Back to Journals » Drug Design, Development and Therapy » Volume 13
A first-in-human study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of KM-819 (FAS-associated factor 1 inhibitor), a drug for Parkinson’s disease, in healthy volunteers
Authors Shin W, Lim KS, Kim MK, Kim HS, Hong J, Jhee S ![]() , Kim J, Yoo S, Chung YT, Lee JM
, Kim J, Yoo S, Chung YT, Lee JM ![]() , Cho DY
, Cho DY ![]()
Received 18 December 2018
Accepted for publication 18 February 2019
Published 29 March 2019 Volume 2019:13 Pages 1011—1022
DOI https://doi.org/10.2147/DDDT.S198753
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Wonsuk Shin,1 Kyoung Soo Lim,1 Min-Kyoung Kim,1 Hyun Sook Kim,2 Jihwa Hong,3 Stanford Jhee,3 Joseph Kim,3 Sungeun Yoo,4 Yeon-Tae Chung,4 Jae Moon Lee,4 Doo-Yeoun Cho1
1Department of Clinical Pharmacology and Therapeutics, CHA Bundang Medical Center, CHA University School of Medicine, Seongnam, Republic of Korea; 2Department of Neurology, CHA Bundang Medical Center, CHA University School of Medicine, Seongnam, Republic of Korea; 3Department of Scientific Affairs, PAREXEL International, Waltham, MA, USA; 4Kainos Medicine Inc., Seongnam, Republic of Korea
Background: KM-819 is a novel FAS-associated factor 1 (FAF1) inhibitor, and a neuroprotective agent, under clinical development for the treatment of Parkinson’s disease as a disease-modifying drug.
Methods: This first-in-human, single and multiple ascending dose study investigated the safety, tolerability, pharmacokinetics, and pharmacodynamics of KM-819 in healthy volunteers. Additionally, the effect of age on safety and pharmacokinetics were assessed. The starting dose was determined considering the no observed adverse effect level based on preclinical studies, and the dose escalations in subsequent cohorts were decided based on safety, tolerability, and pharmacokinetic data from previous dose cohorts.
Results: After a single dose, the KM-819 plasma exposure showed a less than dose-proportional increase across a dose range of 10–400 mg. After repeated dosing, KM-819 plasma exposure increased in an approximately dose-proportional manner across the evaluated dose range (30–400 mg once daily for 7 days). The mean elimination half-life was 1.8 to 4.8 h with the lower KM-819 doses (≤30 mg), which increased to around 9 h with the higher doses (100–400 mg). When administered to the elderly population, KM-819 plasma exposure increased to 102% after a 200 mg once-daily dosing for 7 days. No clear treatment-related effects on the estimated pharmacodynamic variables were observed. Single or multiple doses of KM-819 were generally well tolerated.
Conclusion: The data from this study can be used to guide rational drug dosing and choose therapeutic regimens in subsequent clinical studies.
Keywords: first-in-human, KM-819, pharmacokinetics, pharmacodynamics, safety
Introduction
Parkinson’s disease is a progressive neurodegenerative disease caused by the loss of dopaminergic neurons in the substantia nigra in the midbrain. The symptoms include bradykinesia, resting tremor, rigidity, unstable posture, and postural reflex impairment.1 The available drugs for the development and progression of Parkinson’s disease are limited and not sufficient to cater for the existing medical needs. Although the mechanism for the neuronal loss is unknown, the movement control ability can be temporally improved by the dopamine precursor L-3,4-dihydroxyphenylalanine. The currently available drugs for the treatment of Parkinson’s disease can be classified largely into 4 classes (dopamine replacement, dopamine catechol-O-methyl transferase inhibitors, dopamine agonists, and monoamine oxidase type B inhibitors).2 Although these drugs help to supplement dopamine or aid in relieving symptoms, there is no remedy for the development of the disease, such as inhibiting the death of the dopaminergic neuronal cells. Further, the presently used drugs require increasing dose over time and have serious side effects, such as impulse control disorders, movement impairment, and hallucinations.3–5
FAS-associated factor 1 (FAF1) is a protein related to Fas-mediated apoptosis.6 It has been reported that the expression of FAF1 is increased in the brain in Parkinson’s disease.7 A significant increase in FAF1 expression in the midbrain has been confirmed by animal models of Parkinson’s disease. The cells and animal models of Parkinson’s disease have also demonstrated an inhibition of cell death when FAF1 expression is reduced.8 Therefore, FAF1 is validated as a potential novel target for new drug discovery for Parkinson’s disease.
KM-819 is an innovative new drug that protects the neuronal cells from death through inhibition of FAF1. KM-819 inhibits Parkinson’s disease progression by inhibiting the Fas-mediated cell death pathway.9,10 According to an in vitro study, KM-819 was found to protect the dopaminergic neurons treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in a dose-dependent manner. In an MPTP-treated mice model, KM-819 protected the dopaminergic neuronal cells both in the substantia nigra and striatum, as well as improved the behavioral impairments. This suggested that KM-819 has a potential capability of delaying or stopping the progression of Parkinson’s disease. Therefore, if successful, KM-819 can be considered as a disease-modifying drug (unpublished data). In a subacute toxicity study, KM-819 did not show adverse effects after repeated administration for 4 weeks in Sprague Dawley rats (up to 500 mg/kg/day) and for 2 weeks in beagle dogs (up to 1,000 mg/kg/day).11 Based on these data, a first-in-human trial was performed to investigate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of KM-819 in healthy young male subjects and healthy elderly subjects.
