism of action for IGFBP3 involves its role as a binding protein affecting the downstream signaling of IGF. The half-life of the locally formed complexes is significantly prolonged.24 This effect may lead two potential outcomes: firstly, the complex may protect IGF and reduce its degradation, thus facilitating the downstream effects of IGF; secondly, the complex may restrict the release of IGF, thereby inhibiting its activation of receptors. The former possibility seems more apparent under normal physiological conditions, as the complex extends the duration of IGF action. In studies exploring the latter possibility, IGFBP3 markedly inhibited the binding of IGF-1 to the cell membrane,25 and similarly suppressed receptor activation, akin to IGF-neutralizing antibodies.26 Furthermore, the inhibition of IGFBP3 on the synthesis of proteoglycans in human OA knee chondrocytes could be reversed by mutant forms of IGF-1, such as R3rhIGF-1, Des(1–3)IGF-1, Long R3 IGF-1, which lack affinity for IGFBP3, or by complex formation inhibitors like NBI-31772. This suggests an IGF-1 dependent inhibitory role for IGFBP3. The promoting or inhibitory effects seem to depend on the ratio of IGFBP3 to IGF. IGFBP3 at concentrations exceeding four times that of IGF-1 can completely suppress IGF-1’s promotion of proteoglycan synthesis and proliferation in chondrocytes27,28 (Figure 2). Cells appear to exert diverse effects on cartilage by perceiving and then regulating their expression in different ratios. The mechanisms underlying these effects and whether manipulating their concentration ratios could be a therapeutic approach for OA require further investigation.
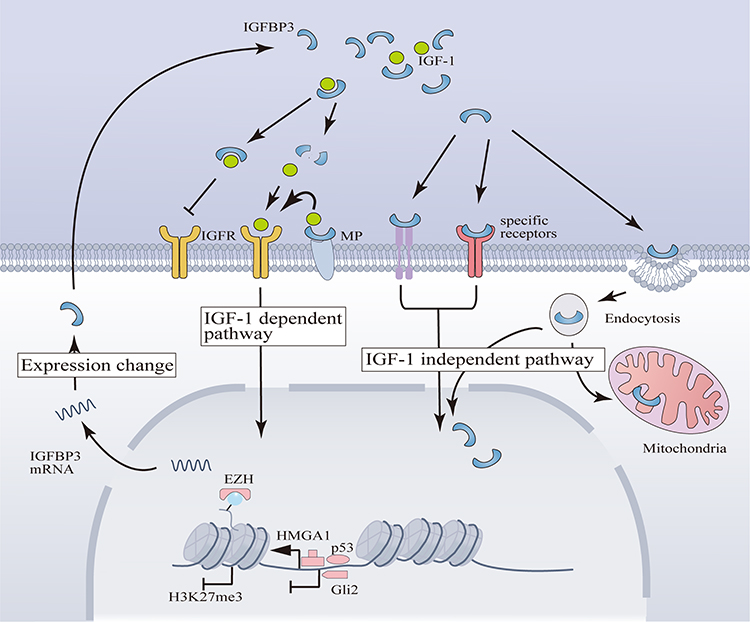 |
Figure 2 IGFBP3’s Mechanisms of Action and Transcriptional Regulation in Chondrocytes. (1) IGF-1 Dependent Mechanisms of IGFBP3: IGFBP3 modulates the release of IGF-1, thereby exerting stimulatory or inhibitory effects on downstream pathways of IGFR. MPs can facilitate the binding of IGFBP3 and the subsequent release of IGF-1. The IGF-1 Independent Mechanisms of IGFBP3 involve activation of signaling pathways through binding to specific receptors or internalization into the nucleus and mitochondria via endocytosis. (2) The transcription of IGFBP3 in chondrocytes is positively regulated by the HMGA1 complex and p53, while Gli2 acts as a transcriptional repressor. EZH inhibits IGFBP3 transcription by promoting H3K27me3. |
IGF-1 Independent Effect
IGFBP3 has been observed to activate cell membrane receptors such as TGF-βR,29 LRP1, and TMEM219,25 or internalize into cells via various pathways, including clathrin-coated pits, the caveolin pathway, and endocytosis,30 thereby exerting IGF-1 independent effects. Despite evidence of IGFBP3’s IGF-1 independent actions in several diseases, its specific involvement in chondrocytes remains equivocal. Some studies indicate that the sole addition of IGFBP3 does not influence chondrocytes.27 Furthermore, experiments employing NBI-31772 to inhibit complex formation have solely demonstrated the IGF-1 dependent effects.31 But, studies show that IGFBP3 mediate Endoplasmic reticulum stress through PERK/ATF4/CHOP cascade.32 However, these experiments did not scrutinize the impact of varied IGFBP3 concentrations or assess whether blocking IGFBP3-IGF binding with NBI-31772 affects potential IGF-1 independent actions of IGFBP3. Consequently, these findings do not definitively exclude the presence of IGFBP3’s IGF-1 independent effects in chondrocytes. Additionally, IGFBP3 has been detected within the nuclei of both normal and OA cartilage.17 IGFBP3 is transported into the nucleus by importin-β protein, performing diverse functions such as activating nuclear hormone receptors,25 participating in DNA damage repair through non-homologous end joining,33 regulating transcription via interaction with RNA polymerase II subunit 3,30 and engaging in epigenetic regulation by binding to histone 3.34 The subcellular distribution of IGFBP3 in chondrocytes constitutes significant evidence of its IGF-1 independent actions. Apart from nuclear translocation, IGFBP3 has been observed to localize to chondrocyte mitochondria.35 However, beyond these observed localizations, further investigation is warranted to elucidate the intricate mechanisms underlying IGFBP3’s IGF-1 independent actions within chondrocytes.
Although the IGF-1 independent effects of IGFBP3 in mature chondrocytes are still debatable, its role in promoting apoptosis in chondrogenic progenitor cells has been definitively established. GGG-IGFBP3, a recombinant IGFBP3 protein with low IGF affinity, has been shown to activate the p21/STAT-1 signaling pathway, resulting in decreased synthetic metabolism and increased apoptosis.36,37 Moreover, IGFBP3 has been found to inhibit the chondrogenic differentiation induced by TGF-β in an IGF-1 independent manner.38 However, it has been noted that the non-IGF-1-dependent pro-apoptotic effect of IGFBP3 is predominantly observed in undifferentiated and early differentiating chondrogenic progenitor cells, but not in late-stage differentiation36 (Figure 2). In summary, the precise nature of IGFBP3’s IGF-1 independent actions in mature chondrocytes remains elusive.
Additionally, IGFBP3 has been implicated in stabilizing mRNA through m6A methylation modifications, thereby impacting disease progression.39 Exploring whether IGFBP3 exerts analogous effects following OA constitutes another prospective avenue for investigating its independent actions.
Factors Influencing the Effects of IGFBP3
Endocrine
IGFBP3 can be transported via the bloodstream or locally secreted by adjacent tissues. Given the occurrence of local hematoma and synovial neovascularization following OA, the predominant source of elevated IGFBP3 levels in local cartilage, whether from local tissue secretion or systemic delivery,40 remains uncertain. Some pharmacological agents have demonstrated the ability to enhance hepatic synthesis of IGF/IGFBP3, thereby augmenting local cartilage concentrations via circulation, consequently promoting cell proliferation41 (Figure 3). These findings highlight the considerable influence of endocrine factors on the augmentation of local IGFBP3 concentrations.
