")
Back to Journals » International Journal of Nanomedicine » Volume 11
A facile route to form self-carried redox-responsive vorinostat nanodrug for effective solid tumor therapy
Authors Han LQ, Wang TQ, Wu JL , Yin XL, Fang H, Zhang N
Received 4 August 2016
Accepted for publication 11 October 2016
Published 11 November 2016 Volume 2016:11 Pages 6003—6022
DOI https://doi.org/10.2147/IJN.S118727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Leiqiang Han, Tianqi Wang, Jingliang Wu, Xiaolan Yin, Hao Fang, Na Zhang
School of Pharmaceutical Science, Shandong University, Ji’nan, Shandong, People’s Republic of China
Abstract: Small molecule-based nanodrugs with nanoparticles (NPs) that are mainly composed of small molecules, have been considered as a promising candidate for a next-generation nanodrug, owing to their unique properties. Vorinostat (SAHA) is a canonical US Food and Drug Administration-approved histone deacetylase (HDAC) inhibitor for the treatment of cutaneous T-cell lymphoma. However, the lack of efficacy against solid tumors hinders its progress in clinical use. Herein, a novel nanodrug of SAHA was developed based on disulfide-linked prodrug SAHA-S-S-VE. SAHA-S-S-VE could self-assemble into 148 nm NPs by disulfide-induced mechanisms, which were validated by molecular dynamics simulations. Under reduced conditions, the redox-responsive behavior of SAHA-S-S-VE was investigated, and the HDAC inhibition results verified the efficient release of free SAHA. With a biocompatible d-a-tocopheryl polyethylene glycol succinate (TPGS) functionalization, the SAHA-S-S-VE/TPGS NPs exhibited low critical aggregation concentration of 4.5 µM and outstanding stability in vitro with drug-loading capacity of 24%. In vitro biological assessment indicated that SAHA-S-S-VE/TPGS NPs had significant anticancer activity against HepG2. Further in vivo evaluation demonstrated that the resulting NPs could be accumulated in the tumor region and inhibit the tumor growth effectively. This approach, which turned SAHA into a self-assembled redox-responsive nanodrug, provided a new channel for the use of HDAC inhibitor in solid tumor therapy.
Keywords: SAHA, HDAC, small molecule, nanoparticles, self-assemble, disulfide bond
Introduction
Small molecule-based nanodrug (SMND) (Figure 1A) is a kind of self-carried nanodrug, whose nanoparticles (NPs) are mainly composed of small molecules with or without a little material as stabilizer. SMND includes amphiphilic self-assembly nanodrug,1–3 nanocrystallized or template-assisted nanodrug,4–7 disulfide-induced self-assembled nanodrug,8–10 and so on. It has attracted great interest as a desirable strategy, because of its unique properties in increased chemical stability and solubility, high drug loading, economical fabrication, relative “green” nanoformulation. Prodrug strategies have been developed which enable selective delivery of the anticancer agents to the tumor tissue, based on elevated enzymes in tumor tissues, hypoxic environment inside the core of solid tumors, and tumor-specific antigens expressed on the surface of tumor cells. Prodrug approach provides a remarkable tool to improve the therapeutic efficacy and to reduce the toxic side effects of the anticancer agents.11–13 In this work, prodrug strategy combined with the SMND system was reported for solid tumor therapy.
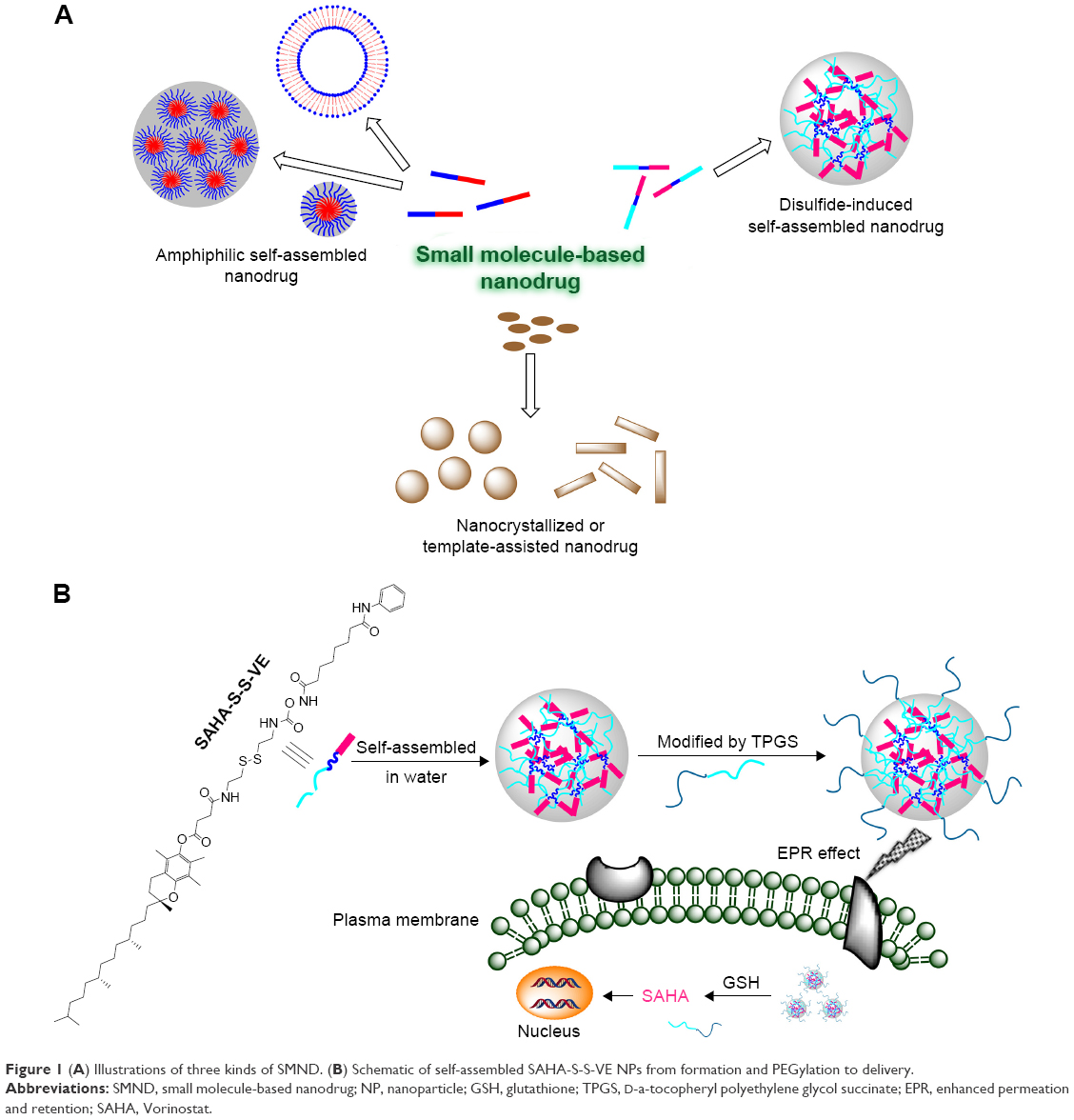 | Figure 1 (A) Illustrations of three kinds of SMND. (B) Schematic of self-assembled SAHA-S-S-VE NPs from formation and PEGylation to delivery. |
Histone deacetylase (HDAC) is one of the epigenetic protein targets overexpressed in cancer cell lines. The HDAC inhibitory effect leads to reacetylation of histones, which, in turn, stimulates tumor suppressor gene expression, inducing differentiation, growth arrest, or apoptosis. It has been proved that chelation of the Zn2+ is crucial for HDAC inhibition.14 The hydroxamic acid has been the preferred Zn2+ binding group due to its strong Zn2+ chelation.15 Vorinostat (SAHA) was the first US Food and Drug Administration-approved HDAC inhibitor for the treatment of cutaneous T-cell lymphoma. However, similar to other hydroxamic acid-based HDAC inhibitors, SAHA shows serious limitations, especially that ineffective in solid tumors, which is hindering its progress in clinical use.14,16,17 This problem emanated from the poor properties of hydroxamic acid, which is an essential pharmacophore of HDAC inhibitor (Figure S1). Due to its strong Zn2+ chelation, it is liable to suffer from off-target effects, as there are many metalloenzymes in the body.18 Metabolic activities in plasma lead to chemical instability and rapid elimination of hydroxamic acid.15,16,18,19 Besides, hydroxamic acid is known to undergo deprotonation due to their acidic nature,20 which could prevent its diffusion through cell membrane.21 All these reasons attribute to its limited concentration of HDAC inhibitor in solid tumor.
In addressing these hurdles, SAHA has been delivered by various means, such as carbamate prodrug,22 clickable pH-responsive prodrug,23 thiol-sensitive prodrug,17 SAHA-loaded solid lipid NPs,24 hyaluronic acid-coated solid lipid NPs,25 SAHA-loaded poly (DL-lactide-co-glycolide) NPs,26 protein-SAHA nanodrug,27 SAHA-loaded poly(ethylene glycol)-b-poly(DL-lactic acid) micelle,28 and so on. Nevertheless, few reports have evaluated their efficacy toward solid tumor in vivo. Recently, SAHA-polymer conjugate NPs constructed by Blanquart et al showed good effect toward malignant mesothelioma by passive tumor targeting.14 Inspired by these, self-assembled SAHA prodrug can not only take the advantage of nanoscale drug delivery system but also increase the drug loading capacity dramatically.
In the present study, a redox-responsive prodrug SAHA-S-S-VE (Scheme 1) with disulfide bond was synthesized through a facile and efficient condensation reaction. Meanwhile, as a control system, a redox unresponsive prodrug without disulfide bond was employed in SAHA-O-VE (Scheme S1). The rationale for this design are as follows. It has been widely investigated that tumor tissue has a more reductive environment with its glutathione (GSH) concentration several times higher than that in normal tissues. Cancer cells have much higher intracellular GSH concentration than normal cells.29–31 All these theoretical factors can trigger drug release at tumor region. In addition, the carbamate prodrug can protect the hydroxamic acid from off-target and improve its cellular permeability. The icing on the cake is that SAHA-S-S-VE formed NPs with the aid of disulfide bond by a simple nanoprecipitated method, which can deliver drug into tumor tissues possibly by passive accumulation via enhanced permeation and retention (EPR) effect. Here, the assemble mechanism of SAHA-S-S-VE NPs at the molecular level has been validated by molecular dynamics simulations. Under reduced conditions, the redox-responsive behavior of SAHA-S-S-VE was investigated and the efficient release of free drug was demonstrated by HDAC inhibition assay. To achieve better biocompatibility in vivo, PEGylated NPs (Figure 1B) were prepared with D-a-tocopheryl polyethylene glycol succinate (TPGS) functionalization.