Material and methods
Subjects
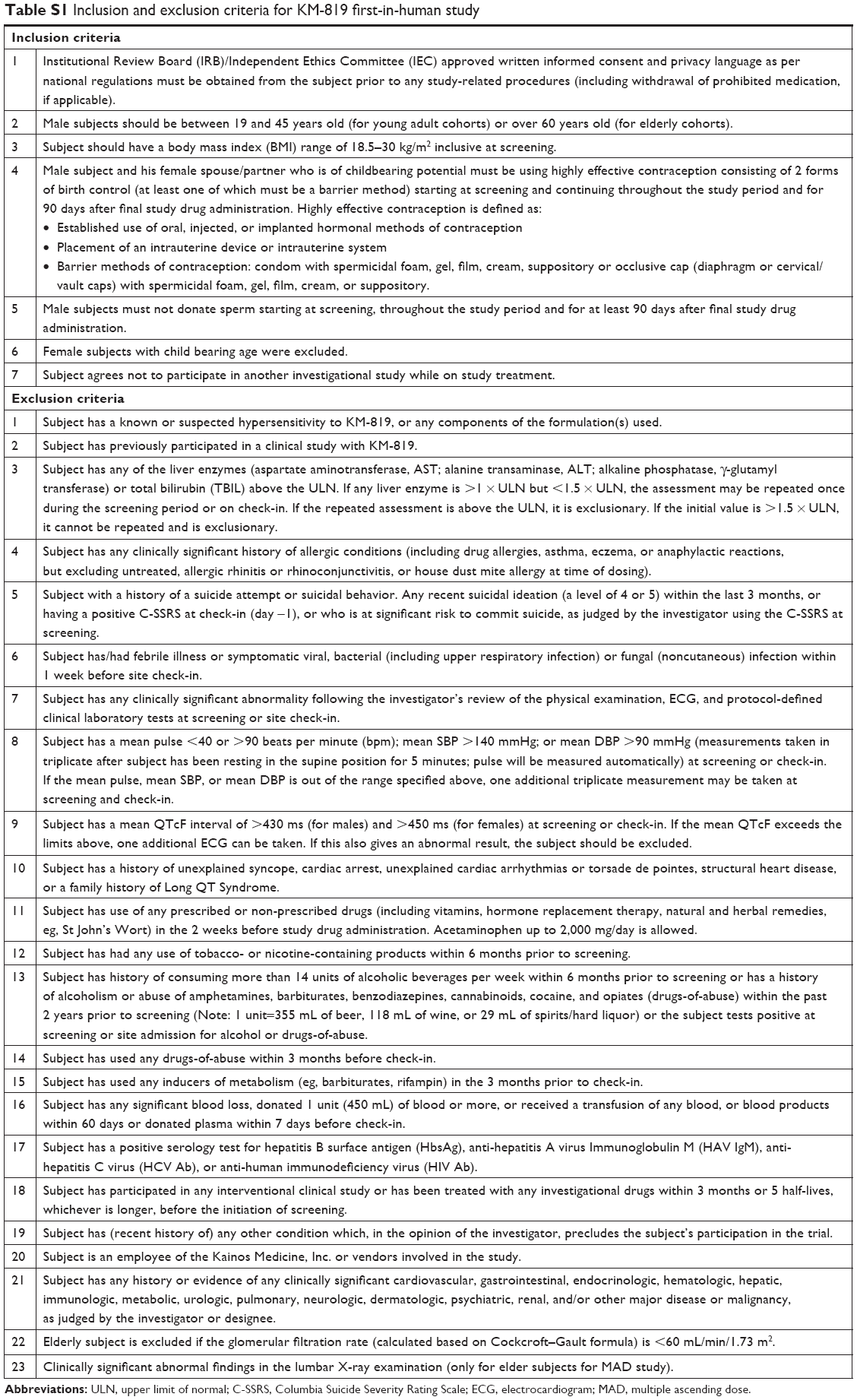
Male subjects aged 19–45 years old (for the young cohorts) or over 60 years old (for the elderly cohorts) with a body mass index (BMI) of 18.5–30.0 kg/m2 were eligible to participate in this study. All the subjects were deemed healthy by a physical examination, medical history, vital signs, clinical laboratory tests (hematology, blood chemistry, and urinalysis), and 12-lead electrocardiogram (ECG) performed up to 4 weeks prior to the first administration of the study drug. The subjects with a history of hepatic, renal, psychiatric, or cardiovascular disorders, those with known hypersensitivity to KM-819 and any components of the formulation, those who reported use of other drugs that might interfere with the study results within 2 weeks of the study drug administration, and those with a history of drug abuse were excluded from the study. Female subjects were excluded because fertility and early embryonic development study was not completed. Specific inclusion and exclusion criteria are presented in Table S1.
Study design
This study was approved by the ethics review board of CHA Bundang Medical Center, Seongnam, Republic of Korea. All the subjects were given detailed written and oral information about the study and written informed consent were obtained before screening for eligibility. The study was conducted at the Clinical Trials Center of CHA Bundang Medical Center, Seongnam, Republic of Korea in compliance with the ethical principles of the Declaration of Helsinki, the Good Clinical Practice Guidelines of the International Conference on Harmonization, and local laws and regulations. This study was registered at ClinicalTrials.gov (https://ClinicalTrials.gov, identifier: NCT03022799).