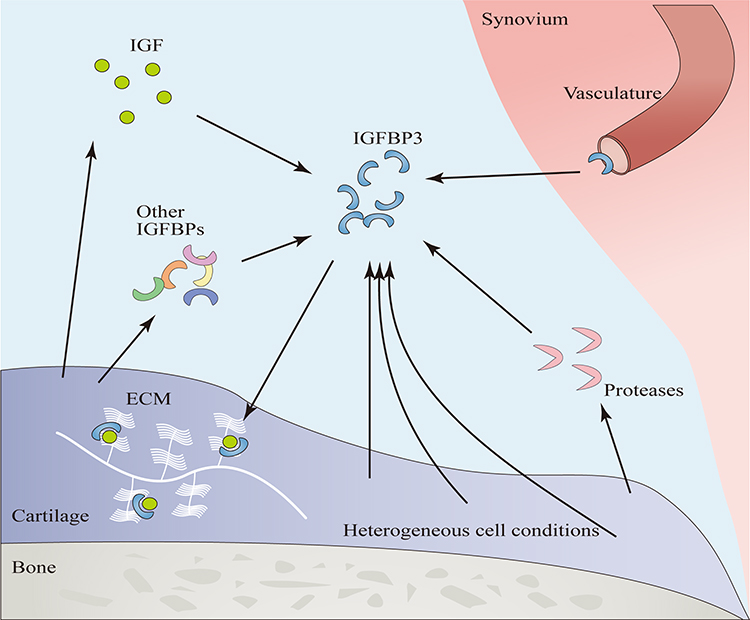 |
Figure 3 Factors Influencing the Function of IGFBP3. Endocrine IGFBP3 enters the joint cavity via the vasculature. In osteoarthritis, tissues simultaneously secrete IGF, proteases, and other IGFBPs. The concentration of IGF affects the IGF-1 dependent actions of IGFBP3, proteases can degrade IGFBP3, and other IGFBPs can exert either synergistic or antagonistic effects relative to IGFBP3. Components within the ECM can bind IGFBP3 to form a reservoir. The heterogeneous cell conditions have different reactions to IGFBP3. |
Different Cellular States
During the developmental stages of the growth plate, IGFBP3 exhibits dual roles: it stimulates chondrocyte proliferation during the proliferative phase but paradoxically induces apoptosis in terminally differentiated chondrocytes through an IGF-1 dependent mechanism.42 Moreover, during the early differentiation of chondroprogenitor cells, IGFBP3 impedes cell proliferation via an IGF-1 independent pathway, whereas this inhibitory effect diminishes in later stages of differentiation.36 The dynamic modulation of IGFBP3 function reflects the intricate regulatory processes governing cellular responses in distinct physiological contexts (Figure 3). Although the precise mechanisms governing IGFBP3 function regulation within cells remain elusive, it is evident that the diverse cellular states observed at different stages following OA will inevitably impact the functional outcomes of IGFBP3.
Concentration Ratio of IGFBP3 to IGF-1
In most cases, changes in IGFBP3 expression are typically accompanied by alterations in the expression levels of IGF and the insulin-like growth factor receptor (IGFR). As a bridging protein between IGF and IGFR, the concentration ratio of IGFBP3 to these molecules inevitably influences downstream biochemical signaling. Studies have indicated that despite a significant increase in IGFR on the surface of OA chondrocytes, these cells exhibit an inability to respond to IGF-1 stimulation.43 This observation indirectly underscores the pivotal role of molar concentration ratios in modulating the IGF-1 dependent effects of IGFBP3. This scenario elucidates why, in the synovial fluid of OA and rheumatoid arthritis (RA) patients, where heightened IGFBP3 concentrations are detected, these fluids still possess the capability to promote chondrocyte proteoglycan synthesis via the IGF signaling pathway.44 The pronounced imbalance in their proportions may underlie the fundamental basis for the IGF-1 dependent inhibitory effects of IGFBP3. IGFBP3 concentrations surpassing four times that of IGF-1 may represent a critical threshold for altered functionality.28 Despite findings indicating a 3.5-fold increase in IGF-1 expression and a 24-fold increase in IGFBP3 among certain OA patients,45 the ratio of IGF-1 to IGFBP3 may also exhibit variability in response to diverse pathological conditions following OA (Figure 3). Therefore, research endeavors should not solely focus on changes in IGFBP3 but should also consider its ratio to IGF-1.
ECM
The C-terminal domain of IGFBP3 contains motifs that facilitating interactions with proteins like heparin, fibronectin, and hyaluronic acid.46 These interactions within the ECM significantly modulates the function of IGFBP3. Within the deep layers of normal cartilage, IGFBP3 and fibronectin co-localize within the pericellular matrix. Notably, fibronectin retards the diffusion of IGFBP3, facilitating the establishment of a stable reservoir of IGF-1 around cells. This reduces concentration fluctuations, thereby maintaining chondrocyte viability.47 Mechanical loading stimulates cell vitality by compressing the ECM, thereby releasing internally sequestered IGFBP3 and IGF-1. Conversely, matrix loss following OA may lead to an increase in extracellular free IGFBP3. However, the presence of matrix proteins may exacerbate the adverse impacts of IGFBP3. With aging, the ECM fosters the excessive accumulation of IGFBP3, hindering chondrocyte synthetic metabolism.48 Furthermore, matrix proteins not only enhance the inhibitory effects of low concentrations of IGFBP3 but also induce the upregulation of IGFBP3 expression in chondrocytes (Figure 3). It is evident that the ECM’s influence on IGFBP3 action manifests duality. Although some studies have attempted to simulate ECM using materials containing heparin to attenuate the impact of IGFBP3 on chondrocyte differentiation, a consensus on the regulation of IGFBP3 action through the ECM remains elusive.
 |
Table 1 The Proteases That Hydrolyze IGFBP3 |
Proteolysis
IGFBP3 is susceptible to cleavage by various proteases, including serine proteinases and metalloproteinases (Table 1). Post-cleavage, IGFBP3 releases IGF, thereby modulating downstream signaling cascades.24 Consequently, the expression levels of local proteases inevitably influence the functionality of IGFBP3. Following OA, the expression of certain proteases escalates. Pro-inflammatory cytokines, such as IL-1α and TNF-α, not only upregulate IGFBP3 expression but also enhance the expression of neutrophil metalloproteinases and fibrinolytic enzymes.55,56 These proteases play pivotal roles in the loss of ECM following OA (Figure 3). However, the precise impact of these proteases on IGFBP3 function and their involvement in cellular perception and regulation of IGFBP3 remain unclear.
Effects of Other IGFBPs
The roles of different IGFBP family members are diverse. IGFBP5 primarily facilitates the activity of IGF-1,57 while IGFBP2 behaves similarly to IGFBP3, enhancing IGF activity at lower concentrations but exhibiting inhibitory effects at higher concentrations.15 Additionally, these proteins are subject to distinct regulatory mechanisms that govern their expression. After cellular stimulation by various hormones and growth factors, IGFBP family members exhibit differential expression patterns.58 For example, chondrocyte stimulation with Dexamethasone prompts increased IGFBP3 synthesis, whereas IGFBP5 expression diminishes.59 In the chondrocyte lysates of OA patients, elevated expression levels are observed for IGFBP-2, −3, and −4, while IGFBP-1, −5, and −6 remain unaffected43 (Figure 3). This intricate interplay of varied roles and independently regulated expressions among IGFBPs constitutes a complex network, yielding synergistic or antagonistic effects that modulate IGFBP3 functionality.