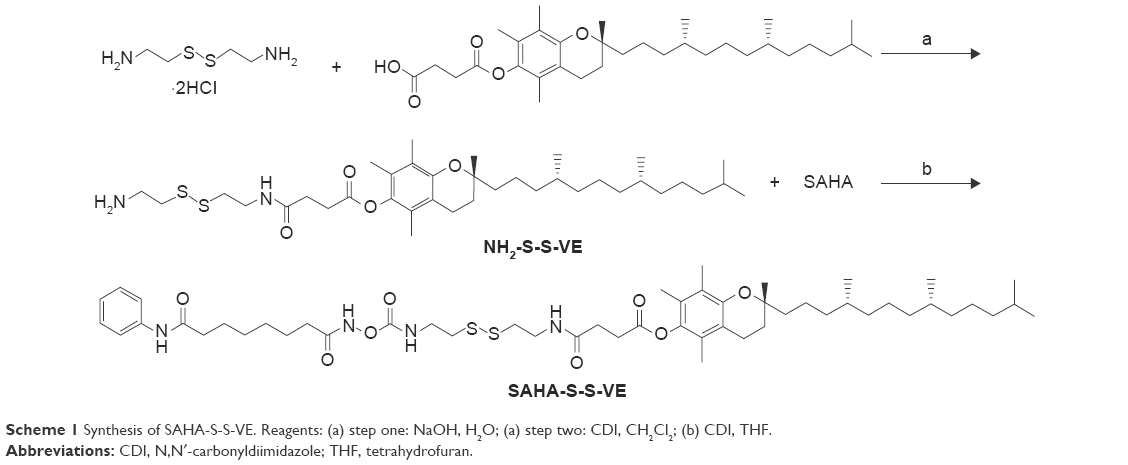 |
Scheme 1 Synthesis of SAHA-S-S-VE. Reagents: (a) step one: NaOH, H2O; (a) step two: CDI, CH2Cl2; (b) CDI, THF. |
After extensive biophysical and biological characterizations, including morphology, particle size, zeta potential, critical aggregation concentration (CAC), plasma stability, antiproliferative activities, tumor accumulation, and anti-tumor efficacy, the developed SAHA-S-S-VE/TPGS NPs bestowed several striking characteristics: 1) simple syntheses and economical process for fabrication; 2) turned SAHA into self-assembled redox-responsive NPs with high drug loading capacity, low CAC and outstanding stability in vitro; and 3) significantly high tumor accumulation and extremely good antitumor activity in solid tumor mouse models. To the best of our knowledge, this delivering strategy has never been applied to HDAC inhibitor and appeared of great interest to circumvent the clinical limitations of this class of compounds. Such SMND platform, combining the prodrug strategy with nanotechnology, provided a new channel for the use of SAHA and similar HDAC inhibitors in solid tumor therapy.
Experimental procedures
Materials
All materials and reagents used in this work were analytical reagents without further purification. SAHA was purchased from Melonepharma (Dalian, People’s Republic of China). Electrospray ionization mass spectrometry was determined on an API 4000 spectrometer. Nuclear Magnetic Resonance (NMR) spectra were obtained on a Bruker DRX spectrometer (400 MHz). The chemical shifts are defined as δ values (parts per million) relative to TMS internal standard. Significant 1H NMR data are reported in the following order: multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet) number of protons. High-resolution mass spectrometer (HRMS) spectrums were conducted on an Agilent 6510 Quadrupole Time-of-Flight LC/MS deliver.
Preparation of SAHA-S-S-VE NPs and SAHA-S-S-VE/TPGS NPs
SAHA-S-S-VE NPs were prepared by nano-precipitation according to the reported method.2,8 Prodrugs were dissolved in dimethyl sulphoxide (DMSO) (50 mg/mL). Then 100 μL was added dropwise to 900 μL ddH2O, under mechanical stirring (800 rpm) at room temperature, to obtain a final concentration of 5 mg/mL prodrug. PEGylated NPs were produced after 50 μL of various concentrations of nonionic surfactant solution was injected to the above formulation. Subsequently, the solution was dialyzed against deionized water for 24 h to remove the organic solvent (molecular weight cutoff =1,000 g/mol). The final prodrug concentration was >5 mg/mL. Then, 5% glucose was added, and it was stored at 4°C for further application.
Characterization of NPs
The morphology of NPs was examined by transmission electron microscopy (TEM) (JEM-1200EX, Japan). Samples were prepared by placing a drop of NP suspension onto a copper grid and air-dried, following negative staining with one drop of 3% aqueous solution of sodium phosphotungstate for contrast enhancement. The air-dried samples were then examined by TEM. The mean particle size and zeta potential were analyzed by dynamic light scattering using Zetasizer (Malvern Instruments, Malvern, UK). The NPs were centrifuged at 4,000 rpm for 10 min by using ultra centrifugal filters. An aliquot of the supernatant was dissolved in equal volume of methanol, and the SAHA-S-S-VE content was quantified by HPLC. The amount of SAHA in prodrug SAHA-S-S-VE is fixed. The drug loading capacity and encapsulation efficiency were determined using the following equation:
|
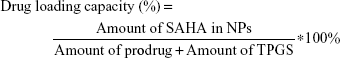
|
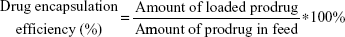
Molecular dynamics simulations of the self-assembly of SAHA-S-S-VE NPs
Herein the SAHA-S-S-VE molecule was optimized by Gaussian09 package32 at the level of HF/6-31g*. Then the partial atomic charges of these molecules were calculated by the restrained electrostatic potential33 charge from the calculation with Gaussian09 package at HF/6-31g* level. The CHARMM force field was applied for this molecule. After setting the parameter, 10 SAHA-S-S-VE molecules were randomly put into a 7×7×7 box, and then the box was filled up with the TIP4P waters. Before the molecular dynamics simulations, 100,000 step steepest descent energy minimization were carried out to optimize the system. Then the system was heated up from 0 K to 300 K gradually under the NVT ensemble for 100 ps. Finally, molecular dynamics simulations were performed under NPT ensemble. The periodic boundary condition was kept and a cutoff of 7 Å was set for both van der Waals and electrostatic interactions. All bonds involving hydrogen atoms were restricted by the SHAKE algorithm with a cutoff value of 10−5. All the simulations were done by Gromacs 4.5.34 The initial state and final state were as shown in Figures S2 and S3.
GSH-responsive rate
The concentration of remaining SAHA-S-S-VE was quantified by reverse-phase HPLC (SPD-10Avp Shimadzu pump, LC-10Avp Shimadzu UV–vis detector, Shimadzu, Kyoto, Japan) analysis on a C18 column (4.6×250 mm reverse phase stainless steel column packed with 5 μm particles Inertdil® ODS-3, GL Sciences Inc., Tokyo, Japan) with methanol as eluting solution at a flow rate of 1.0 mL/min. GSH was diluted with DMSO/phosphate-buffered saline (PBS) solution. Then different equivalent of SAHA-S-S-VE in DMSO was added and the final clear solution (DMSO/PBS solution, 60/40, v/v, pH =7.4) was incubated at 37°C. At specified time point, 0.2 mL of the solution was collected and subject to HPLC analysis directly or after 1:1 dilution with methanol to quantify the drug content (λabs =288 nm, Figures S4 and S5). The samples were filtered through a 0.22 μm filter before being detected by HPLC. The samples were prepared in triplicates. The prodrug relative degradation was calculated according to the following formula:
|
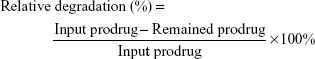
In vitro HDACs assay
Assays were performed according to the kit instruction. HDACs came from HeLa cell nucleus extracts, mainly including HDAC1 and HDAC2, and the substrate was an acetylated histone peptide. The required stock solution of thiol was prepared and used immediately prior to assay from the corresponding disulfide analogs using 1.5 equiv tris(2-carboxyethyl)phosphine hydrochloride (TCEP) as the reducing agent, in a 9:1 DMSO/water solution according to the general procedure described by Baud et al.35 Stock solutions of test compounds and the control drug SAHA were diluted to various concentrations. On the 96-well plate, HDACs (15 μL/well) were incubated at 37°C with 10 μL of various concentrations of samples and 25 μL of substrate. After reacting for 30 min, the mixture (50 μL/well) of Color de Lys Developer and TSA were added. Then, after 30 min, the ultraviolet absorption of the wells was measured on a microtiter-plate reader at 405 nm. The inhibition rates were calculated from the ultraviolet absorption readings of inhibited wells related to those of control wells. Finally, the half maximal inhibitory concentration (IC50) values were determined using a regression analysis of the concentration and inhibition data.
CAC measurement
To determine the CAC of SAHA-S-S-VE/TPGS NPs, pyrene method was used.1 Different concentrations of SAHA-S-S-VE/TPGS NPs were equilibrated with a fixed concentration of pyrene, 6.0×10−7 M. The emission spectra were scanned from 350 to 500 nm at a fixed excitation wavelength of 334 nm using HITACHI F-7000 Fluorescence spectrometer. I1 and I3 were the emission intensities of the first and third bands in the fluorescence spectrum of pyrene, respectively. The I3/I1 values of all the solution were calculated and plotted against the logarithmic concentration of SAHA-S-S-VE/TPGS NPs. The CAC was obtained from the threshold concentration of self-assembled NPs.
Stability of SAHA-S-S-VE/TPGS NPs in human plasma
The stability study of SAHA-S-S-VE/TPGS NPs in human plasma was performed according to the reported procedure.2 Human plasma was provided by Qilu hospital (Jinan, People’s Republic of China). This study was approved by the Research Ethics Committee of Qilu Hospital, Shandong University. In brief, SAHA-S-S-VE/TPGS NPs were incubated in 50% human plasma (final concentration 1 mg/mL, SAHA-S-S-VE equivalent) in vitro. Samples (1 mL) were withdrawn at 0, 4, 8, 12, and 24 h and measured by dynamic light scattering.
In vitro cytotoxicity
In vitro cytotoxicity assays were performed according to the previously reported method.36 MDA-MB-231, PC-3, HepG2, and A549 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum at 37°C in 5% CO2 humidified incubator. Cell proliferation was determined by the MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) assay. Briefly, cells were plated in a 96-well plate at 5,000 cells per well, cultured for 8 h in complete growth medium, then treated by different SAHA formulations of varied concentrations for 48 h. Then, 0.5% MTT solution was added to each well. After further incubation for 4 h, formazan formed from MTT was extracted by adding 150 μL of DMSO and mixing for 10 min. Optical density was read with a microtiter-plate reader at 570 nm.
Animals and tumor models
The in vivo experiments were performed in H22-bearing Kunming mice. H22 (murine hepatocellular carcinoma cells) were extracted from the abdominal dropsy of mice bearing cancer cells and diluted with normal saline to achieve a cell concentration of 2×106/mL, and 0.1 mL cell suspension was subcutaneously injected into right armpit of each mouse, resulting in the formation of tumor per mouse. This study was approved by the Institutional Animal Care and Use Committee of Shandong University, and all animal handling procedures were in accordance with the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China).
Tumor accumulation study
The near-infrared fluorescence (NIRF) dye DiR was used to investigate the in vivo targeting effect of SAHA-S-S-VE/TPGS NPs in H22-bearing Kunming mice. When the tumor reached to 200–300 mm3, mice were randomly grouped into DiR solution group and DiR-loaded NPs (SAHA-S-S-VE/TPGS NPs) group (5 mice per group). DiR-loaded NPs (SAHA-S-S-VE/TPGS NPs) were prepared following the similar reported method.1 Each mouse was injected with different DiR formulation at 0.5 mg/kg intraperitoneally (i.p.). Images were acquired at 24 h post-injection (p.i.). After the 24 h imaging, mice were sacrificed, and tumors were excised, followed by washing the surface with 0.9% NaCl for ex vivo fluorescence imaging. In vivo and ex vivo fluorescence imaging were performed with a NIRF imaging system (IVIS Kinetic; Caliper Life Sciences, Waltham, MA, USA), and the fluorescence intensity of the tumor defined as radiance (p/s/cm2/sr) was analyzed and quantified with Living Image software 3.1 (Caliper Life Sciences).