This was a first-in-human, randomized, single and multiple-dose, placebo-controlled dose escalation study in healthy subjects. In the single ascending dose (SAD) study, 40 healthy young male subjects and 8 healthy elderly male subjects were enrolled and randomized to receive either KM-819 or placebo. Each of the 5 dose escalation cohorts consisted of 8 healthy young male subjects; 6 subjects received a single dose of 10, 30, 100, 200, or 400 mg KM-819 and 2 subjects received placebo. After completion of the 5 dose escalation cohorts, 8 elderly male subjects were enrolled into an additional cohort, in which 6 subjects received 200 mg KM-819 and 2 subjects received placebo. In each of the 6 single-dose cohorts, there were 2 sentinel subjects for each dose escalation cohort. Two subjects were dosed on the first day (1 subject received KM-819 and 1 subject received placebo) and the remaining 6 subjects were dosed at least 24 hours after the first 2 subjects. All the subjects underwent a 3-day confinement period during which they were hospitalized for study-related activities (day −1 to day 3). The subjects were required to return for outpatient visits on day 4, day 7, and for a follow-up visit on day 14 (Figure 1A).
 | Figure 1 Study Design of the single ascending dose (SAD) study (A), and multiple ascending dose (MAD) study (B). |
In the multiple ascending dose (MAD) study, 32 healthy young male subjects and 8 healthy elderly male subjects were enrolled and randomized to receive either KM-819 or placebo. Each of the 4 dose escalation cohorts consisted of 8 healthy young male subjects; 6 subjects received 30, 100, 200 or 400 mg of KM-819 once daily (QD) for 7 days and 2 subjects received placebo. The cohorts were dosed sequentially with escalating doses. After completion of the 4 dose escalation cohorts, 8 elderly male subjects were enrolled into an additional cohort, in which 6 subjects received 200 mg KM-819 QD for 7 days and 2 subjects received placebo. All the subjects underwent an 8-day confinement period during which they were hospitalized for study-related activities (day −1 to day 8). The subjects were required to return for a follow-up visit on day 14 (Figure 1B).
An escalation to the next dose level was decided only after the safety and tolerability data for all subjects in the cohort and available plasma PKs from the previously administered dose cohort were reviewed. The results of exploratory PD assessments and the central nervous system scales were not included in the review for dose escalation decisions.
Rationale for dose selection
The United States Food and Drug Administration (US FDA) guidance document for estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers12 and allometric scaling modeling and simulation was used for the initial calculation of the starting human dose. Based on the US FDA guidance document and no observed adverse effect level (NOAEL) of 500 mg/kg/day obtained from the 4-week rat toxicology study,11 the human equivalent dose (HED) was estimated to be 5,600 mg for a human subject weighing 70 kg. A 10-fold safety factor was applied to the HED, resulting in an estimated maximum recommended starting dose (MRSD) of 560 mg per person for a 70 kg subject. In addition to the US FDA guidance document, an allometric scaling method was used to predict the human PKs. The human PK parameters were predicted as follows: apparent oral clearance (CL/F)=2.93 L/h, apparent volume of distribution (Vz/F)=39.2 L, and apparent terminal elimination half-life (t1/2)=9.27 h. Using these predicted human PK parameters, a simulation was performed based on the assumption that the human PK behaves like a one-compartment model with first-order absorption and first-order elimination rate. The bioavailability (F) was assumed to be 35% (mean F from animal data). Based on a simulated human PK profile and animal efficacy data, a lower dose was selected as the starting dose. The study had an initial starting dose of 10 mg, which is 56-fold below the estimated MRSD derived from the rat NOAEL.
PK assessments
In the SAD study, blood samples (6 mL) for the PK study of KM-819 were obtained on day 1 predose and 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 72 hours postdose. The urine samples for the possible analysis of KM-819 and qualitative analysis of any metabolites were collected at day 1 predose and from 0 to 24 hours postdose. In the MAD study, the blood samples for the PK study were collected on day 1 predose and 0.25, 0.5, 1, 2, 4, 6, 8, and 12 hours postdose, on days 2, 3, 4, 5, and 6 predose, and on day 7 predose and 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 hours postdose. The cerebrospinal fluid (CSF) samples for the PK analysis were collected on day 1 predose and on day 7 (at 1 hour after the last dosing). The CSF (approximately 2 mL) samples were collected by lumbar puncture between the third and fourth lumbar vertebrae.
The blood samples were centrifuged at 3,000 rpm for 10 min at 4°C, and the obtained plasma samples (0.8 mL) were transferred into 3 polypropylene tubes (1.5 mL). Similarly, the urine (1 mL) and CSF samples were (0.5 mL) were transferred into 3 polypropylene tubes (1.5 mL). All the tubes were immediately stored in a freezer at −70°C until analysis.
The KM-819 concentrations in plasma and urine were analyzed by an ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The system comprised of a Waters ACQUITY UPLC™ (Waters Corporation, Milford, MA, USA) coupled with a Waters Xevo™ TQ MS (Waters Corporation, Milford, MA, USA). Briefly, a sample of either plasma or urine was mixed with dimethyl sulfoxide in the presence of an internal standard (KM-819-d5) in a Master Block 96 well plate (Greiner Bio-One International GmbH, Kremsmünster, Austria). The mixture was then vortexed for 3 min and centrifuged for 1 min at 4,000 rpm. Subsequently, 0.400 mL of methanol was added, vortexed for 3 min, and centrifuged for 1 min at 4,000 rpm. After transferring 0.250 mL of the supernatant to another 96 well plate, 0.150 mL of 0.1% (w/v) ammonium acetate was added, vortexed for 3 min, and centrifuged for 1 min at 4,000 rpm. Then, a 2 μL of the aliquot was injected into the UPLC-MS/MS system for analysis. The quantification was performed using multiple reaction monitoring of the transitions m/z 460.1→214.2 and m/z 467.1→219.2 for KM-819 and KM-819-d5, respectively. The calibration curves were linear over the range of 2–20,000 ng/mL for plasma and 0.5–1,000 ng/mL for urine. The limits of quantification for plasma and urine were 2 ng/mL and 0.5 ng/mL, respectively.