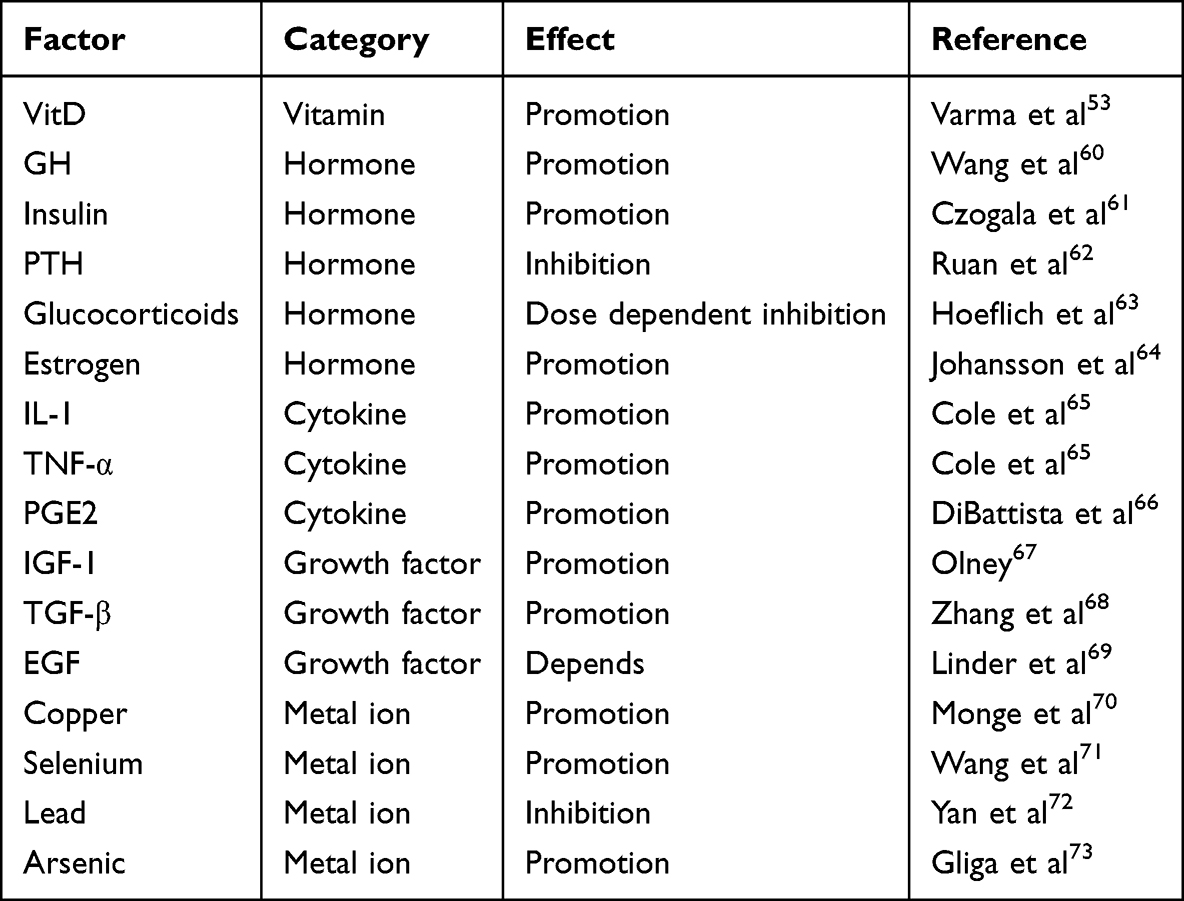 |
Table 2 Factors Influence Expression of IGFBP3 |
Regulation of IGFBP3 Expression
Transcription Factors
Various agents have been observed to induce alterations in cellular IGFBP3 expression in previous investigations (Table 2). These agents activate specific transcription factors that orchestrate changes in IGFBP3 expression. Several transcription factors implicated in the regulation of IGFBP3 have been identified, including testis-specific protein Y-linked 1,74 KN motif and ankyrin repeat domain 1,75 and Myb-like protein 2.76 However, investigations into the regulatory mechanisms governing IGFBP3 at the transcriptional level in cartilage remain relatively sparse.
Gasparini et al77 conducted ChIP experiments revealing that high mobility group protein A1 (HMGA1) can bind directly to the promoter region of IGFBP3, leading to increased IGFBP3 levels upon HMGA1 overexpression. These findings underscore HMGA1’s role as a transcription factor in driving IGFBP3 expression within chondrocytes. Moreover, this investigation highlighted the potential formation of transcriptional complexes between HMGA1 and transcript factor Sp1 and CCAAT/enhancer-binding protein beta, further enhancing IGFBP3 transcription.77 Additionally, another study identified a highly correlated single nucleotide polymorphism site, rs788748, linked to IGFBP3 expression, which interacts with CCAAT/enhancer-binding protein beta.6 These findings collectively support the involvement of the HMGA1 complex in regulating IGFBP3 transcription in chondrocytes. Nonetheless, in Gasparini’s work, the use of ChIP data from human liver cells rather than OA chondrocytes to demonstrate promoter binding relationships may lack direct applicability. Furthermore, while gene silencing and overexpression experiments in chondrocyte lines validated the correlation between HMGA1 and IGFBP3 expression, they do not conclusively rule out alternative mechanisms by which HMGA1 may indirectly regulate IGFBP3.
Previously, tumor protein p53 (p53) was identified as a transcriptional regulator of IGFBP3 in cancer cells.57 Its regulatory elements are located upstream of the IGFBP3 gene promoter and within an intronic region.78 Additionally, Zinc finger protein Gli2 (Gli2) has been shown to bind to a conserved sequence approximately 1.2kb upstream of IGFBP3.79 A study examining the roles of p53 and Gli2 in IGFBP3 regulation in growth plate chondrocytes revealed that p53 positively regulates IGFBP3 expression, while Gli2 exerts a negative regulatory effect. Their transcriptional activities operate independently.79 (Figure 2). However, this study employed fetal mouse growth plates as the experimental model, and it remains uncertain if similar dynamics occur in mature articular cartilage. Nevertheless, given p53’s pivotal role in regulating cell senescence80 and apoptosis81 in OA, it warrants investigation whether it also mediates the dysregulation of IGFBP3 expression in these contexts.
The expression of IGFBP3 is subject to modulation by various factors, including the cell type, its physiological state, and the surrounding microenvironment. The intricate diversity observed in IGFBP3 expression profiles, intimately intertwined with the myriad pathological processes ensuing in OA, underscores the putative involvement of a plethora of transcriptional regulators. However, a comprehensive understanding of the transcriptional regulation of IGFBP3 in OA remains elusive and warrants further investigation. Transcription factors capable of modulating IGFBP3 transcription, such as hypoxia-inducible factor 1-alpha82 and yes-associated protein,83 which also serve as pivotal mediators in the pathological cascades following OA, may potentially exert their regulatory influence upon translocation into the cellular nucleus, thereby contributing to the dysregulation of IGFBP3 expression.
Epigenetic Regulation
Epigenetic regulation plays a pivotal role in governing gene expression dynamics. The IGFBP3 gene is a direct transcriptional target of the methyltransferase EZH2,84 which catalyzes H3K27me3, a hallmark of repressive chromatin states. Studies using EZH1/2 knockout models in mice have shown a marked reduction in H3K27me3 enrichment within the IGFBP3 promoter region in chondrocytes, concomitant with heightened IGFBP3 expression85 (Figure 2). Nevertheless, the landscape of epigenetic regulation is multifaceted, involving various histone modifications and chromatin remodeling events beyond mere histone methylation. The overarching epigenetic mechanisms orchestrating the aberrant changes of IGFBP3 post-OA remain elusive. Additional layers of epigenetic regulation, including microRNA-mediated control,86 DNA methylation patterns,78,87 and histone acetylation dynamics,88 warrant exploration to decipher the intricate regulatory networks governing IGFBP3 expression alterations following OA.