In vivo therapeutic efficacy
The in vivo antitumor efficacy was evaluated in H22-bearing Kunming mice. When the maximum diameter of tumor reached 5 mm, mice were randomly grouped into Vehicle group, SAHA group, and SAHA-S-S-VE/TPGS NPs group (10 mice per group). Vehicle group (0.2 mL/day, 40% DMSO), SAHA group (50 mg/kg, first SAHA was dissolved in DMSO, then added with normal saline, and the final concentration of DMSO was 40%), and SAHA-S-S-VE/TPGS NPs group (208 mg/kg, an equivalent amount of SAHA), respectively, received i.p. injection on days 1, 3, 5, 7, 9, 11, and 13. Tumor volume (V) was monitored on days 1, 3, 5, 7, 9, 11, 13, and 17 and calculated by the following formula: V = W2 (width) × L (length)/2. Each animal was weighed at the time of tumor size measurement.
Statistical analysis
Statistical analysis was carried out with GraphPad Prism. Studies involving two groups were subjected to unpaired Student’s t-test. The significance level was set at a probability of P<0.05.
Results and discussion
Synthesis and characterization of SAHA-S-S-VE
The synthetic route of prodrug SAHA-S-S-VE was depicted in Scheme 1 (details in Method S1). The key intermediate compound NH2-S-S-VE was synthesized from the reaction of D-α-tocopherol succinate and cystamine. Then, SAHA was activated with 1,1′-carbonyldiimidazole in THF. The target prodrug SAHA-S-S-VE was obtained from the reaction of 1,4,2-dioxazol-5-one of SAHA and NH2-S-S-VE. Meanwhile, as a control system, an uncleavable linker was employed in SAHA-O-VE (Scheme S1 and Method S2).
Compared with the 1H-NMR spectrum (Figure 2) of SAHA and SAHA-S-S-VE, the peak at 10.33 ppm related to the hydroxyl proton of SAHA disappeared completely. 1H-NMR spectrum of SAHA-S-S-VE showed signals at 7.94 (1), 9.84 (2) ppm attributable to the two amide protons of SAHA, and five proton signals at 7.49 (a), 7.26 (b), 7.05 (c) ppm belonging to the phenyl ring of SAHA. The amide proton signals of cystamine were identified at 6.23 (3), 6.70 (4) ppm, and the cystamine methylene (1′+2′) were confirmed at 3.46–3.53 ppm (Figure 2C). The signals at 0.83–2.95 ppm, especially the methyl peaks (3′~10′), were part of VE (Figure 2C). Moreover, the experimental value for [M+H]+ from HRMS is very similar to the calculated value (Figure 2B). These results suggested the successful synthesis of SAHA-S-S-VE.
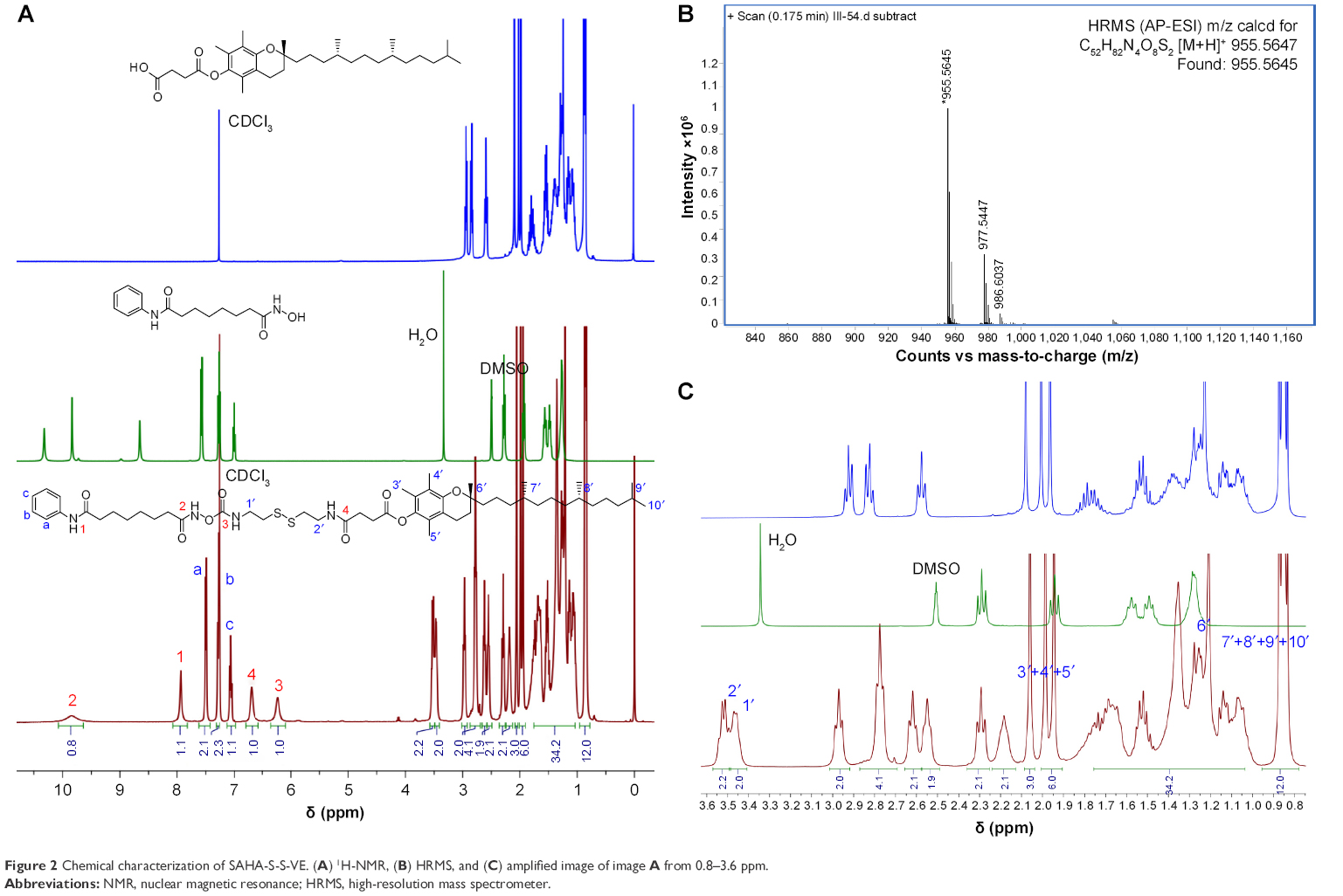 |
Figure 2 Chemical characterization of SAHA-S-S-VE. (A) 1H-NMR, (B) HRMS, and (C) amplified image of image A from 0.8–3.6 ppm. |
Formulating SAHA-S-S-VE NPs
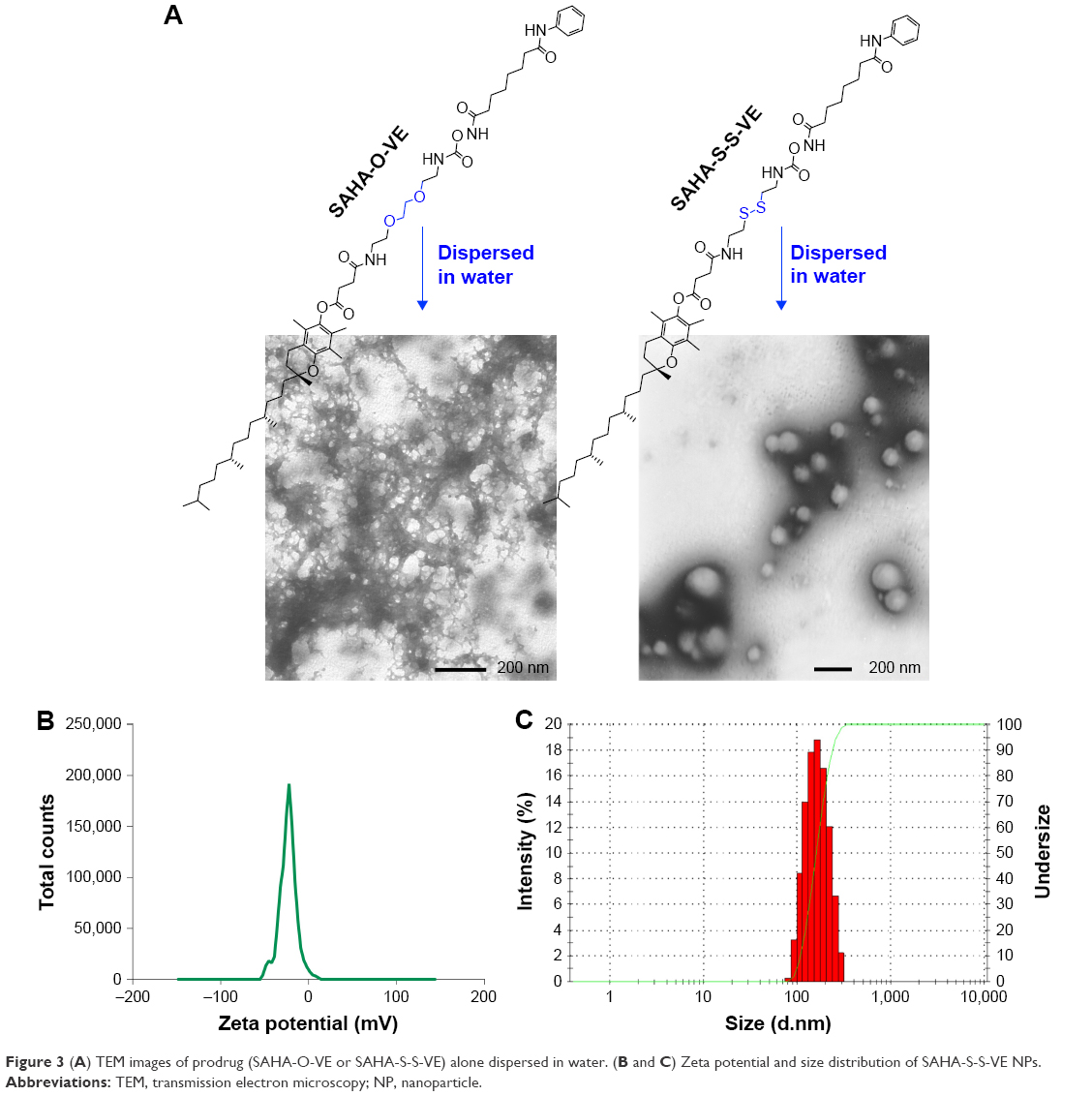
When a disulfide-bond was inserted between SAHA and the hydrophobic D-α-tocopherol succinate material, prodrug SAHA-S-S-VE assembled into spherical NPs (Figure 3) alone in aqueous solution, with a particle size 148.2±6.3 nm, polydispersity index (PDI) 0.111±0.038, and zeta potential −25.7±4.1 mV. However, when SAHA was conjugated with D-α-tocopherol succinate with a hydrophilic dioxaoctane linker, prodrug, SAHA-O-VE failed to form NPs during self-assembly (Figure 3). Molecular dynamics simulations were performed to help understand how the molecules would interact with each other in aqueous solution. As shown in Figure 4A, initially, when the SAHA-S-S-VE molecules were in a dispersed state, their interactions were small. After 2 ns, the assembly phenomena emerged, and they spontaneously clustered. Within 20 ns, the clusters gathered closer and closer and ultimately formed a stable integration. Finally, only movement of the whole NPs could be observed. Considering the driving forces, a tetrameric of SAHA-S-S-VE was picked out from the final NPs for analysis (Figure 4B). It was likely to be held together by three factors. First, a number of amide and hydroxamic acid groups existed in the tetrameric, which could form strong hydrogen bonds. Second, the van der Waals force between the aromatic rings and hydrophobic chain including π-π stacking and CH-π interactions played important roles in the structure stabilization. The increase in the hydrophobic tail than its comparator should promote the self-assembling process.37 Third and most importantly, as is known to all, disulfide bond plays an important role in guiding the precise folding of a protein into its native conformation and stabilizing its tertiary and quaternary structures.38 As shown in Figure 4B, the disulfide bond showed a preference for dihedral angles approaching 90°, and SAHA-S-S-VE molecules could fold at the disulfide bond position, which synergistically favored the former two aspects to generate a stable conformation.