The PK parameters of KM-819 were estimated using non-compartmental methods in the Phoenix WinNonlin 6.3 software (Certara USA Inc., Princeton, NJ, USA). The maximum plasma concentration after a single dose (Cmax), the maximum plasma concentration at steady-state (Cmax,ss), time to Cmax (Tmax), and Cmax,ss (Tmax,ss) were determined directly from the concentration-time curve. The terminal phase rate-constant (λz) was estimated by a linear regression of the data points included in the terminal phase of the log-linear plot of the concentration-time curve, and the elimination half-life (t1/2) was calculated as 0.693/λz. The area under the plasma concentration-time curve (AUC) from time 0 to 24 hours postdose (AUC0–24), the AUC from zero extrapolated to infinity (AUCinf), the AUC from time 0 hour to the last quantifiable concentration (AUClast), and the AUC over the dosing interval at steady-state (AUCtau) were calculated using the linear trapezoidal rule. Any BLQ (below the limit of quantification) values occurring prior to Cmax were assigned a value of zero; BLQ values after the last quantifiable concentration were treated as missing. The apparent oral clearance (CL/F) was calculated using the ratio of the dose to the AUCtau, and the accumulation index was calculated as the ratio of the AUCtau and Cmax,ss on day 7 to the AUC0–24 and Cmax on day 1.
PD assessments
In the SAD study, Bond and Lader visual analog scale (VAS), profile of mood states (POMS), and Korean Wechsler Adult Intelligence Scale-IV (K-WAIS-IV) were used on day −1, day 1 predose and at 3, 6, 12, 24, 48, and 72 hours postdose.13–15 The Columbia Suicide Severity Rating Scale (C-SSRS) was used at screening and on day 3. In the MAD study, Bond and Lader VAS, POMS and K-WAIS-IV were used on day −1, day 1 predose, day 7 predose, and at 3, 6, 12, and 24 hours postdose. The C-SSRS was used at screening, on day 3 and day 8. The samples for estimation of alpha-synuclein oligomer (blood and CSF), total tau (CSF), and phosphor-tau (CSF) were collected on day 1 predose and on day 7 (1 hour after the last dosing). Protein concentrations were estimated at the University of California at San Diego (ADCS Biomarker Core).
Safety assessments
Safety was assessed throughout the study based on vital signs, clinical laboratory tests, 12-lead ECGs, physical examinations, and adverse events (AEs) reported by the subjects or observed by the medical investigators. All adverse events were coded using MedDRA version 19.1.
Statistical analyses
All demographic data, PK parameters, PD parameters, and safety data were summarized using descriptive statistics for continuous variables and using the number and percentage of subjects for categorical variables. The dose proportionality was assessed for Cmax, AUClast, and AUCinf in the SAD study and for Cmax, AUC0–24 (on day 1), Cmax,ss, Cmin,ss, and AUCtau, (on day 7) in the MAD study, using a power model with the following equation:
|
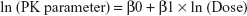
The estimate of the slope of the regression line (β1) and the corresponding 95% CI were calculated. All statistical analyses were performed in the SAS® version 9.3 software (SAS Institute Inc., Cary, NC, USA) and a P-value <0.05 was considered statistically significant.
Results
Subjects
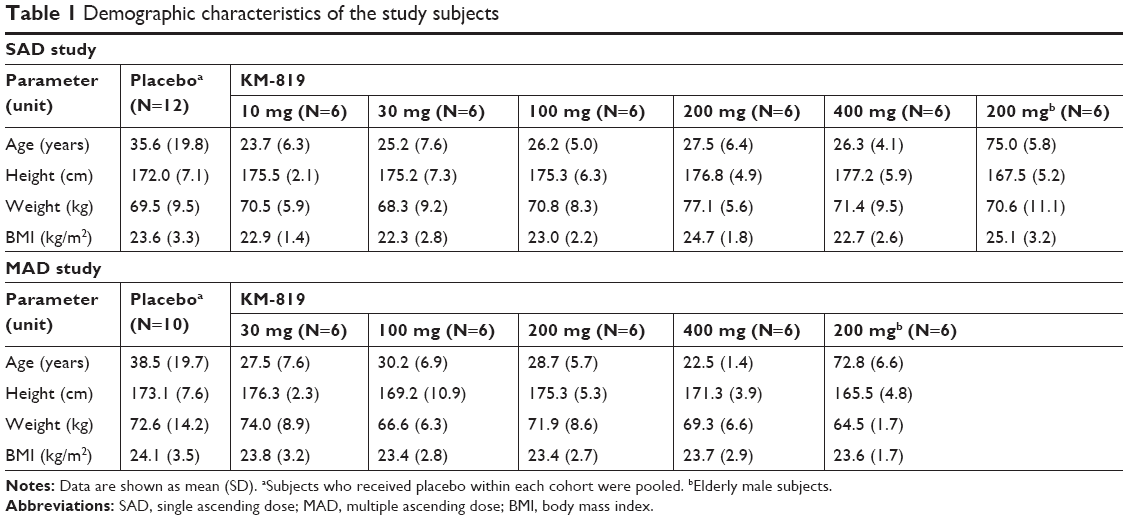
A total of 88 subjects were enrolled, 66 subjects received KM-819 (36 in the SAD study and 30 in the MAD study), and 22 subjects received placebo (12 in the SAD study and 10 in the MAD study). Two subjects in the MAD study who received KM-819 dropped out before completion of the study by withdrawing consent, and the remaining 86 subjects completed the study as per protocol. The safety analysis included all the randomized subjects in the SAD (N=48) or MAD (N=40) studies. The PK analysis included 66 subjects who received KM-819 in the SAD (N=36) or MAD (N=30) studies, and the PD analysis included 64 subjects who received KM-819 in the SAD (N=36) or MAD (N=28) studies. The demographic characteristics of the enrolled subjects are enumerated in Table 1.