Post-Transcription and Post-Translational Modifications
Modifications occurring post-transcriptionally and post-translationally are pivotal factors influencing protein expression. Experimental findings from rat primary chondrocytes derived from growth plates, subjected to specific stimuli, have demonstrated parallel alteration in both IGFBP3 mRNA and protein levels, suggesting minimal post-transcriptional and post-translational modifications affecting IGFBP3.89 However, such conclusions may be somewhat limited, as stability-related modifications could still be at play. Notably, chordin-like protein 1 has been identified as a facilitator of IGFBP3 stabilization through post-translational modifications, thereby mitigating its degradation.73 Consequently, a more nuanced exploration and discussion concerning post-transcriptional and post-translational modifications of IGFBP3 following OA are warranted.
Effect of IGFBP3 on Cell Interactions
IGFBP3 exhibits expression across diverse cell types and contributes to numerous physiological processes. Consequently, following OA, a sophisticated network of cellular interactions may arise within the joint via IGFBP3 involvement.
Macrophages
Macrophages have been identified as potential sources of IGFBP3 expression, particularly in conditions such as RA, where synovial macrophages are believed to be the primary producers of IGFBP3.90 Moreover, M2 macrophages exhibit heightened levels of IGFBP3 expression compared to their M1 counterparts,91 suggesting a potential involvement of IGFBP3 in the tissue repair functions attributed to M2 macrophages. Given the role of IGF-1 as a crucial growth factor secreted by M2 macrophages for tissue repair and its pivotal role in their activation,92 IGFBP3, as a key modulator of IGF-1, likely influences the activation status of M2 macrophages. Notably, IGFBP3 not only influences macrophage differentiation but also impacts their functional activity.93 Furthermore, pro-inflammatory M1 macrophages have been shown to stimulate IGFBP3 expression in other cell types94 (Figure 4). Therefore, following OA, the activation of M1 macrophages may induce IGFBP3 expression in adjacent cells, while elevated IGFBP3 levels may impede the transition of macrophages into the reparative M2 phenotype, thereby impeding cartilage repair. This mechanistic interplay could contribute to the exacerbation of OA.
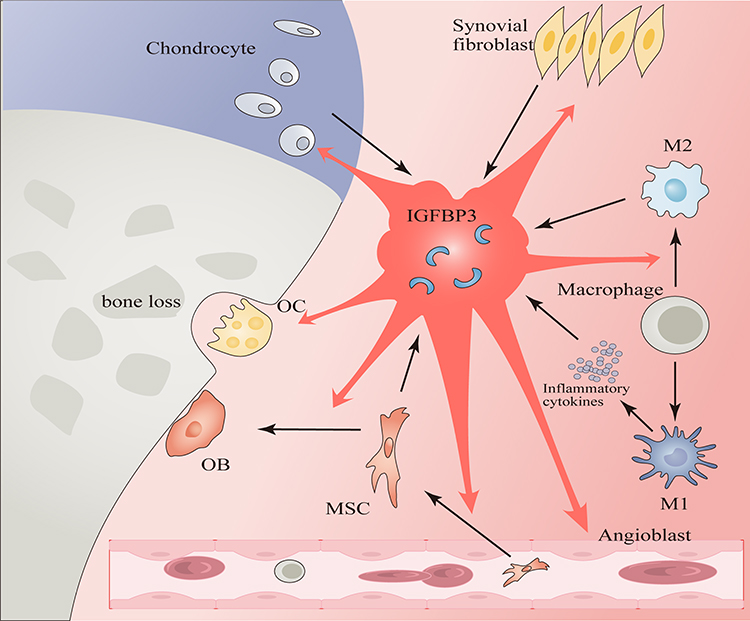 |
Figure 4 Cell Interactions Mediated by IGFBP3. (1) Synovial fibroblasts, chondrocytes, M2 macrophages, and MSCs can produce IGFBP3. M1 macrophages can stimulate other cells to produce IGFBP3 through secreting inflammatory cytokines. (2) IGFBP3 can induce phenotypic changes in chondrocytes, facilitate angiogenesis, inhibit synovial inflammation, and trigger apoptosis of synovial fibroblasts. It also facilitates macrophages transition into M2 phenotype, enhances the homing of MSCs and their differentiation into OBs, and modulates the function of OC. |
Synovial Fibroblasts and Angioblasts
The expression of IGFBP3 is closely linked to the condition of the synovium. Inflammatory factors induce an increase in the secretion of IGFBP3 and IGF-1 by synovial fibroblasts.90 Notably, IGFBP3 serves as a key regulator of angiogenesis.95 Elevated levels of IGFBP3 not only facilitate angiogenesis within the synovium of RA,90 but also suppress the activation of NF-κB pathway and the expression of inflammation-associated genes in synovial fibroblasts through mechanisms independent of IGF-1. This action leads to the apoptosis of synovial fibroblasts, consequently mitigating synovial inflammation and hyperplasia96 (Figure 4). Localized inflammation and hyperplasia in the synovium are key pathological features following OA. A clear understanding of the interplay between IGFBP3 and these processes holds promise for refining future therapeutic modalities targeting IGFBP3.
Mesenchymal Stem Cells (MSCs)
IGFBP3 acts as a critical regulatory factor secreted by MSCs.97 In this context, human bone marrow MSCs notably secrete abundant IGFBP3, which, upon deposition in the ECM, enhances both MSC homing and osteogenic differentiation capacities.98 Beyond its autocrine effects on MSC differentiation,99 IGFBP3 secretion by MSCs also exerts paracrine effects, influencing the phenotype of neighboring cells26 (Figure 4). Consequently, bone marrow MSCs establish a complex multicellular interaction network, modulating both their own behavior and that of adjacent cells through IGFBP3 secretion, thereby impacting the repair process in OA.
Osteoblast (OB) and Osteoclast (OC)
Perspectives on the impact of IGFBP3 on bone health are heterogeneous. In RA, IGFBP3 exhibits a bone-protective effect by inhibiting RANKL expression, thereby attenuating osteoclastogenesis and bone resorption.100 However, in murine models with IGFBP3 overexpression, there is an observed increase in OC formation exceeding that of OB, resulting in diminished bone density101 (Figure 4). These discrepant findings may arise from the dual actions of IGFBP3, whether dependent or independent of IGF-1 signaling, or variations in the IGF-1 concentration ratios. Notably, bone degradation in the later stages of OA is a hallmark manifestation, and intercellular interactions mediated by IGFBP3 are likely pivotal in influencing osteoclastic and osteoblastic activities.
IGFBP3 in Diabetic OA
Diabetic OA is increasingly regarded as a metabolically distinct OA subtype characterized by insulin resistance, chronic low-grade inflammation and impaired growth factor signaling. The IGF axis is tightly linked to glucose metabolism, and systemic IGFBP3 levels are frequently altered in diabetes.102 Hyperglycemia and advanced glycation end products attenuate chondrocyte responsiveness to IGF-1,103 while elevated IGFBP3 may further restrict IGF-1 bioavailability, thereby exacerbating anabolic resistance in diabetic cartilage.
Beyond IGF-dependent actions, IGFBP3 may contribute to mitochondrial dysfunction, oxidative stress and chondrocyte senescence under high-glucose conditions through IGF-independent nuclear and mitochondrial pathways. In contrast to primary OA, diabetic OA is likely influenced more strongly by endocrine-derived IGFBP3,40 reflecting systemic metabolic dysregulation rather than purely local cartilage pathology. However, direct evidence defining the specific role of IGFBP3 in diabetic OA remains limited. Elucidating whether IGFBP3 represents a shared mediator or a subtype-specific regulator will be critical for developing targeted therapies for metabolically driven OA.