 | Figure 3 (A) TEM images of prodrug (SAHA-O-VE or SAHA-S-S-VE) alone dispersed in water. (B and C) Zeta potential and size distribution of SAHA-S-S-VE NPs. |
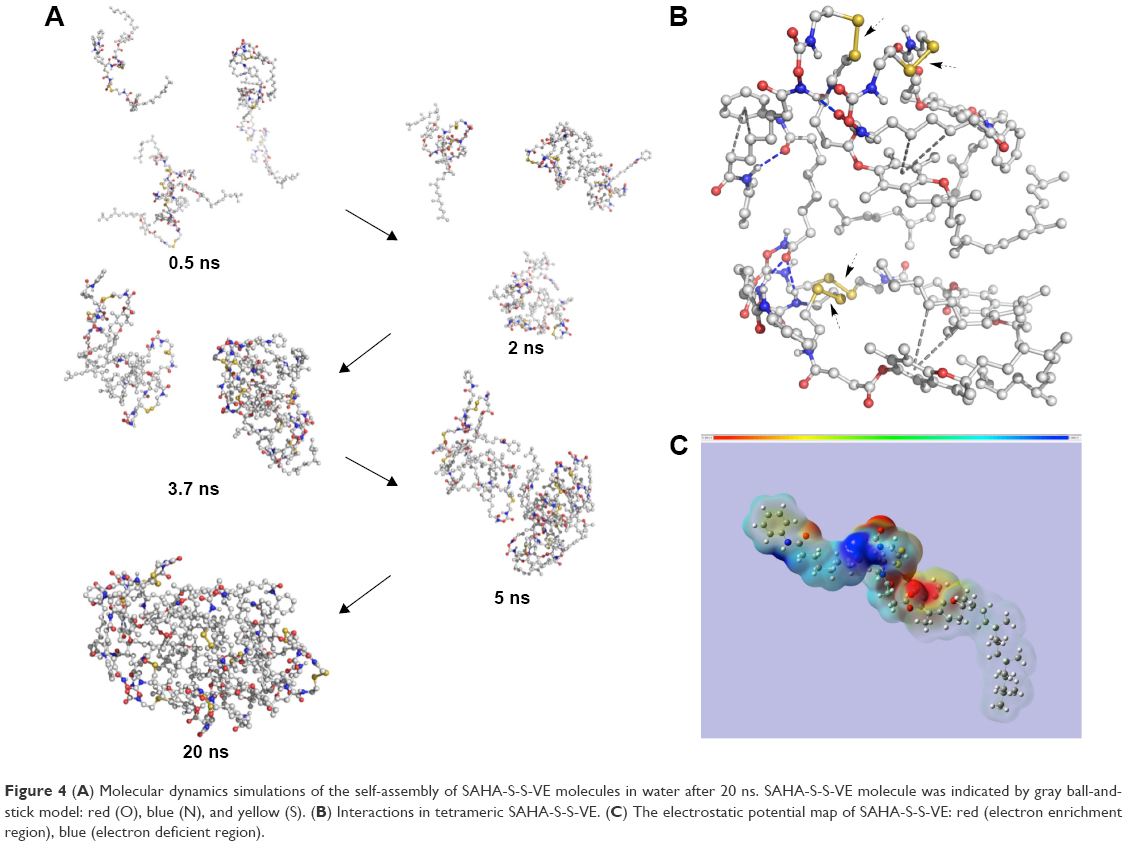 |
Figure 4 (A) Molecular dynamics simulations of the self-assembly of SAHA-S-S-VE molecules in water after 20 ns. SAHA-S-S-VE molecule was indicated by gray ball-and-stick model: red (O), blue (N), and yellow (S). (B) Interactions in tetrameric SAHA-S-S-VE. (C) The electrostatic potential map of SAHA-S-S-VE: red (electron enrichment region), blue (electron deficient region). |
Moreover, the charge density of SAHA-S-S-VE was calculated, as shown in Figure 4C. The negative charge mainly came from oxygen, which was concentrated near the disulfide bond. VE and the two sulfur atoms contributed little to the negative charge. As mentioned earlier, the SAHA-S-S-VE molecule could fold or cross to establish a favorable conformation at the disulfide bond position and simultaneously exposing the high density of negative charge on the NP surface and contributing to stabilize the particle-solution interface. This explained why SAHA-S-S-VE NPs showed a negative zeta potential −25.7±4.1 mV, an important property for the colloidal stability.
GSH-responsive rate
Disulfide bonds are fairly stable in normal biological conditions but are rapidly cleaved through thiol-disulfide exchange reactions facilitated by intracellular reducing molecules, especially GSH.8,39,40 It has been demonstrated that GSH concentration in tumor cells (2–10 mM) is much higher than that in blood plasma (1–2 μM).8,41,42 The GSH-responsive rate of SAHA-S-S-VE at molecular level was expressed by the relative degradation of SAHA-S-S-VE. As shown in Figure 5A, with 0 mM, 2.5 mM (10 eq) of GSH, the prodrug degradation was slow and only 20% of SAHA-S-S-VE was degraded in 24 h, which meant that the prodrug could alleviate the side effects toward normal tissues. In the presence of 2.5 mM (68 eq) GSH, the degradation of SAHA-S-S-VE was enhanced, and ~80% of SAHA-S-S-VE was degraded in 24 h. And in 10 mM (136 eq) GSH, the degradation of SAHA-S-S-VE showed a significant increase, and nearly 100% of the prodrug was degraded in only 6 h. This indicated an advantage of SAHA-S-S-VE for controlled release of parent drug.
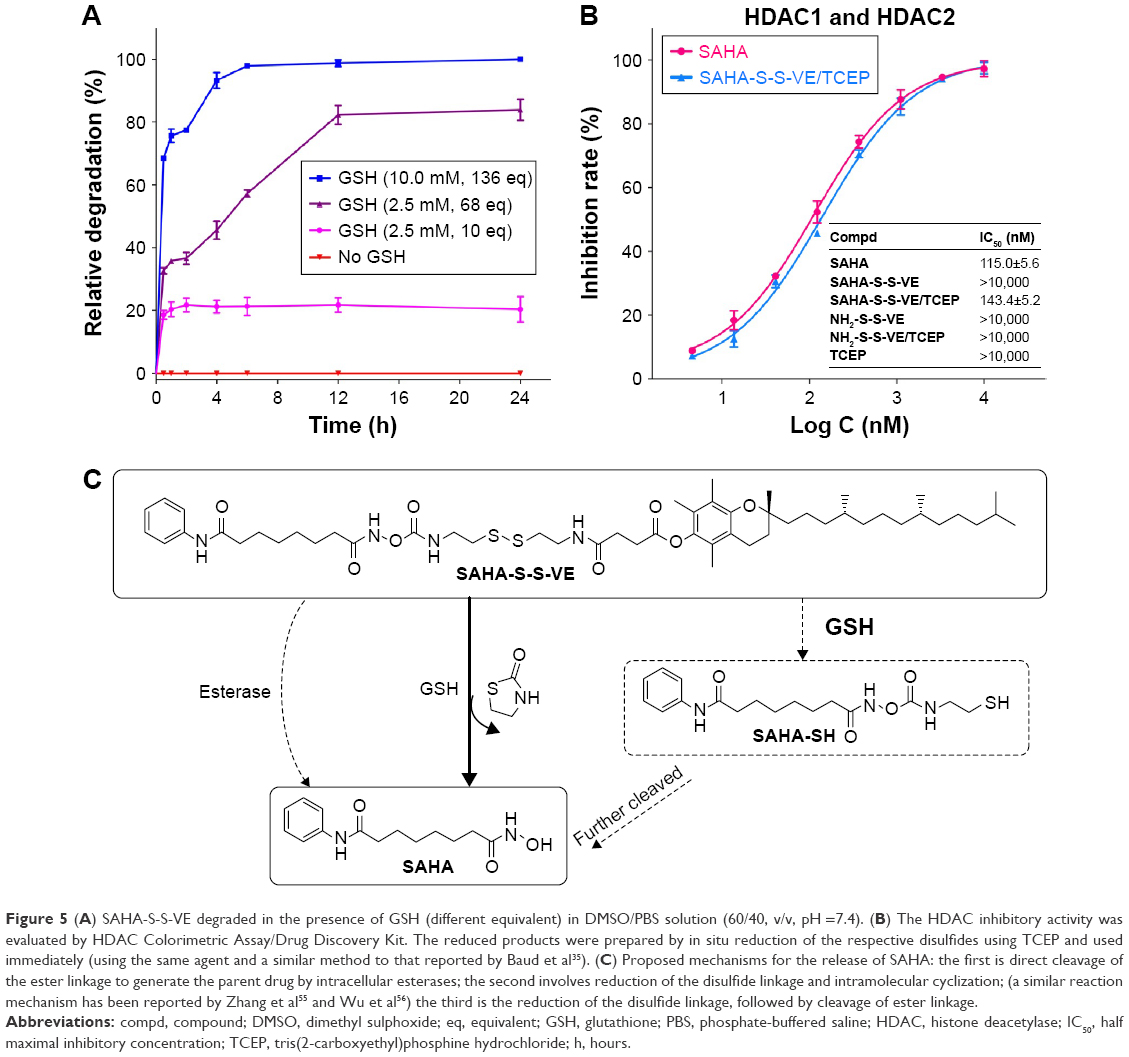 | Figure 5 (A) SAHA-S-S-VE degraded in the presence of GSH (different equivalent) in DMSO/PBS solution (60/40, v/v, pH =7.4). (B) The HDAC inhibitory activity was evaluated by HDAC Colorimetric Assay/Drug Discovery Kit. The reduced products were prepared by in situ reduction of the respective disulfides using TCEP and used immediately (using the same agent and a similar method to that reported by Baud et al35). (C) Proposed mechanisms for the release of SAHA: the first is direct cleavage of the ester linkage to generate the parent drug by intracellular esterases; the second involves reduction of the disulfide linkage and intramolecular cyclization; (a similar reaction mechanism has been reported by Zhang et al55 and Wu et al56) the third is the reduction of the disulfide linkage, followed by cleavage of ester linkage. |
In vitro HDAC assay
HDAC inhibitory activities of SAHA-S-S-VE, NH2-S-S-VE and their reduced products SAHA-S-S-VE/TCEP, NH2-S-S-VE/TCEP (TCEP which is known to reduce the S-S bond and have no influence on this assay condition35) were evaluated by the Color de Lys assay. The results were tabulated as IC50 values in Figure 5B. According to the data, the reduced products SAHA-S-S-VE/TCEP exhibited similar HDAC inhibitory activity compared with SAHA. In contrast, both the material parts (NH2-S-S-VE, NH2-S-S-VE/TCEP) and the prodrug (SAHA-S-S-VE) alone showed poor inhibition against HDACs. Based on the above redox-responsive results, it suggested that the prodrug system could restore the HDACs inhibitory activity efficiently through the release of SAHA, after taken up by cells through the second way (Figure 5C).