 | Table 1 Demographic characteristics of the study subjects |
Pharmacokinetics
The mean plasma concentration-time profiles of KM-819 in the SAD and MAD studies are presented in Figure 2. In the SAD study, the KM-819 concentrations were quantifiable at less than 0.5 hour following administration of all doses including the elderly male cohort. The median Tmax values were 1–4 hours following administration of all doses and a similar Tmax was observed in the elderly cohort. The mean t1/2 was 1.8–4.8 hours for KM-819 doses ≤30 mg and increased to around 9 hours for higher doses. There was a moderate degree of variability for the major PK parameters (AUCinf and Cmax), with a coefficient of variation (CV%) ranging up to 55%. After administering a single dose of 200 mg KM-819, Cmax was found to be similar between the young and the elderly subjects. The elderly subjects, however, showed a 17% higher t1/2 and a 49% higher AUC compared to the young subjects (Table 2). The concentrations of KM-819 in urine were very low, and 24-hour recovery of KM-819 in urine (percent of dose) was less than 1% for all subjects.
 | Figure 2 Mean plasma concentration-time profile of KM-819 in the single ascending dose (SAD) (A) and multiple ascending dose (MAD) (B) studies. |
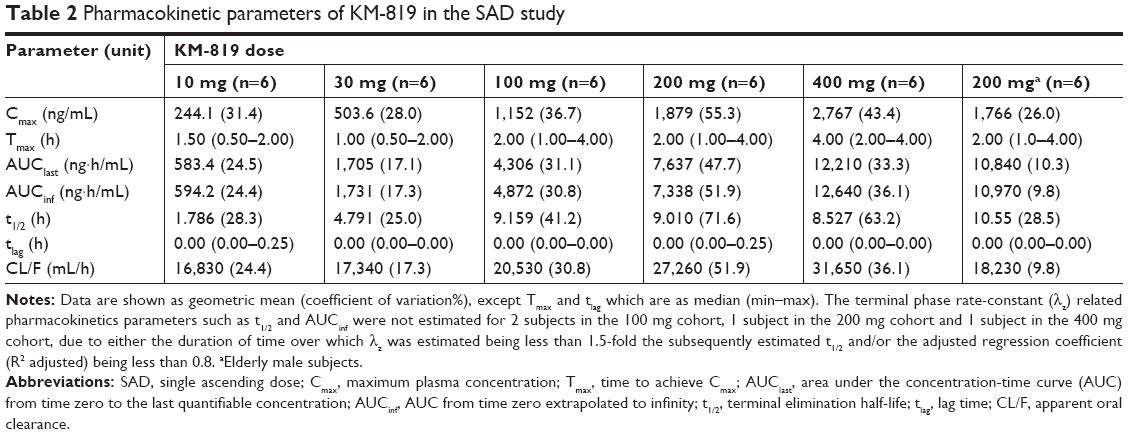 | Table 2 Pharmacokinetic parameters of KM-819 in the SAD study |
In the MAD study, the single dose PK was similar to that observed in the SAD study. The mean variability (CV%) for the major PK parameters (AUCtau and Cmax,ss) were moderate to high ranging up to 96%. The geometric mean accumulation ratio for KM-819 AUC and Cmax ranged from 0.63–1.49 for all doses indicating no significant accumulation. After 200 mg QD dosing for 7 days, the day 7 KM-819 Cmax,ss and AUCtau were increased to 191% and 102%, respectively in the elderly population (Table 3). Consistent with the estimated t1/2 (around 9 hours in the SAD study), the steady-state trough concentrations were achieved after 5 days of repeated dose in each group (data not shown).
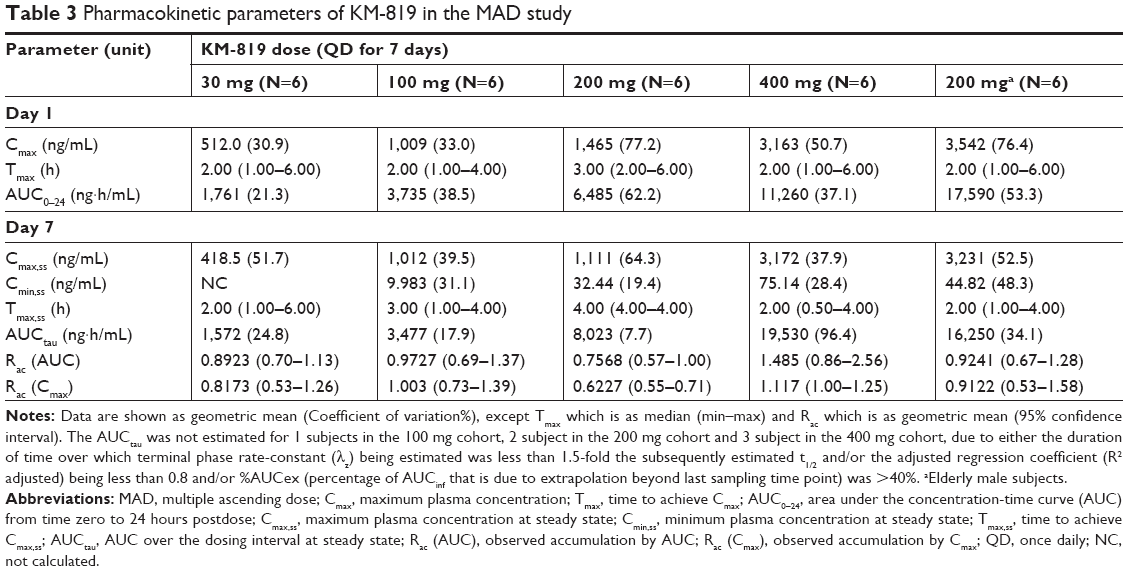 | Table 3 Pharmacokinetic parameters of KM-819 in the MAD study |
Based on the power model, KM-819 AUC and Cmax showed a less than dose-proportional increase after a single dose administration of KM-819 in both the SAD and MAD studies, as the upper boundary of 95% CI of the slope was below 1.0. After repeat dosing of KM-819 for 7 days in the MAD study, KM-819 AUC on day 7, however, had a dose-proportional increase, but Cmax showed a less than dose-proportional increase, and Cmin had a slightly more than dose-proportional increase (Table 4).