Conclusion and Perspectives
IGFBP3, as a key component of the IGF system, exerts both IGF-dependent and IGF-independent effects in cartilage and joint tissues. Accumulating evidence demonstrates that IGFBP3 expression is dynamically altered in OA in a stage- and region-specific manner. However, existing studies report inconsistent functional outcomes, with IGFBP3 showing both inhibitory and protective roles in chondrocyte metabolism. These discrepancies may be attributed to differences in disease stage, cartilage zone, experimental models, and particularly the molar ratio of IGFBP3 to IGF-1. Beyond its classical role in modulating IGF-1 signaling, IGFBP3 may function independently through nuclear and mitochondrial localization, participating in transcriptional regulation and apoptosis. Nevertheless, most mechanistic evidence is derived from non-articular or progenitor cell models, and its precise role in mature articular chondrocytes remains insufficiently defined.
IGFBP3 is also involved in intercellular communication within the joint microenvironment, including interactions with macrophages, synovial fibroblasts, MSCs, and bone cells. Such multicellular regulatory networks suggest that IGFBP3 may serve as a molecular hub linking cartilage degeneration with inflammation, angiogenesis, and osteochondral remodeling.
Current evidence is largely based on experimental models, and IGF-independent mechanisms and IGFBP family interactions remain unresolved. Despite these limitations, IGFBP3 emerges as a context-dependent regulator of OA. Future multi-omics and cell-specific studies are required to define its mechanisms and therapeutic potential.
Abbreviations
ECM, extracellular matrix; HMGA1, high mobility group protein A1; IGF, insulin-like growth factor; IGF-1, insulin-like growth factor 1; IGFR, insulin-like growth factor receptor; IGFBP3, insulin-like growth factor binding protein 3; MSCs, Mesenchymal Stem Cells; OA, Osteoarthritis; RA, rheumatoid arthritis; p53, tumor protein p53; Gli2, Zinc finger protein Gli2; MP, Membrane protein; OB, Osteoblast; OC, Osteoclast.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analyzed in this study.
Acknowledgments
During the preparation of this manuscript, the authors used ChatGPT-5.2 for the purposes of language polishing and Nanobanana 2.0 in assisting with the graphical optimization and refinement. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Author Contributions
JC: Conceptualization, Writing – original draft, Writing – review and editing, Data curation, Visualization. XL: Conceptualization Writing – original draft, Writing – review and editing, Data curation, Visualization. YG: Conceptualization, Writing – review and editing, Data curation, Visualization. HZ: Writing – review and editing, Data curation, Visualization. YY: Writing – review and editing, Data curation. ZL: Funding acquisition, Supervision, Writing – review and editing, Data curation. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Joint Fund for Innovation and Development of Natural Science Foundation of Hubei Province (No. 2025AFD655), the Hubei Provincial Health Commission medical research achievements transformation project (No. WJ2023ZH0025).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sanchez-Lopez E, Coras R, Torres A, et al. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. 2022;18(5):258–15. doi:10.1038/s41584-022-00749-9
2. Matsuoka K, Bakiri L, Bilban M, et al. Metabolic rewiring controlled by c-Fos governs cartilage integrity in osteoarthritis. Ann Rheum Dis. 2023;82(9):1227–1239. doi:10.1136/ard-2023-224002
3. Xiang J, Yang X, Tan M, et al. NIR-enhanced Pt single atom/g-C3N4 nanozymes as SOD/CAT mimics to rescue ATP energy crisis by regulating oxidative phosphorylation pathway for delaying osteoarthritis progression. Bioact Mater. 2024;36:1–13. doi:10.1016/j.bioactmat.2024.02.018
4. Xie Y, Zinkle A, Chen L, et al. Fibroblast growth factor signalling in osteoarthritis and cartilage repair. Nat Rev Rheumatol. 2020;16(10):547–564. doi:10.1038/s41584-020-0469-2
5. Eleftheriou A, Petry CJ, Hughes IA, et al. The High-Risk Type 1 Diabetes HLA-DR and HLA-DQ Polymorphisms Are Differentially Associated With Growth and IGF-I Levels in Infancy: the Cambridge Baby Growth Study. Diabetes Care. 2021;44(8):1852–1859. doi:10.2337/dc20-2820
6. Evans DS, Cailotto F, Parimi N, et al. Genome-wide Association and Functional Studies Identify a Role for IGFBP3 in Hip Osteoarthritis. Ann Rheum Dis. 2015;74(10):1861–1867. doi:10.1136/annrheumdis-2013-205020
7. Bittner N, Shi C, Zhao D, et al. Primary osteoarthritis chondrocyte map of chromatin conformation reveals novel candidate effector genes. Ann Rheum Dis. 2024;83(8):1048–1059. doi:10.1136/ard-2023-224945
8. Li H, Miao L, Wu J, et al. Identification and validation of cellular senescence-related genes and immune cell infiltration characteristics in intervertebral disc degeneration. Front Immunol. 2025;16:1589849. doi:10.3389/fimmu.2025.1589849
9. Tang Y, Hong F, Ding S, et al. METTL3-mediated m6A modification of IGFBP7-OT promotes osteoarthritis progression by regulating the DNMT1/DNMT3a-IGFBP7 axis. Cell Rep. 2023;42(6):112589. doi:10.1016/j.celrep.2023.112589
10. Klinder A, Kussauer S, Hiemer B, et al. Influence of Conditioned Media on the Re-Differentiation Capacity of Human Chondrocytes in 3D Spheroid Cultures. J Clin Med. 2020;9(9):2798. doi:10.3390/jcm9092798
11. Kiepe D, Ciarmatori S, Haarmann A, et al. Differential expression of IGF system components in proliferating vs. differentiating growth plate chondrocytes: the functional role of IGFBP-5. Am J Physiol Endocrinol Metab. 2006;290(2):E363–E371. doi:10.1152/ajpendo.00363.2005
12. Parker EA, Hegde A, Buckley M, et al. Spatial and temporal regulation of GH-IGF-related gene expression in growth plate cartilage. J Endocrinol. 2007;194(1):31–40. doi:10.1677/JOE-07-0012
13. Wang D, Yang H, Zhang M, et al. Insulin-like growth factor-1 engaged in the mandibular condylar cartilage degeneration induced by experimental unilateral anterior crossbite. Arch Oral Biol. 2019;98:17–25. doi:10.1016/j.archoralbio.2018.11.002
14. Liu Z, Liu H, Li D, et al. Comprehensive analysis of m6A RNA methylation modification patterns and the immune microenvironment in osteoarthritis. Front Immunol. 2023;14:1128459. doi:10.3389/fimmu.2023.1128459
15. de Los Rios P, Hill DJ. Cellular localization and expression of insulin-like growth factors (IGFs) and IGF binding proteins within the epiphyseal growth plate of the ovine fetus: possible functional implications. Can J Physiol Pharmacol. 1999;77(4):235–249. doi:10.1139/y99-015
16. Zhang C, Xiao D, Shu L, et al. Single-cell RNA sequencing uncovers cellular heterogeneity and the progression of heterotopic ossification of the elbow. Front Pharmacol. 2024;15:1434146. doi:10.3389/fphar.2024.1434146
17. Hunziker EB, Kapfinger E, Martin J, et al. Insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) is closely associated with the chondrocyte nucleus in human articular cartilage. Osteoarthritis Cartilage. 2008;16(2):185–194. doi:10.1016/j.joca.2007.06.008
18. Coates E, Fisher JP. Gene expression of alginate-embedded chondrocyte subpopulations and their response to exogenous IGF-1 delivery. J Tissue Eng Regen Med. 2012;6(3):179–192. doi:10.1002/term.411
19. Hu C, Xie D, Li J, et al. Nanozyme-Integrated 3D-Printed Gradient Scaffold Rescues Redox Homeostasis for Enhanced Osteochondral Repair. Small Struct. 2026;7(1):e202500413. doi:10.1002/sstr.202500413
20. Hajjar D, Santos MF, Kimura ET. Mandibular repositioning modulates IGFBP-3, −4, −5 and −6 expression in the mandibular condylar cartilage of young rats. Biorheology. 2006;43(3):311–321.