In order to enhance the biocompatibility and solubility of the as-prepared SAHA-S-S-VE NPs for facilitating further in vivo applications, surface modification is essential. Through screening, TPGS was chosen as a suitable stabilizer43–45 to the formulation, compared with F68 (Table 1). When F68 was added to the formulation, no NPs were formed because its large structure might influence the formation of the NPs, whereas TPGS could anchor to the surface of the NPs (Figures 1B and 6). The optimal mass ratio of TPGS/SAHA-S-S-VE was 15:100, with high SAHA loading capacity (24%) and encapsulation efficiency (99%). The mean particle size and PDI (Table 1) indicated that the SAHA-S-S-VE/TPGS NPs were small enough and with a narrow size distribution, which were favorable for passive tumor targeting. The negative zeta potential also meets the necessary surface characteristic for injectable nanodrug.46
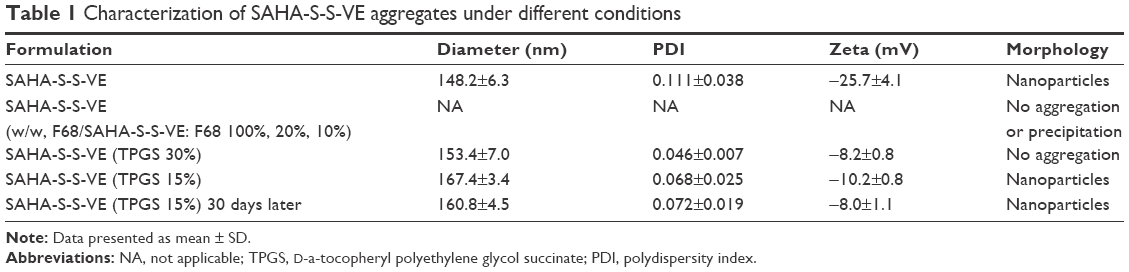 | Table 1 Characterization of SAHA-S-S-VE aggregates under different conditions |
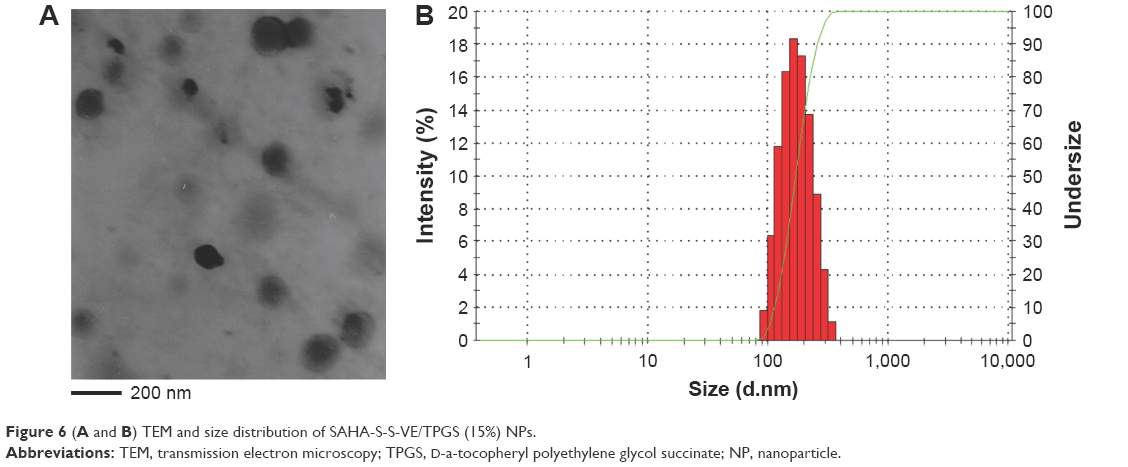 | Figure 6 (A andB) TEM and size distribution of SAHA-S-S-VE/TPGS (15%) NPs. |
Stability of SAHA-S-S-VE/TPGS NPs
The emission intensity ratio (I3/I1) of pyrene is very sensitive to the surrounding environmental polarity. The higher is the polarity of the medium; the lower is the intensity ratio.1 Based on that, the CAC of SAHA-S-S-VE/TPGS NPs was determined from the crossover point by plotting the I3/I1 versus logarithm concentration of the NPs. The CAC was at the low concentration range, 4.3 μg/mL (4.5 μM) (Figure 7A), which was similar to the reported nanodrug delivery systems.1 This low CAC value was important for the in vivo application. Next, to investigate whether SAHA-S-S-VE/TPGS NPs could survive even in the presence of human plasma and thereby benefit from the EPR effect; dynamic light scattering measurements were used to study the stability of the SAHA-S-S-VE/TPGS NPs. As shown in Figure 7B and C, the size of SAHA-S-S-VE/TPGS NPs remained unchanged over 24 h and the PDI showed a bit of increase after 12 h. These stable characteristics indicated that the SAHA-S-S-VE/TPGS NPs might be delivered to solid tumor through EPR effect. In addition, the NPs exhibited excellent storage stability, with no significant change in the size over 1 month at 4°C (Table 1).
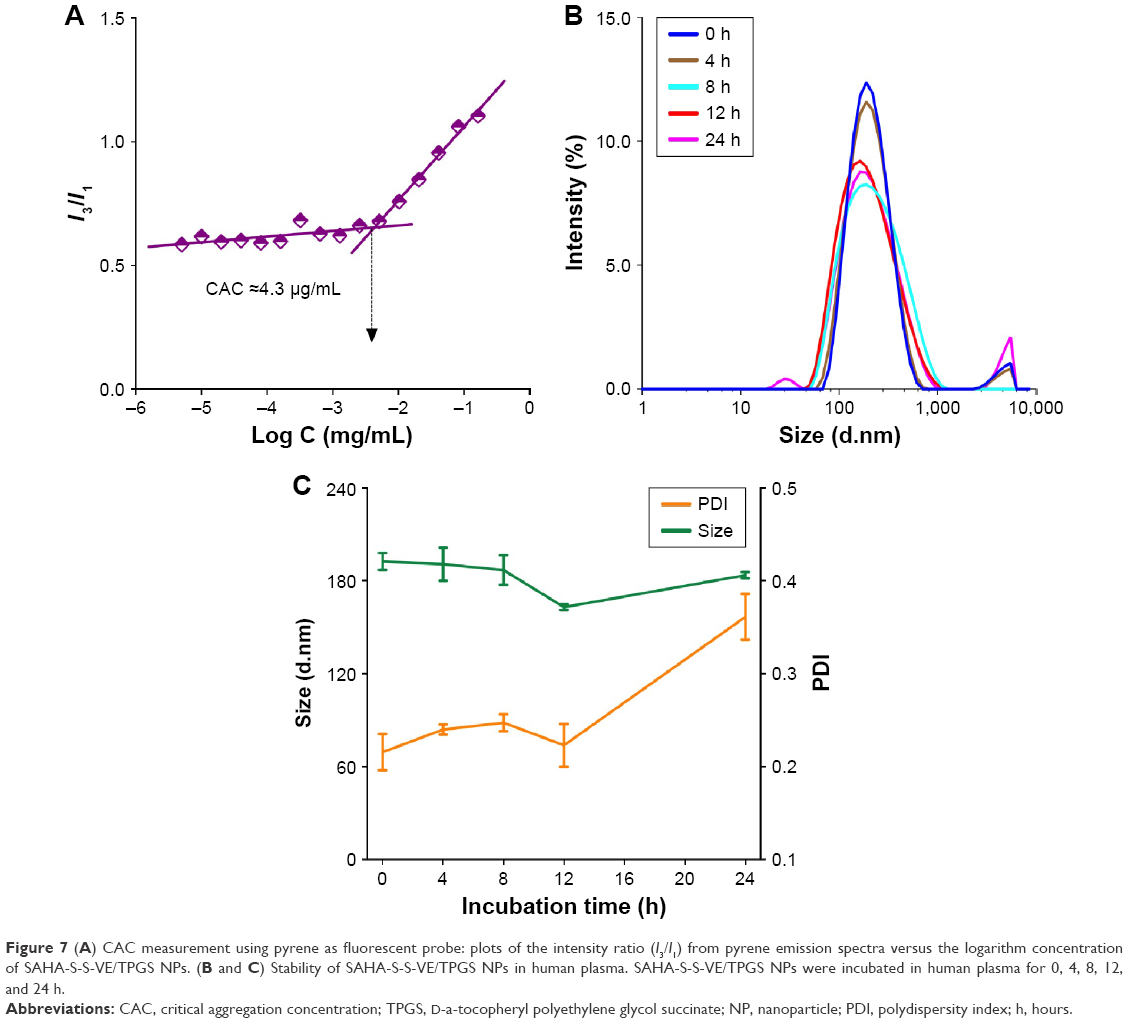 | Figure 7 (A) CAC measurement using pyrene as fluorescent probe: plots of the intensity ratio (I3/I1) from pyrene emission spectra versus the logarithm concentration of SAHA-S-S-VE/TPGS NPs. (B and C) Stability of SAHA-S-S-VE/TPGS NPs in human plasma. SAHA-S-S-VE/TPGS NPs were incubated in human plasma for 0, 4, 8, 12, and 24 h. |
In vitro cytotoxicity
The proliferation inhibition of SAHA-S-S-VE/TPGS NPs was evaluated against HDAC inhibitor sensitive cell lines: MDA-MB-231 (human breast cancer cell),47 PC-3 (human prostatic cancer),48 HepG2 (human hepatocellular cancer),49 and A549 (human lung cancer),50 compared with free SAHA, SAHA-S-S-VE, SAHA-O-VE, and the conjugate material. The IC50 values were summarized in Table 2. Both SAHA-S-S-VE and SAHA-S-S-VE/TPGS NPs showed enhanced antiproliferative activities (lower IC50 values) compared with free SAHA in all the tested cell lines, whereas the NH2-S-S-VE showed little toxicity in the given concentration range after 48 h. Among these four cell lines, SAHA-S-S-VE/TPGS NPs revealed the most obvious antiproliferative activity (P<0.01, compared to free SAHA) in HepG2 cell line. The enhanced cytotoxic might be due to one or two of the following mechanisms: 1) the lipophilic nature of SAHA-S-S-VE (higher log P; Table 2) might facilitate intracellular uptake11,14,51 and 2) when the concentration of SAHA-S-S-VE/TPGS NPs was higher than the CAC, part of the self-assembled SAHA-S-S-VE/TPGS NPs might keep intact for the low GSH concentration in the cell culture medium,38 which resulted in the greater cellular uptake through the endocytic pathway and higher cytotoxic efficiency.1 Besides, the GSH-insensitive SAHA-O-VE appeared to have a significantly lower proliferation inhibitory efficacy than SAHA-S-S-VE, which might be resulted from the slow release of SAHA.