 | Table 4 Assessment of dose proportionality of KM-819 pharmacokinetic parameters (power model) |
In the MAD study, after repeat dose of KM-819 30 mg for 7 days in the 30 mg cohort, no CSF KM-819 concentrations were measurable at 1 hour postdose on day 7. In the higher dose cohorts (≥100 mg QD), KM-819 concentrations were, however, measurable in more than half of the CSF samples after repeat dose and the ratio of CSF/Cmax was calculated for those subjects with measurable CSF KM-819 concentrations on day 7. The mean CSF/Cmax ratios (%) were 0.067%, 0.074%, 0.049%, and 0.032% for 100 mg, 200 mg, 400 mg cohort, and 200 mg elderly cohort, respectively.
Pharmacodynamics
No signs of KM-819 treatment-related effects on the Bond and Lader VAS were observed. There were no apparent treatment or cohort differences in the POMS study results. There were no clear treatments differences in the K-WAIS-IV study results, but the day 7 values were slightly higher than the day 1 and baseline values, possibly pointing to a training effect. This effect was less pronounced in the placebo cohorts, and was less likely to be significantly different. Likewise, there were no apparent treatment or cohort differences in the C-SSRS study results.
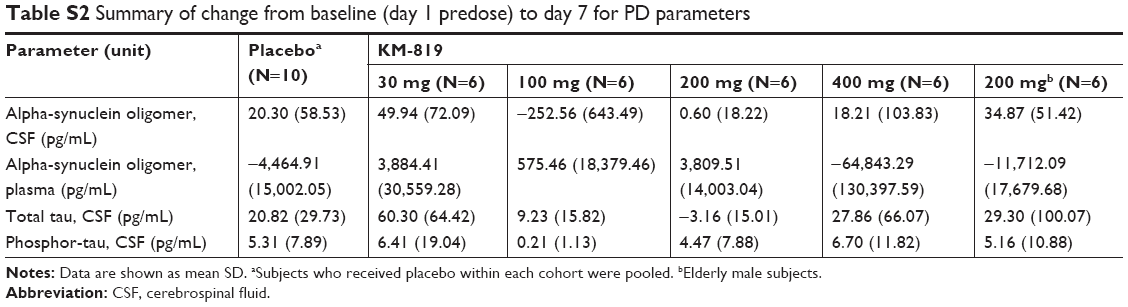
No clear treatment- or dose-related changes from day 1 to day 7 were apparent in the alpha-synuclein oligomer, total tau, and phosphor tau estimations in the CSF or alpha-synuclein oligomer estimation in the plasma. The results of these parameters were highly variable, without any discernable trend (Table S2).
Safety and tolerability
In total, 17 treatment-emergent adverse events (TEAEs) were reported from 12 of the 48 subjects in the SAD study (Table 5). Ten of these were considered to be related to the KM-819 by the investigator. No treatment- or dose-related trend in the incidence of TEAEs was observed. There were no serious adverse events (SAEs) reported, no subjects were withdrawn from the SAD study due to AEs, and there were no TEAEs leading to death.
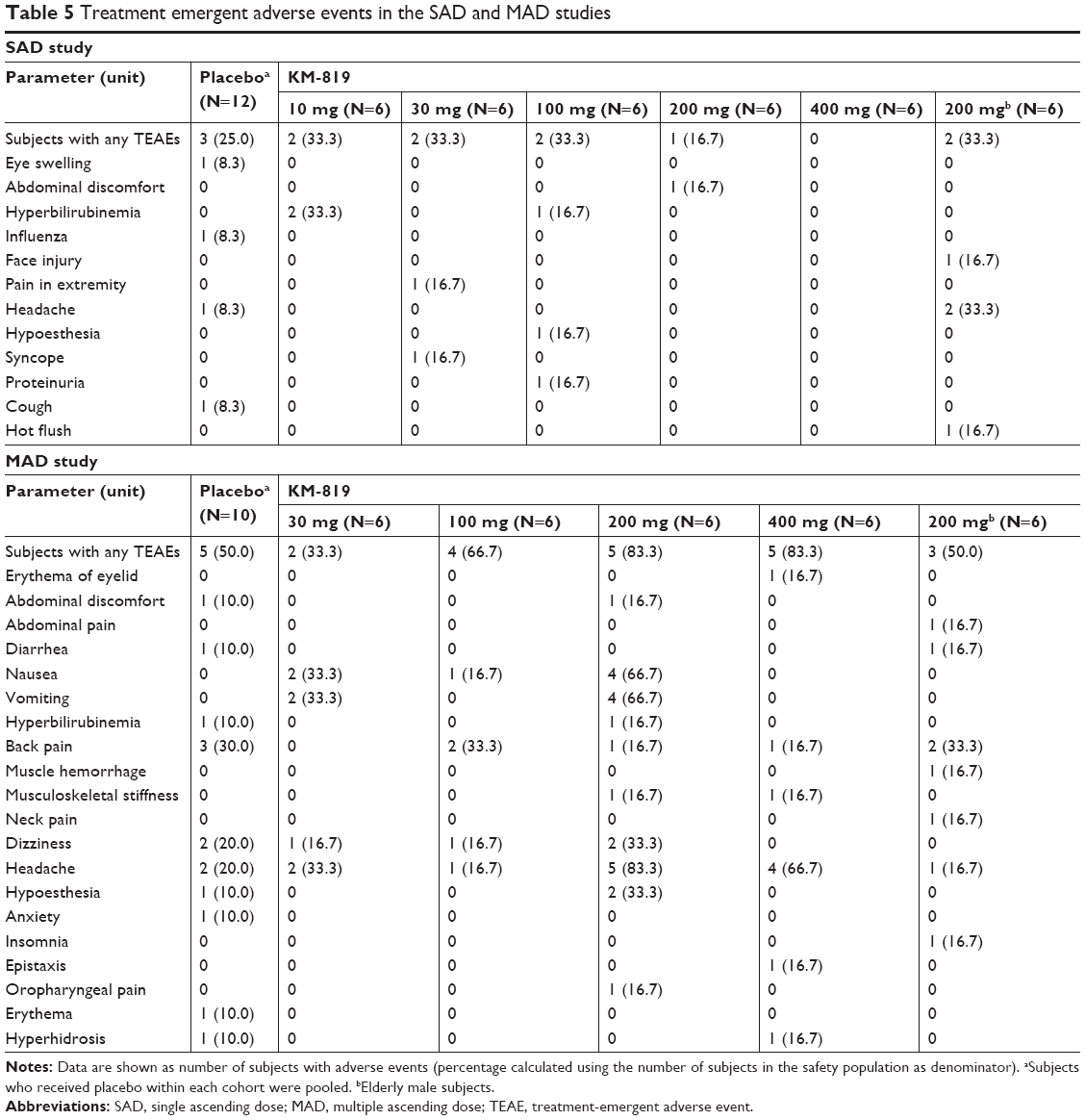 | Table 5 Treatment emergent adverse events in the SAD and MAD studies |
The incidence of TEAEs was notably higher in the MAD study than in the SAD study. In total, 82 TEAEs were reported from 24 of the 40 subjects in the MAD study (Table 5). The lumbar puncture procedure performed in the MAD study was most probably attributable to the higher incidence of TEAEs. Sixty-eight out of the 82 reported TEAEs were considered by the investigator to be related to KM-819. No treatment-related trend in the incidence of TEAEs was detected, but the incidence of TEAEs in the 200 mg KM-819 treatment group where young males were dosed, was notably higher than that in the other treatment groups. There was 1 SAE reported (a headache which was considered unlikely to be related to KM-819, and probably related to the lumbar puncture procedure), and 2 subjects were withdrawn from the MAD study due to AEs (headache). There were no TEAEs leading to death.
In the SAD study for the first two dose cohorts (10 mg and 30 mg KM-819), an increasing trend of bilirubin levels was apparent on day 2, with bilirubin levels returning to normal on day 7. At higher doses, this effect was not observed. No other treatment- or dose-related or clinically relevant trends were observed for the clinical laboratory parameters, vital signs measurements, 12-lead ECG, or physical examination evaluations during the study. Overall, no remarkable or significant safety concerns were observed from the safety data in the study.