21. van de Lest CH, van den Hoogen BM, van Weeren PR, et al. Loading-induced changes in synovial fluid affect cartilage metabolism. Biorheology. 2000;37(1–2):45–55.
22. Deschner J, Rath-Deschner B, Reimann S, et al. Regulatory effects of biophysical strain on rat TMJ discs. Ann Anat Anat Anz Off Organ Anat Ges. 2007;189(4):326–328. doi:10.1016/j.aanat.2007.02.004
23. Li K, Ding K, Zhu Q, et al. Extracellular matrix stiffness aggravates urethral stricture through Igfbp3/Smad pathway. Sci Rep. 2023;13(1):14315. doi:10.1038/s41598-023-41584-6
24. Kim H, Fu Y, Hong HJ, et al. Structural basis for assembly and disassembly of the IGF/IGFBP/ALS ternary complex. Nat Commun. 2022;13(1):4434. doi:10.1038/s41467-022-32214-2
25. Baxter RC. Signaling Pathways of the Insulin-like Growth Factor Binding Proteins. Endocr Rev. 2023;44(5):753–778. doi:10.1210/endrev/bnad008
26. Liu QW, Ying YM, Zhou JX, et al. Human amniotic mesenchymal stem cells-derived IGFBP-3, DKK-3, and DKK-1 attenuate liver fibrosis through inhibiting hepatic stellate cell activation by blocking Wnt/β-catenin signaling pathway in mice. Stem Cell Res Ther. 2022;13(1):224. doi:10.1186/s13287-022-02906-z
27. Matsumoto T, Tsurumoto T, Goldring MB, et al. Differential effects of IGF-binding proteins, IGFBP-3 and IGFBP-5, on IGF-I action and binding to cell membranes of immortalized human chondrocytes. J Endocrinol. 2000;166(1):29–37. doi:10.1677/joe.0.1660029
28. De Ceuninck F, Caliez A. A simple and reliable assay of proteoglycan synthesis by cultured chondrocytes. Methods Mol Med. 2004;100:209–218. doi:10.1385/1-59259-810-2:209
29. Sechrist ZR, Belcher DJ, Patel NR, et al. Ablation of tumor-derived IGFBP-3 attenuates cancer-associated skeletal muscle wasting in murine pancreatic cancer. Am J Physiol Cell Physiol. 2026;330(2):C467–C481. doi:10.1152/ajpcell.00421.2025
30. Cai Q, Dozmorov M, Oh Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells. 2020;9(5):1261. doi:10.3390/cells9051261
31. De Ceuninck F, Caliez A, Dassencourt L, et al. Pharmacological disruption of insulin-like growth factor 1 binding to IGF-binding proteins restores anabolic responses in human osteoarthritic chondrocytes. Arthritis Res Ther. 2004;6(5):R393–403. doi:10.1186/ar1201
32. Zhang Z, Cheng D, Dang J, et al. Decoding Endoplasmic Reticulum Stress on Chondrocyte Driving Osteoarthritis Development through Integrating Single-Cell and Transcriptomic Profiling. Int J Med Sci. 2025;22(15):3906–3923. doi:10.7150/ijms.119573
33. de Silva HC, Lin MZ, Phillips L, et al. IGFBP-3 interacts with NONO and SFPQ in PARP-dependent DNA damage repair in triple-negative breast cancer. Cell Mol Life Sci CMLS. 2019;76(10):2015–2030. doi:10.1007/s00018-019-03033-4
34. Bhardwaj A, Pathak KA, Shrivastav A, et al. Insulin-Like Growth Factor Binding Protein-3 Binds to Histone 3. Int J Mol Sci. 2021;22(1):407. doi:10.3390/ijms22010407
35. Wang SH, Chen YL, Hsiao JR, et al. Insulin-like growth factor binding protein 3 promotes radiosensitivity of oral squamous cell carcinoma cells via positive feedback on NF-κB/IL-6/ROS signaling. J Exp Clin Cancer Res CR. 2021;40(1):95. doi:10.1186/s13046-021-01898-7
36. Spagnoli A, Hwa V, Horton WA, et al. Antiproliferative effects of insulin-like growth factor-binding protein-3 in mesenchymal chondrogenic cell line RCJ3.1C5.18. relationship to differentiation stage. J Biol Chem. 2001;276(8):5533–5540. doi:10.1074/jbc.M005088200
37. Longobardi L, Torello M, Buckway C, et al. A novel insulin-like growth factor (IGF)-independent role for IGF binding protein-3 in mesenchymal chondroprogenitor cell apoptosis. Endocrinology. 2003;144(5):1695–1702. doi:10.1210/en.2002-220959
38. Spagnoli A, Longobardi L, O’Rear L. Cartilage disorders: potential therapeutic use of mesenchymal stem cells. Endocr Dev. 2005;9:17–30.
39. Lai A, Sun J, Dai Z, et al. Unraveling IGFBP3-mediated m6A modification in fracture healing. Pathol Res Pract. 2024;255:155220. doi:10.1016/j.prp.2024.155220
40. Zhang Y, Ching K, Zhang L, et al. Endothelial Dysfunction Contributes to Chondrocyte Senescence Associated With Insulin-Like Growth Factor-Binding Protein-6 in a Spontaneously Hypertensive Rat Model. J Am Heart Assoc. 2026;15(1):e042132. doi:10.1161/JAHA.124.042132
41. Kim OK, Yun JM, Lee M, et al. A Mixture of Humulus japonicus Increases Longitudinal Bone Growth Rate in Sprague Dawley Rats. Nutrients. 2020;12(9):2625. doi:10.3390/nu12092625
42. Kiepe D, Ulinski T, Powell DR, et al. Differential effects of insulin-like growth factor binding proteins-1, −2, −3, and −6 on cultured growth plate chondrocytes. Kidney Int. 2002;62(5):1591–1600. doi:10.1046/j.1523-1755.2002.00603.x
43. Tardif G, Reboul P, Pelletier JP, et al. Normal expression of type 1 insulin-like growth factor receptor by human osteoarthritic chondrocytes with increased expression and synthesis of insulin-like growth factor binding proteins. Arthritis Rheum. 1996;39(6):968–978. doi:10.1002/art.1780390614
44. Neidel J, Blum WF, Schaeffer HJ, et al. Elevated levels of insulin-like growth factor (IGF) binding protein-3 in rheumatoid arthritis synovial fluid inhibit stimulation by IGF-I of articular chondrocyte proteoglycan synthesis. Rheumatol Int. 1997;17(1):29–37. doi:10.1007/PL00006847
45. Olney RC, Tsuchiya K, Wilson DM, et al. Chondrocytes from osteoarthritic cartilage have increased expression of insulin-like growth factor I (IGF-I) and IGF-binding protein-3 (IGFBP-3) and −5, but not IGF-II or IGFBP-4. J Clin Endocrinol Metab. 1996;81(3):1096–1103. doi:10.1210/jcem.81.3.8772582
46. Buhrmann C, Honarvar A, Setayeshmehr M, et al. Herbal Remedies as Potential in Cartilage Tissue Engineering: an Overview of New Therapeutic Approaches and Strategies. Molecules. 2020;25(13):3075. doi:10.3390/molecules25133075
47. Martin JA, Miller BA, Scherb MB, et al. Co-localization of insulin-like growth factor binding protein 3 and fibronectin in human articular cartilage. Osteoarthritis Cartilage. 2002;10(7):556–563. doi:10.1053/joca.2002.0791
48. Tang X, Zhong J, Luo H, et al. Efficacy of Naringenin against aging and degeneration of nucleus pulposus cells through IGFBP3 inhibition. Sci Rep. 2025;15(1):6780. doi:10.1038/s41598-025-90909-0
49. Srinivasan S, Kryza T, Bock N, et al. A PSA SNP associates with cellular function and clinical outcome in men with prostate cancer. Nat Commun. 2024;15(1):9587. doi:10.1038/s41467-024-52472-6
50. Wetterau LA, Moore MG, Lee KW, et al. Novel aspects of the insulin-like growth factor binding proteins. Mol Genet Metab. 1999;68(2):161–181. doi:10.1006/mgme.1999.2920
51. Conover CA, Oxvig C. The IGF System and Aging. Endocr Rev. 2025;46(2):214–223. doi:10.1210/endrev/bnae029
52. Cai Q, Kim M, Harada A, et al. Alpha-1 Antitrypsin Inhibits Tumorigenesis and Progression of Colitis-Associated Colon Cancer through Suppression of Inflammatory Neutrophil-Activated Serine Proteases and IGFBP-3 Proteolysis. Int J Mol Sci. 2022;23(22):13737. doi:10.3390/ijms232213737
53. Varma Shrivastav S, Bhardwaj A, Pathak KA, et al. Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3): unraveling the Role in Mediating IGF-Independent Effects Within the Cell. Front Cell Dev Biol. 2020;8:286. doi:10.3389/fcell.2020.00286
54. Conover CA, Oxvig C. The Pregnancy-Associated Plasma Protein-A (PAPP-A) Story. Endocr Rev. 2023;44(6):1012–1028. doi:10.1210/endrev/bnad017
55. AI Martín, T Priego, Á Moreno-Ruperez, et al. IGF-1 and IGFBP-3 in Inflammatory Cachexia. Int J Mol Sci. 2021;22(17):9469.