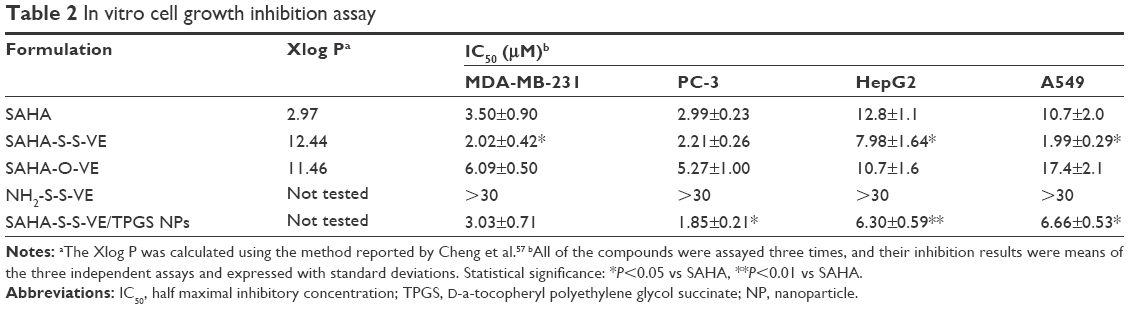 | Table 2 In vitro cell growth inhibition assay |
Tumor accumulation study
Particles in the 60–400 nm size range usually extravasate and accumulate in a wide range of tumor types through a passive tumor targeting mechanism referred to as the EPR effect.52 To verify the applicability of SAHA-S-S-VE/TPGS NPs for passive tumor targeting, DiR solution and DiR-loaded NPs were administered into H22 tumor-bearing mice. Due to i.p. injection, both the groups showed noticeable fluorescence signal in abdominal cavity. As shown in Figure 8A, after 24 h, the signals of free DiR were extremely weaker than DiR-loaded NPs, indicating that free DiR has been quickly cleared from the bloodstream. On the other hand, the DiR fluorescence signal of DiR-loaded NPs was clearly detected at the tumor site. However, little signal was displayed at the tumor site for free DiR. This pronounced intratumoral distribution could be attributed to the nanosized particles by taking the advantage of EPR effect. Moreover, the stability of the DiR-loaded NPs also contributed to reach the tumor tissues. And the PEG corona in NPs further prevented DiR from opsonization. Ex vivo imaging was performed and analyzed to acquire the fluorescent signal of internal tumors. As shown in Figure 8B, compared to DiR, DiR-loaded NPs showed higher fluorescence signal. The fluorescent intensities of tumors treated with DiR solution and DiR-loaded NPs were calculated as (0.59±0.18) ×107 p/s/cm2/sr and (2.50±0.39) ×107 p/s/cm2/sr, respectively, and were statistically significant (Figure 8C, P<0.005, n=5). As compared to free DiR, the DiR-loaded NPs acquired higher fluorescent intensity at the tumor region even after 24 h. These results indicated that NPs were effective nanodrug platform to deliver HDAC inhibitor to solid tumor.
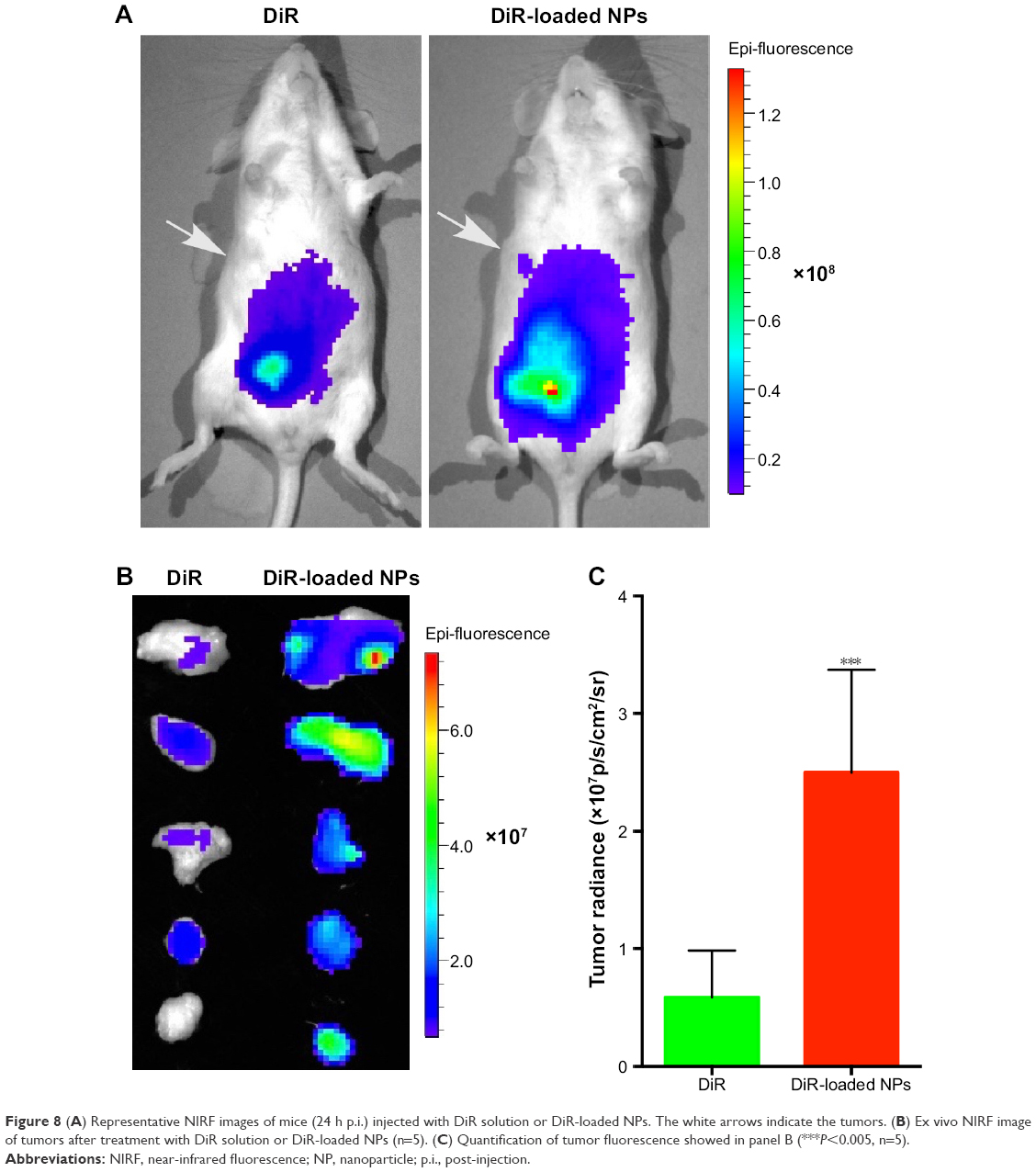 |
Figure 8 (A) Representative NIRF images of mice (24 h p.i.) injected with DiR solution or DiR-loaded NPs. The white arrows indicate the tumors. (B) Ex vivo NIRF image of tumors after treatment with DiR solution or DiR-loaded NPs (n=5). (C) Quantification of tumor fluorescence showed in panel B (***P<0.005, n=5). |
In vivo therapeutic efficacy
To illustrate the advantages of SAHA-S-S-VE/TPGS NPs for solid tumor therapy, the antitumor efficacies toward female Kunming mice bearing (H22) hepatoma tumors were studied. As shown in Figure 9A, tumors in vehicle group showed rapid uncontrolled growth 9 days later; SAHA group showed a modest effect in inhibiting the tumor growth at a dose of 50 mg/kg every other day. However, delivery of SAHA-S-S-VE/TPGS NPs at the same dose led to an enhanced antitumor activity (P<0.05 vs SAHA, P<0.01 vs vehicle) (Figure 9A), presumably because of its enhanced NP accumulation at tumor tissue and triggered SAHA release in tumor cells due to the strongly reductive intracellular environments. Moreover, the perfusion in solid tumor is poor, which may be the primary reason that many nanodrugs failed.53 However, when the redox-responsive nanodrug accumulated at the tumor site, higher GSH concentration in the tumor environment could trigger drug release. Free drug might subsequently penetrate deep into the tumor. On the other hand, the lower antitumor efficacy of free SAHA should be attributed to the destruction of hydroxamic acid under physiological conditions.18,19,54 In addition, the changes in the body weight of mice were monitored during the period of study (Figure 9B). There were no obvious changes of body weight in all the treatment groups.
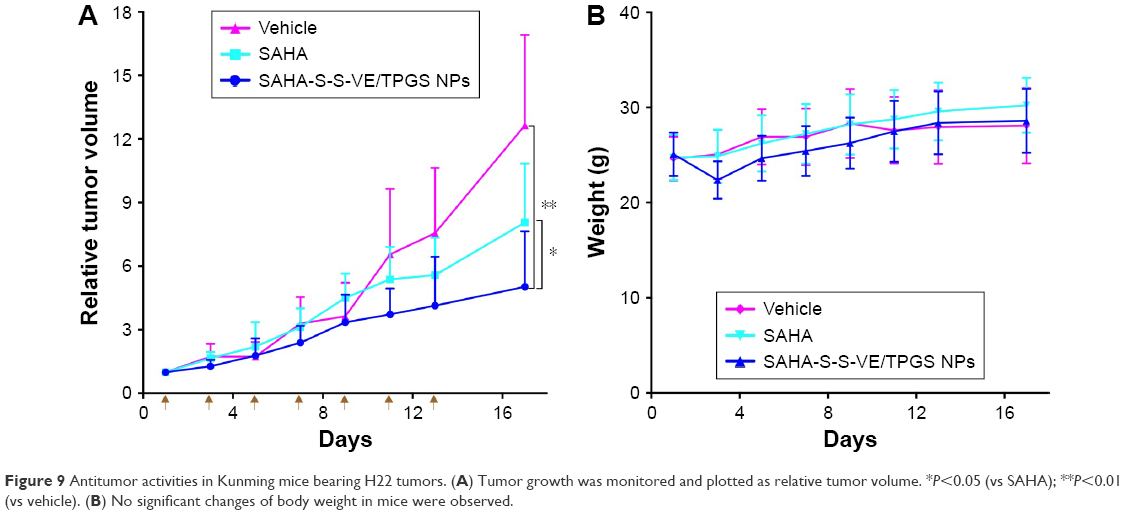 |
Figure 9 Antitumor activities in Kunming mice bearing H22 tumors. (A) Tumor growth was monitored and plotted as relative tumor volume. *P<0.05 (vs SAHA); **P<0.01 (vs vehicle). (B) No significant changes of body weight in mice were observed. |
Conclusion
In conclusion, the objective was to develop self-assembling SAHA prodrug with high drug loading by taking advantage of the EPR effect of NPs and naturally difference of GSH between the tumor and normal tissues. Prodrug SAHA-S-S-VE was well designed and facilely synthesized, with redox trigger-responsive property and comparable HDAC inhibitory activity after reduction. The prodrug was able to self-assemble into spherical NPs with nanometer size, unimodal size distribution, whose mechanisms were validated through molecular dynamics simulations. With a biocompatible TPGS functionalization, the SAHA-S-S-VE/TPGS NPs showed outstanding stability in vitro and enhanced antiproliferative activities. Moreover, in vivo animal experiments confirmed that SAHA-S-S-VE/TPGS NPs possessed significant passive tumor-targeting property and tumor inhibition effect. Collectively, our studies demonstrated that the improved SAHA delivery in solid tumor was made possible, through this redox-responsive nanodrug delivery system. This nanodrug strategy opened a new window to deliver HDAC inhibitor efficiently for solid tumor therapy.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81573368). The authors are very grateful to Dr Livesey David Olerile (School of Pharmaceutical Science, Shandong University) for the English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
Huang P, Wang D, Su Y, et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748–11756. | ||
Wang H, Xie H, Wang J, et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor efficacy. Adv Funct Mater. 2015;25(31):4956–4965. | ||
Jin Y, Xin R, Tong L, Du L, Li M. Combination anti-HIV therapy with the self-assemblies of an asymmetric bolaamphiphilic zidovudine/didanosine prodrug. Mol Pharm. 2011;8(3):867–876. | ||
Zhang J, Li Y, An FF, Zhang X, Chen X, Lee CS. Preparation and size control of sub-100 nm pure nanodrugs. Nano Lett. 2015;15(1):313–318. | ||
Zhao Y, Chen F, Pan Y, et al. Nanodrug formed by coassembly of dual anticancer drugs to inhibit cancer cell drug resistance. ACS Appl Mater Interfaces. 2015;7(34):19295–19305. | ||
Chen F, Zhao Y, Pan Y, et al. Synergistically enhanced therapeutic effect of a carrier-free HCPT/DOX nanodrug on breast cancer cells through improved cellular drug accumulation. Mol Pharm. 2015;12(7):2237–2244. | ||
Zhang J, Li S, An FF, et al. Self-carried curcumin nanoparticles for in vitro and in vivo cancer therapy with real-time monitoring of drug release. Nanoscale. 2015;7(32):13503–13510. | ||
Wang Y, Liu D, Zheng Q, et al. Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett. 2014;14(10):5577–5583. | ||
Pei Q, Hu X, Li Z, Xie Z, Jing X. Small molecular nanomedicines made from a camptothecin dimer containing a disulfide bond. RSC Adv. 2015;5(99):81499–81501. | ||
Song Q, Wang X, Wang Y, et al. Reduction responsive self-assembled nanoparticles based on disulfide-linked drug–drug conjugate with high drug loading and antitumor efficacy. Mol Pharm. 2016;13(1):190–201. | ||
Mahato R, Tai W, Cheng K. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63(8):659–670. | ||
Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63(1–2):3–23. | ||
Sinhababu AK, Thakker DR. Prodrugs of anticancer agents. Adv Drug Deliv Rev. 1996;19(2):241–273. | ||
Denis I, El Bahhaj F, Collette F, et al. Vorinostat-polymer conjugate nanoparticles for acid-responsive delivery and passive tumor targeting. Biomacromolecules. 2014;15(12):4534–4543. | ||
Patil V, Sodji QH, Kornacki JR, Mrksich M, Oyelere AK. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J Med Chem. 2013;56(9):3492–3506. | ||
Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4(4):505–524. | ||
Daniel KB, Sullivan ED, Chen Y, et al. Dual-mode HDAC prodrug for covalent modification and subsequent inhibitor release. J Med Chem. 2015;58(11):4812–4821. | ||
el Bahhaj F, Dekker FJ, Martinet N, Bertrand P. Delivery of epidrugs. Drug Discov Today. 2014;19(9):1337–1352. | ||
Flipo M, Charton J, Hocine A, Dassonneville S, Deprez B, Deprez-Poulain R. Hydroxamates: relationships between structure and plasma stability. J Med Chem. 2009;52(21):6790–6802. | ||
Brink CP, Fish LL, Crumbliss AL. Temperature-dependent acid dissociation constants (Ka, .DELTA.Ha, .DELTA.Sa) for some C-aryl hydroxamic acids: the influence of carbon and nitrogen substituents on hydroxamate anion solvation in aqueous solution. J Org Chem. 1985;50(13):2277–2281. | ||