Discussion
The existing standard of care for Parkinson’s disease is symptomatic treatment by dopamine replacement, dopamine agonists, or analogous mechanisms. A disease-modifying treatment is one of the major unmet medical needs for halting disease progression.2 KM-819 is an orally active small molecule drug developed as an inhibitor for FAF1, a proapoptotic protein, involved in Parkinson’s disease.7,8
This first-in-human study investigated the safety, tolerability, PKs, and PDs of single and multiple escalating doses of KM-819 in healthy volunteers. The effect of age on safety and PKs were also assessed. The KM-819 steady-state exposure increased in an approximately dose-proportional manner across the evaluated dose range (30–400 mg once daily for 7 days) with the maximum KM-819 concentrations occurring between 1 and 4 hours postdose. Consistent with the estimated t1/2, the steady-state concentrations of KM-819 were achieved after 5 days of dosing. When administered to the elderly population, KM-819 Cmax and AUC increased to 191% and 102% after 200 mg QD dosing for 7 days. The administration of KM-819 was safe and well tolerated and resulted in no SAEs or deaths.
The terminal phase t1/2 was <5 hours for the lower doses (10 mg and 30 mg) and increased to 8.5–9.2 hours for the higher doses of KM-819. The dose-dependent nature of t1/2 may be the result of unmasking the true t1/2 values as the concentrations in the terminal phase increased with higher doses. Because KM-819 plasma concentrations declined in a bi-exponential manner, t1/2 may be a good predictor of drug accumulation and fluctuation, and support once daily or twice daily dosing of KM-819.
This study showed that the exposure of the elderly subjects to KM-819 was generally higher than that observed in the young subjects. At steady state, Cmax and AUC increased on average to 191% and 102% in the elderly subjects, whereas no differences in tlag and Tmax were observed between the young and the elderly subjects. It is believed that there is no difference in the absorption of KM-819 between these two populations (young and elderly), but the elimination of KM-819 might have been inhibited and resulted in increased t1/2 and PK exposures in the elderly population. Considering that Parkinson’s disease prevalence is increasing with age and Parkinson’s disease affects about 1% of the population above 60 years,16 these findings are ideal for a drug targeting Parkinson’s disease in the elderly population.
The mean CSF/Cmax ratios (%) of KM-819 were less than 0.1% in each cohort and no dose-dependent trend was observed. The CSF sample after administrated KM-819 was only obtained on day 7 at 1 hour after the last dosing in this study. Therefore, the estimated drug concentration in CSF could not reflect the maximum CSF concentration on day 7. The transport across the blood–brain barrier (BBB) requires time,17,18 and the included subjects in this study were all healthy with an intact BBB, which limited the penetration of KM-819 in the CSF. BBB dysfunctions have been reported in Parkinson’s disease,19–21 the CSF concentrations of KM-819 in patients with Parkinson’s disease are expected to be higher than observed in this study.