56. Ueng SWN, Hsu YH, Lin YC, et al. Therapeutic blockade of platelet-derived growth factor receptor-like protein attenuates cartilage degeneration and modulates cytokines in a spontaneous osteoarthritis mouse model. Int Immunopharmacol. 2025;164:115294. doi:10.1016/j.intimp.2025.115294
57. Zhang Y, Katkhada K, Meng LZ, et al. Myogenic IGFBP5 levels in rhabdomyosarcoma are nourished by mesenchymal stromal cells and regulate growth arrest and apoptosis. Cell Commun Signal. 2025;23(1):184. doi:10.1186/s12964-025-02171-6
58. Houtman E, Tuerlings M, Riechelman J, et al. Elucidating mechano-pathology of osteoarthritis: transcriptome-wide differences in mechanically stressed aged human cartilage explants. Arthritis Res Ther. 2021;23(1):215. doi:10.1186/s13075-021-02595-8
59. Koedam JA, Hoogerbrugge CM, Van Buul-Offers SC. Differential regulation of IGF-binding proteins in rabbit costal chondrocytes by IGF-I and dexamethasone. J Endocrinol. 2000;165(3):557–567. doi:10.1677/joe.0.1650557
60. Wang L, Yan R, Yang Q, et al. Role of GH/IGF axis in arsenite-induced developmental toxicity in zebrafish embryos. Ecotoxicol Environ Saf. 2020;201:110820. doi:10.1016/j.ecoenv.2020.110820
61. Czogała W, Strojny W, Tomasik P, et al. The Insight into Insulin-Like Growth Factors and Insulin-Like Growth-Factor-Binding Proteins and Metabolic Profile in Pediatric Obesity. Nutrients. 2021;13(7):2432. doi:10.3390/nu13072432
62. Ruan X, Jin X, Sun F, et al. IGF signaling pathway in bone and cartilage development, homeostasis, and disease. FASEB J off Publ Fed Am Soc Exp Biol. 2024;38(17):e70031.
63. Hoeflich A, Fitzner B, Walz C, et al. Reduced Fragmentation of IGFBP-2 and IGFBP-3 as a Potential Mechanism for Decreased Ratio of IGF-II to IGFBPs in Cerebrospinal Fluid in Response to Repeated Intrathecal Administration of Triamcinolone Acetonide in Patients With Multiple Sclerosis. Front Endocrinol. 2020;11:565557. doi:10.3389/fendo.2020.565557
64. Johansson H, Macis D, Oliva M, et al. Predictive Effect of IGFBP-3 on Low-Dose Tamoxifen Efficacy in Noninvasive Breast Cancer in the Phase III Tam-01 Trial. Clin Cancer Res. 2025;31(10):1841–1846. doi:10.1158/1078-0432.CCR-24-2987
65. Cole CL, Bachman JF, Ye J, et al. Increased myocellular lipid and IGFBP-3 expression in a pre-clinical model of pancreatic cancer-related skeletal muscle wasting. J Cachexia, Sarcopenia Muscle. 2021;12(3):731–745. doi:10.1002/jcsm.12699
66. DiBattista JA, Doré S, Morin N, et al. Prostaglandin E2up-regulates insulin-like growth factor binding protein-3 expression and synthesis in human articular chondrocytes by a c-AMP-independent pathway: role of calcium and protein kinase A and C. J Cell Biochem. 1996;63(3):320–333. doi:10.1002/(SICI)1097-4644(19961201)63:3<320::AID-JCB7>3.0.CO;2-Z
67. Olney RC, Smith RL, Kee Y, et al. Production and hormonal regulation of insulin-like growth factor binding proteins in bovine chondrocytes. Endocrinology. 1993;133(2):563–570. doi:10.1210/endo.133.2.7688290
68. Zhang X, Wang G, Gong Y, et al. IGFBP3 induced by the TGF-β/EGFRvIII transactivation contributes to the malignant phenotype of glioblastoma. iScience. 2023;26(5):106639. doi:10.1016/j.isci.2023.106639
69. Linder M, Hecking M, Glitzner E, et al. EGFR controls bone development by negatively regulating mTOR-signalingta during osteoblast differentiation. Cell Death Differ. 2018;25(6):1094–1106. doi:10.1038/s41418-017-0054-7
70. Escobedo-Monge MF, Barrado E, Parodi-Román J, et al. Copper and Copper/Zn Ratio in a Series of Children with Chronic Diseases: a Cross-Sectional Study. Nutrients. 2021;13(10):3578. doi:10.3390/nu13103578
71. Wang S, Zhao G, Shao W, et al. The Importance of Se-Related Genes in the Chondrocyte of Kashin-Beck Disease Revealed by Whole Genomic Microarray and Network Analysis. Biol Trace Elem Res. 2019;187(2):367–375. doi:10.1007/s12011-018-1404-0
72. Yan R, Ding J, Yang Q, et al. Lead acetate induces cartilage defects and bone loss in zebrafish embryos by disrupting the GH/IGF-1 axis. Ecotoxicol Environ Saf. 2023;253:114666. doi:10.1016/j.ecoenv.2023.114666
73. Gliga AR, Engström K, Kippler M, et al. Prenatal arsenic exposure is associated with increased plasma IGFBP3 concentrations in 9-year-old children partly via changes in DNA methylation. Arch Toxicol. 2018;92(8):2487–2500. doi:10.1007/s00204-018-2239-3
74. Tu W, Yang B, Leng X, et al. Testis-specific protein, Y-linked 1 activates PI3K/AKT and RAS signaling pathways through suppressing IGFBP3 expression during tumor progression. Cancer Sci. 2019;110(5):1573–1586. doi:10.1111/cas.13984
75. Li M, Wu W, Deng S, et al. TRAIP modulates the IGFBP3/AKT pathway to enhance the invasion and proliferation of osteosarcoma by promoting KANK1 degradation. Cell Death Dis. 2021;12(8):767. doi:10.1038/s41419-021-04057-0
76. Fan X, Wang Y, Jiang T, et al. B-Myb Mediates Proliferation and Migration of Non-Small-Cell Lung Cancer via Suppressing IGFBP3. Int J Mol Sci. 2018;19(5):1479. doi:10.3390/ijms19051479
77. Gasparini G, De Gori M, Paonessa F, et al. Functional relationship between high mobility group A1 (HMGA1) protein and insulin-like growth factor-binding protein 3 (IGFBP-3) in human chondrocytes. Arthritis Res Ther. 2012;14(5):R207. doi:10.1186/ar4045
78. Hanafusa T, Shinji T, Shiraha H, et al. Functional promoter upstream p53 regulatory sequence of IGFBP3 that is silenced by tumor specific methylation. BMC Cancer. 2005;5(1):9. doi:10.1186/1471-2407-5-9
79. Ho L, Stojanovski A, Whetstone H, et al. Gli2 and p53 Cooperate to Regulate IGFBP-3- Mediated Chondrocyte Apoptosis in the Progression from Benign to Malignant Cartilage Tumors. Cancer Cell. 2009;16(2):126–136. doi:10.1016/j.ccr.2009.05.013