Silhar P, Eubanks LM, Seki H, et al. Targeting botulinum A cellular toxicity: a prodrug approach. J Med Chem. 2013;56(20):7870–7879. | ||
Schlimme S, Hauser AT, Carafa V, et al. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem. 2011;6(7):1193–1198. | ||
Delatouche R, Denis I, Grinda M, et al. Design of pH responsive clickable prodrugs applied to histone deacetylase inhibitors: a new strategy for anticancer therapy. Eur J Pharm Biopharm. 2013;85(3 Pt B):862–872. | ||
Tran TH, Ramasamy T, Truong DH, et al. Development of vorinostat-loaded solid lipid nanoparticles to enhance pharmacokinetics and efficacy against multidrug-resistant cancer cells. Pharm Res. 2014;31(8):1978–1988. | ||
Tran TH, Choi JY, Ramasamy T, et al. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr Polym. 2014;114:407–415. | ||
Sankar R, Ravikumar V. Biocompatibility and biodistribution of suberoylanilide hydroxamic acid loaded poly (DL-lactide-co-glycolide) nanoparticles for targeted drug delivery in cancer. Biomed Pharmacother. 2014;68(7):865–871. | ||
Chandran P, Kavalakatt A, Malarvizhi GL, et al. Epigenetics targeted protein-vorinostat nanomedicine inducing apoptosis in heterogeneous population of primary acute myeloid leukemia cells including refractory and relapsed cases. Nanomedicine. 2014;10(4):721–732. | ||
Mohamed EA, Zhao Y, Meshali MM, et al. Vorinostat with sustained exposure and high solubility in poly(ethylene glycol)-b-poly(DL-lactic acid) micelle nanocarriers: characterization and effects on pharmacokinetics in rat serum and urine. J Pharm Sci. 2012;101(10):3787–3798. | ||
McMahon BK, Gunnlaugsson T. Selective detection of the reduced form of glutathione (GSH) over the oxidized (GSSG) form using a combination of glutathione reductase and a Tb(III)-cyclen maleimide based lanthanide luminescent ‘switch on’ assay. J Am Chem Soc. 2012;134(26):10725–10728. | ||
Lee MH, Yang Z, Lim CW, et al. Disulfide-cleavage-triggered chemosensors and their biological applications. Chem Rev. 2013;113(7):5071–5109. | ||
Niu LY, Guan YS, Chen YZ, Wu LZ, Tung CH, Yang QZ. BODIPY-based ratiometric fluorescent sensor for highly selective detection of glutathione over cysteine and homocysteine. J Am Chem Soc. 2012;134(46):18928–18931. | ||
Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A1. Wallingford (CT): Gaussian Inc; 2009. | ||
Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–10280. | ||
Pronk S, Pall S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. | ||
Baud MG, Leiser T, Haus P, et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J Med Chem. 2012;55(4):1731–1750. | ||
Yao Y, Su Z, Liang Y, Zhang N. pH-Sensitive carboxymethyl chitosan-modified cationic liposomes for sorafenib and siRNA co-delivery. Int J Nanomedicine. 2015;10:6185–6197. | ||
Sharma VD, Ilies MA. Heterocyclic cationic gemini surfactants: a comparative overview of their synthesis, self-assembling, physicochemical, and biological properties. Med Res Rev. 2014;34(1):1–44. | ||
Brülisauer L, Gauthier MA, Leroux J-C. Disulfide-containing parenteral delivery systems and their redox-biological fate. J Control Release. 2014;195:147–154. | ||
Hong R, Han G, Fernandez JM, Kim BJ, Forbes NS, Rotello VM. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers. J Am Chem Soc. 2006;128(4):1078–1079. | ||
Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–1502. | ||
Wang C, Wesener SR, Zhang H, Cheng YQ. An FAD-dependent pyridine nucleotide-disulfide oxidoreductase is involved in disulfide bond formation in FK228 anticancer depsipeptide. Chem Biol. 2009;16(6):585–593. | ||
Hu YW, Du YZ, Liu N, et al. Selective redox-responsive drug release in tumor cells mediated by chitosan based glycolipid-like nanocarrier. J Control Release. 2015;206:91–100. | ||
Duhem N, Danhier F, Pourcelle V, et al. Self-assembling doxorubicin–tocopherol succinate prodrug as a new drug delivery system: synthesis, characterization, and in vitro and in vivo anticancer activity. Bioconjug Chem. 2014;25(1):72–81. | ||
Zhang Z, Tan S, Feng S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Wu Y, Chu Q, Tan S, et al. D-alpha-tocopherol polyethylene glycol succinate-based derivative nanoparticles as a novel carrier for paclitaxel delivery. Int J Nanomedicine. 2015;10:5219–5235. | ||
Hama S, Itakura S, Nakai M, et al. Overcoming the polyethylene glycol dilemma via pathological environment-sensitive change of the surface property of nanoparticles for cellular entry. J Control Release. 2015;206:67–74. | ||
Suzuki J, Chen YY, Scott GK, et al. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin Cancer Res. 2009;15(9):3163–3171. | ||
Butler LM, Agus DB, Scher HI, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60(18):5165–5170. | ||
Anestopoulos I, Voulgaridou GP, Georgakilas AG, Franco R, Pappa A, Panayiotidis MI. Epigenetic therapy as a novel approach in hepatocellular carcinoma. Pharmacol Ther. 2015;145:103–119. | ||
Singh T, Prasad R, Katiyar SK. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 2013;8(1):54–65. | ||
Yang S, Zhang B, Gong X, Wang T, Liu Y, Zhang N. In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer. Int J Nanomedicine. 2016;11:2329–2343. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110(46):18632–18637. | ||
Sanderson L, Taylor GW, Aboagye EO, et al. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab Dispos. 2004;32(10):1132–1138. | ||
Zhang X, Liu K, Huang Y, et al. Reduction-sensitive dual functional nanomicelles for improved delivery of paclitaxel. Bioconjug Chem. 2014;25(9):1689–1696. | ||
Wu X, Sun X, Guo Z, et al. In vivo and in situ tracking cancer chemotherapy by highly photostable NIR fluorescent theranostic prodrug. J Am Chem Soc. 2014;136(9):3579–3588. | ||
Cheng T, Zhao Y, Li X, et al. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47(6):2140–2148. |
Supplementary materials
Method S1: synthesis of SAHA-S-S-VE
Synthesis of NH2-S-S-VE: cystamine dihydrochloride (3.0 g, 13.0 mmol) was first stirred with NaOH (1.2 g, 30 mmol) in 30 mL ice water for 30 min and then extracted with ether (15 mL3) to get the cystamine solution. D-α-Tocopherol succinate (1.4 g, 2.6 mmol) was dissolved in dry CH2Cl2 (15 mL) under inert atmosphere. Next, 1,1′-carbonyldiimidazole (0.42 g, 2.6 mmol) was added in one portion at 0°C and stirred for 15 min (the conversion was monitored by thin layer chromatography (TLC); indicator: bromocresol green). The resulting solution was slowly dropped to the corresponding cystamine solution for 20 min at 0°C, then stirred for 4 h at room temperature. After removing the solvents, the resulting residue was dissolved in ethyl acetate (50 mL) and washed with water (50 mL), saturated sodium bicarbonate (50 mL), and saturated sodium chloride (50 mL) three times, respectively (the excessive cystamine was eliminated and monitored by TLC; indicator: triketohydrindene hydrate). Evaporate the solvent. The crude product was used for next step without further purification.
Synthesis of SAHA-S-S-VE
SAHA (0.25 g, 0.95 mmol) was added to the solution of 1,1′-carbonyldiimidazole (0.15 g, 0.95 mmol) in anhydrous THF (20 mL) at 0°C. The resulting solution was stirred for 30 min at room temperature. Then NH2-S-S-VE (0.63 g, 0.95 mmol) was added to the above solution and stirred for 4 h (the reaction was monitored by TLC). After removing the solvents, the resulting residue was dissolved in ethyl acetate (50 mL), washed with HCl (0.5 M, 55 mL) and H2O (55 mL3). The crude product can be purified by column chromatography (CH2Cl2/CH3OH, 150:1). SAHA-S-S-VE was obtained as a white waxy solid with the yield of 60%. 1H NMR (CDCl3): δ 0.83–0.87 (m, 12H), 1.04–1.75 (m, 34H), 1.95 (s, 3H), 1.99 (s, 3H), 2.06 (s, 3H), 2.18 (s, 2H), 2.28 (t, J =7.2 Hz, 2H), 2.55 (s, 2H), 2.60 (t, J =6.8 Hz, 2H), 2.76–2.79 (m, 4H), 2.95 (t, J =6.8 Hz, 2H), 3.46–3.53 (m, 4H), 6.23 (s, 1H), 6.70 (s, 1H), 7.05 (t, J =7.8 Hz, 1H), 7.26 (t, J =7.8 Hz, 2H), 7.49 (d, J =7.6 Hz, 2H), 7.94 (s, 1H), 9.84 (s, 1H). 13C NMR (CDCl3): 11.81, 12.13, 12.98, 19.64, 19.75, 20.59, 21.03, 22.62, 22.71, 24.45, 24.79, 25.17, 27.98, 28.05, 28.26, 29.23, 30.84, 31.02, 32.72, 32.80, 37.13, 37.30, 37.46, 37.92, 38.04, 38.63, 39.38, 40.55, 75.15, 117.52, 119.90, 123.10, 124.07, 124.89, 126.61, 128.89, 138.21, 140.47, 149.49, 155.33, 171.77, 171.90, 172.26. MS (ESI) m/z: 978.0 [M+Na]+. HRMS (AP-ESI) m/z: calculated for C52H82N4O8S2 [M+H]+ 955.5647, found: 955.5645.