The AUCtau was not estimated for 6 subjects in the MAD study, due to either the duration of time over which terminal phase rate-constant (λz) was estimated being less than 1.5-fold the subsequently estimated t1/2 and/or the adjusted regression coefficient (R2 adjusted) being less than 0.8 and/or %AUCex (percentage of AUCinf that is due to extrapolation beyond last sampling time point) was >40%. Therefore smaller sample size would affect the slope estimate of AUCtau in power model at day 7 in the MAD study.
This study was performed in healthy young and elderly subjects who were not on any concomitant medication. In the real-life clinical settings, many elderly patients will likely be on a cocktail of medications. Therefore, the current data should be extrapolated with caution to the medicated elderly subjects on multiple medications.
Conclusion
KM-819, a FAF 1 inhibitor, exhibited favorable pharmacokinetic and safety profiles in this first-in-human study. These results support dose selection and further clinical evaluation of KM-819 for a phase II study.
Acknowledgments
This study was funded by Kainos Medicine, Inc., Seongnam, Republic of Korea and supported by a grant of the Korea Health Technology R & D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant no: HI14C2750).
The abstract of this paper was presented at the 2018 Annual Meeting of American Society for Clinical Pharmacology and Therapeutics as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in Clinical Pharmacology and Therapeutics.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Data availability
The raw data of this study will not be shared because of confidentiality.
Disclosure
S Yoo, YT Chung, and JM Lee are the employees of Kainos Medicine Inc. J Hong, S Jhee and J Kim are the employees of PAREXEL, which performed work for this study under contract to Kainos Medicine Inc. The other authors report no conflicts of interest related to this work.
References
Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. doi:10.1136/jnnp.2007.124958 | ||
Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. 2014;311(16):1670–1683. doi:10.1001/jama.2014.3654 | ||
Weintraub D, Siderowf AD, Potenza MN, et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol. 2006;63(7):969–973. doi:10.1001/archneur.63.7.969 | ||
Jiménez-Urbieta H, Gago B, de la Riva P, Delgado-Alvarado M, Marin C, Rodriguez-Oroz MC. Dyskinesias and impulse control disorders in Parkinson’s disease: from pathogenesis to potential therapeutic approaches. Neurosci Biobehav Rev. 2015;56:294–314. doi:10.1016/j.neubiorev.2015.07.010 | ||
Robottom BJ. Efficacy, safety, and patient preference of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease. Patient Prefer Adherence. 2011;5:57–64. doi:10.2147/PPA.S11182 | ||
Ryu S-W, Kim E. Apoptosis induced by human Fas-associated factor 1, hFAF1, requires its ubiquitin homologous domain, but not the Fas-binding domain. Biochem Biophys Res Commun. 2001;286(5):1027–1032. doi:10.1006/bbrc.2001.5505 | ||
Betarbet R, Anderson LR, Gearing M, et al. Fas-associated factor 1 and Parkinson’s disease. Neurobiol Dis. 2008;31(3):309–315. doi:10.1016/j.nbd.2008.05.006 | ||
Sul J-W, Park M-Y, Shin J, et al. Accumulation of the parkin substrate, FAF1, plays a key role in the dopaminergic neurodegeneration. Hum Mol Genet. 2013;22(8):1558–1573. doi:10.1093/hmg/ddt006 | ||
Suh J, Yi KY, Lee Y-S, Kim E, Yum EK, Yoo S-E. Synthesis and biological evaluation of 3-substituted-benzofuran-2-carboxylic esters as a novel class of ischemic cell death inhibitors. Bioorg Med Chem Lett. 2010;20(22):6362–6365. doi:10.1016/j.bmcl.2010.09.102 | ||
Jung SH, Suh JH, Kim EH, Kim JT, Yoo S-E, Kang NS. The discovery of inhibitors of Fas-mediated cell death pathway using the combined computational method. Bioorg Med Chem Lett. 2013;23(18):5155–5164. doi:10.1016/j.bmcl.2013.07.018 | ||
Jeong J-W, Yu C, Lee J-H, et al. Subacute toxicity evaluation of KR-33493, FAF1 inhibitor for a new anti-Parkinson’s disease agent, after oral administration in rats and dogs. Regul Toxicol Pharmacol. 2016;81:387–396. doi:10.1016/j.yrtph.2016.09.022 | ||
US Food and Drug Administration. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf. Accessed May 31, 2016. | ||
Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211–218. doi:10.1111/j.2044-8341.1974.tb02285.x | ||
McNair D, Lorr M, Droppleman L. Profile of Mood States. San Diego: Educational and Industrial Testing Service; 1971. | ||
Hwang ST, Kim JH, Park GB, et al. Korean-Wechlser Adult Intelligence Scale-IV (KWAIS-IV). Daegu: Korea Psychology Co. Ltd; 2011. | ||
Tysnes O-B, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm. 2017;124(8):901–905. doi:10.1007/s00702-017-1686-y | ||
Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5(6):556–569. doi:10.3109/13550289909021285 | ||
Hammarlund-Udenaes M. The use of microdialysis in CNS drug delivery studies. Pharmacokinetic perspectives and results with analgesics and antiepileptics. Adv Drug Deliv Rev. 2000;45(2–3):283–294. doi:10.1016/S0169-409X(00)00109-5 | ||
Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi:10.1016/j.neuron.2007.12.027 | ||
Bartels AL, Willemsen AT, Kortekaas R, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm. 2008;115(7):1001–1009. doi:10.1007/s00702-008-0030-y | ||
Kortekaas R, Leenders KL, van Oostrom JCH, et al. Blood–brain barrier dysfunction in Parkinsonian midbrain in vivo. Ann Neurol. 2005;57(2):176–179. doi.org/10.1002/ana.20369 |
Supplementary materials
 | Table S1 Inclusion and exclusion criteria for KM-819 first-in-human study |
 | Table S2 Summary of change from baseline (day 1 predose) to day 7 for PD parameters |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.