80. Zhao LD, Bie LY, Hu L, et al. IGF-1 induces cellular senescence in rat articular chondrocytes via Akt pathway activation. Exp Ther Med. 2020;20(5):49. doi:10.3892/etm.2020.9177
81. Ma W, Tan X, Xie Z, et al. P53: a Key Target in the Development of Osteoarthritis. Mol Biotechnol. 2024;66(1):1–10. doi:10.1007/s12033-023-00736-9
82. Li H, Wu J, Dai H, et al. PDZRN4 suppresses lung adenocarcinoma progression via inhibiting ubiquitin-mediated HIF-1A degradation. Oncogene. 2026;45(4):577–590. doi:10.1038/s41388-025-03670-z
83. Zhang X, Cai D, Zhou F, et al. Targeting downstream subcellular YAP activity as a function of matrix stiffness with Verteporfin-encapsulated chitosan microsphere attenuates osteoarthritis. Biomaterials. 2020;232:119724. doi:10.1016/j.biomaterials.2019.119724
84. Chen L, Alexe G, Dharia NV, et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128(1):446–462. doi:10.1172/JCI90793
85. Lui JC, Garrison P, Nguyen Q, et al. EZH1 and EZH2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy. Nat Commun. 2016;7(1):13685. doi:10.1038/ncomms13685
86. Xiong LL, Xue LL, Du RL, et al. Vi4-miR-185-5p-Igfbp3 Network Protects the Brain From Neonatal Hypoxic Ischemic Injury via Promoting Neuron Survival and Suppressing the Cell Apoptosis. Front Cell Dev Biol. 2020;8:529544. doi:10.3389/fcell.2020.529544
87. Beck A, Trippel F, Wagner A, et al. Overexpression of UHRF1 promotes silencing of tumor suppressor genes and predicts outcome in hepatoblastoma. Clin Clin Epigenet. 2018;10(1):27. doi:10.1186/s13148-018-0462-7
88. Yaqoob U, Luo F, Greuter T, et al. GIPC-Regulated IGFBP-3 Promotes HSC Migration In Vitro and Portal Hypertension In Vivo Through a β1-Integrin Pathway. Cell Mol Gastroenterol Hepatol. 2020;10(3):545–559. doi:10.1016/j.jcmgh.2020.05.005
89. Zhong K, Luo W, Li N, et al. CDK12 regulates angiogenesis of advanced prostate cancer by IGFBP3. Int J Oncol. 2024;64(2):20. doi:10.3892/ijo.2024.5608
90. Suzuki S, Morimoto S, Fujishiro M, et al. Inhibition of the insulin-like growth factor system is a potential therapy for rheumatoid arthritis. Autoimmunity. 2015;48(4):251–258. doi:10.3109/08916934.2014.976631
91. Mohanraj L, Kim HS, Li W, et al. IGFBP-3 inhibits cytokine-induced insulin resistance and early manifestations of atherosclerosis. PLoS One. 2013;8(1):e55084. doi:10.1371/journal.pone.0055084
92. Chen L, Zhang L, Jin G, et al. Synergy of 5-aminolevulinate supplement and CX3CR1 suppression promotes liver regeneration via elevated IGF-1 signaling. Cell Rep. 2023;42(8):112984. doi:10.1016/j.celrep.2023.112984
93. Park J, Jung MJ, Chung WY. The downregulation of IGFBP3 by TGF-β signaling in oral cancer contributes to the osteoclast differentiation. Biochem Biophys Res Commun. 2021;534:381–386. doi:10.1016/j.bbrc.2020.11.073
94. Rouault C, Marcelin G, Adriouch S, et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia. 2021;64(1):240–254. doi:10.1007/s00125-020-05307-0
95. Zhao HJ, Klausen C, Zhu H, et al. Bone morphogenetic protein 2 promotes human trophoblast cell invasion and endothelial-like tube formation through ID1-mediated upregulation of IGF binding protein-3. FASEB J off Publ Fed Am Soc Exp Biol. 2020;34(2):3151–3164.
96. Wang X, Wang Y, Lei P, et al. IGFBP5 regulates fibrocartilage differentiation and cartilage injury induced by T-2 toxin via blocking IGF-1/IGF-1R signalling. Rheumatology. 2025;64(6):4051–4060. doi:10.1093/rheumatology/keaf084
97. Zhang X, Huang Y, Liu Y, et al. Local transplantation of mesenchymal stem cells improves encephalo-myo-synangiosis-mediated collateral neovascularization in chronic brain ischemia. Stem Cell Res Ther. 2023;14(1):233. doi:10.1186/s13287-023-03465-7
98. Deng M, Luo K, Hou T, et al. IGFBP3 deposited in the human umbilical cord mesenchymal stem cell-secreted extracellular matrix promotes bone formation. J Cell Physiol. 2018;233(8):5792–5804. doi:10.1002/jcp.26342
99. Ushakov RE, Skvortsova EV, Vitte MA, et al. Chondrogenic differentiation followed IGFBP3 loss in human endometrial mesenchymal stem cells. Biochem Biophys Res Commun. 2020;531(2):133–139. doi:10.1016/j.bbrc.2020.07.064
100. Ye C, Hou W, Chen M, et al. IGFBP7 acts as a negative regulator of RANKL-induced osteoclastogenesis and oestrogen deficiency-induced bone loss. Cell Prolif. 2020;53(2):e12752. doi:10.1111/cpr.12752
101. Ding M, Li B, Chen H, et al. Human breastmilk-derived Bifidobacterium longum subsp. infantis CCFM1269 regulates bone formation by the GH/IGF axis through PI3K/AKT pathway. Gut Microbes. 2024;16(1):2290344. doi:10.1080/19490976.2023.2290344
102. Lay AC, Hale LJ, Stowell-Connolly H, et al. IGFBP-1 expression is reduced in human type 2 diabetic glomeruli and modulates β1-integrin/FAK signalling in human podocytes. Diabetologia. 2021;64(7):1690–1702. doi:10.1007/s00125-021-05427-1
103. Mu W, Liang G, Feng Y, et al. The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment. Pharmaceuticals. 2022;15(10):1274. doi:10.3390/ph15101274
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Ferroptosis in Osteoarthritis: Current Understanding
Liu Y, Zhang Z, Fang Y, Liu C, Zhang H
Journal of Inflammation Research 2024, 17:8471-8486
Published Date: 7 November 2024
Disulfidptosis-Induced Chondrocyte-Macrophage Crosstalk via GYS1/CCND1/NOD2 Axis Promotes Osteoarthritis Progression
Sun Q, Wei Z, Shao G, Yang M, Zeng X, Hong X, Gou R, Zhang H, Zhang Y, Dong S, Qu Y, Liu Y
Journal of Inflammation Research 2026, 19:568766
Published Date: 6 January 2026