Method S2: synthesis of SAHA-O-VE
NH2-O-VE was synthesized following the similar procedure of NH2-S-S-VE without neutralizing to get 1,8-diamino-3,6-dioxaoctane (yield: 80%). Then it was used for the next step, and SAHA-O-VE was synthesized following the same procedure of SAHA-S-S-VE. White powder, yield: 75%. 1H NMR (CDCl3): δ 0.83–0.87 (m, 12H), 1.02–1.85 (m, 34H), 1.95 (s, 3H), 2.01 (s, 3H), 2.06 (s, 3H), 2.15 (s, 2H), 2.27 (t, J =7.2 Hz, 2H), 2.53 (s, 2H), 2.61 (t, J =7.8 Hz, 2H), 2.95 (t, J =7.8 Hz, 2H), 3.35–3.38 (m, 2H), 3.43–3.46 (m, 2H), 3.54–3.58 (m, 8H), 6.19 (s, 1H), 6.68 (s, 1H), 7.04 (t, J =7.8 Hz, 1H), 7.27 (t, J =7.8 Hz, 2H), 7.50 (d, J =7.8 Hz, 2H), 7.92 (s, 1H), 9.59 (s, 1H). 13C NMR (CDCl3): 11.81, 12.12, 12.98, 18.43, 19.63, 19.64, 20.50, 20.57, 22.63, 22.64, 24.45, 24.51, 24.80, 25.01, 27.78, 27.98, 28.00, 29.23, 30.72, 31.02, 32.41, 32.71, 32.79, 36.81, 36.98, 37.01, 39.36, 39.37, 41.40, 58.46, 69.35, 69.91, 70.32, 75.09, 76.71, 117.43, 119.78, 123.01, 123.93, 124.95, 126.67, 128.85, 138.30, 140.46, 149.41, 155.17, 171.69, 171.83, 172.22. MS (ESI) m/z: 969.0 [M+NH4]+.
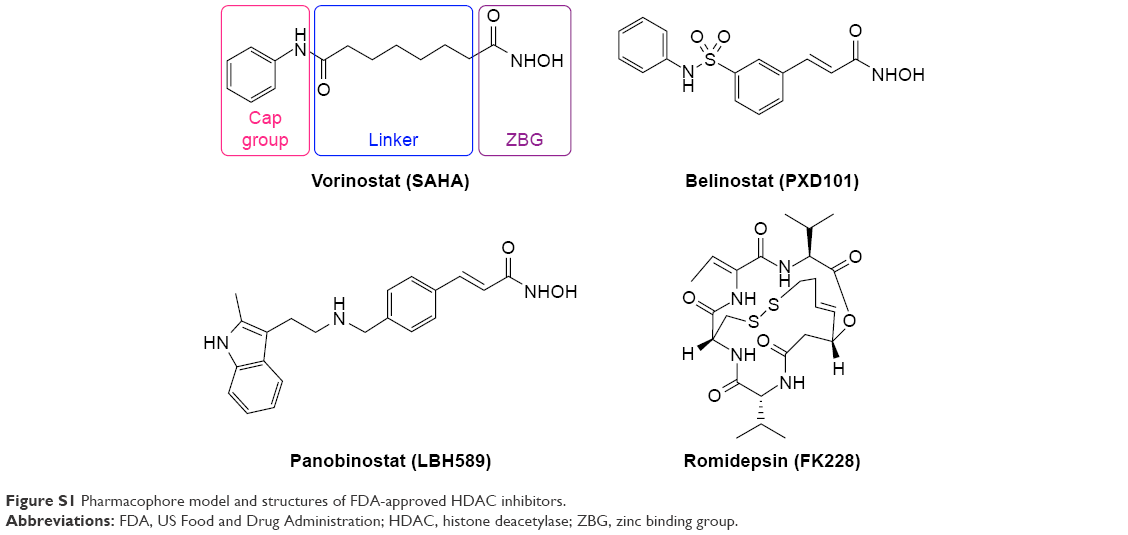 | Figure S1 Pharmacophore model and structures of FDA-approved HDAC inhibitors. |
 | Figure S2 Molecular dynamics simulations of the self-assembly of SAHA-S-S-VE molecules (initial status). |
 | Figure S3 Molecular dynamics simulations of the self-assembly of SAHA-S-S-VE molecules (final status). |
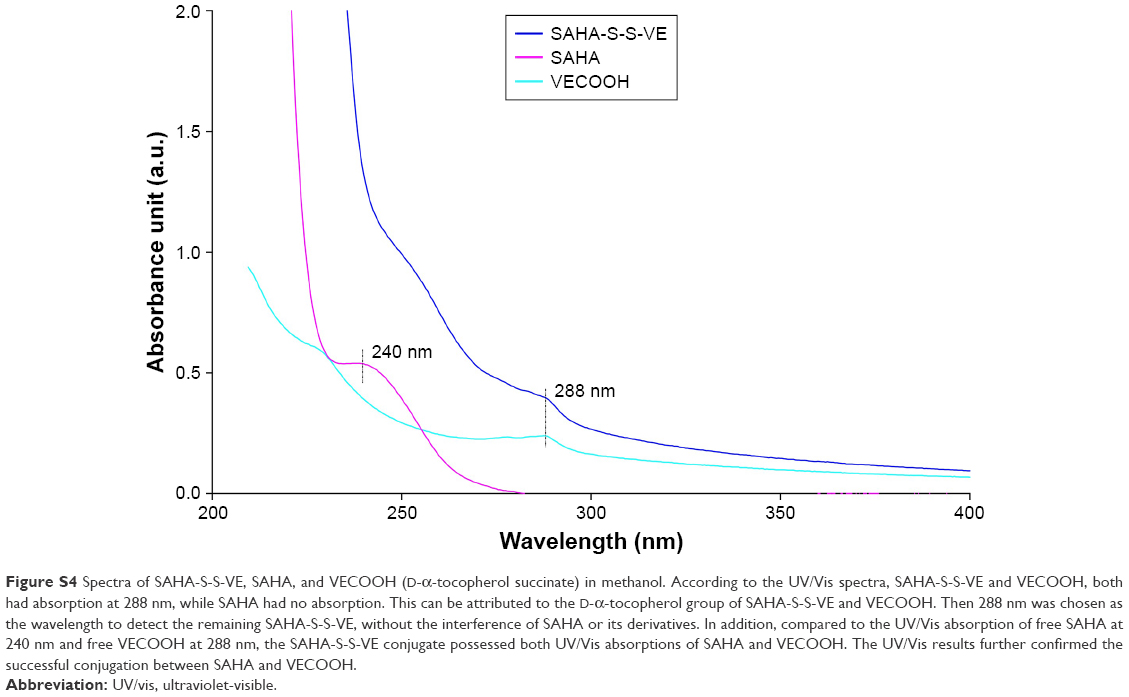 | Figure S4 Spectra of SAHA-S-S-VE, SAHA, and VECOOH (D-α-tocopherol succinate) in methanol. According to the UV/Vis spectra, SAHA-S-S-VE and VECOOH, both had absorption at 288 nm, while SAHA had no absorption. This can be attributed to the D-α-tocopherol group of SAHA-S-S-VE and VECOOH. Then 288 nm was chosen as the wavelength to detect the remaining SAHA-S-S-VE, without the interference of SAHA or its derivatives. In addition, compared to the UV/Vis absorption of free SAHA at 240 nm and free VECOOH at 288 nm, the SAHA-S-S-VE conjugate possessed both UV/Vis absorptions of SAHA and VECOOH. The UV/Vis results further confirmed the successful conjugation between SAHA and VECOOH. |
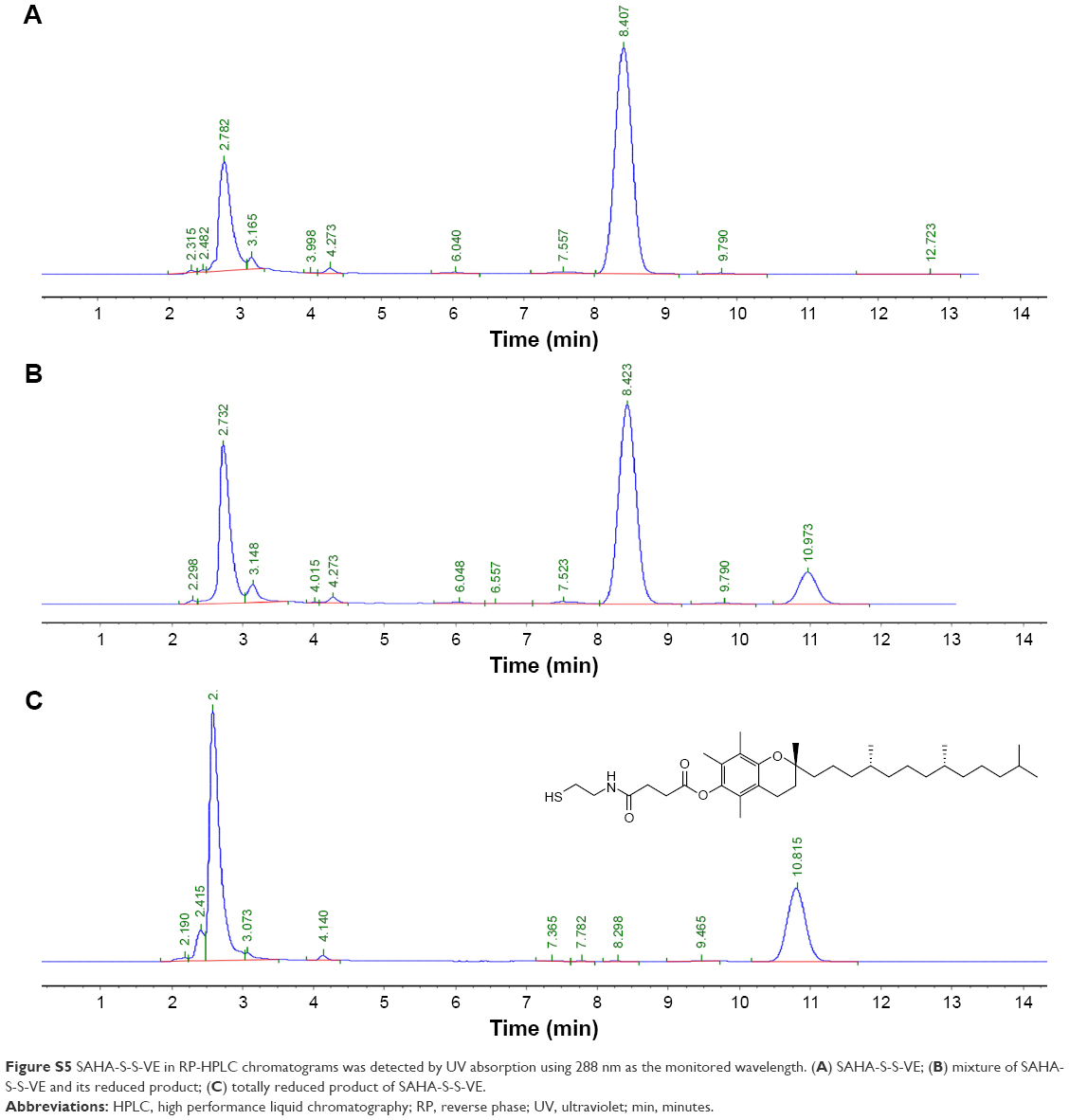 | Figure S5 SAHA-S-S-VE in RP-HPLC chromatograms was detected by UV absorption using 288 nm as the monitored wavelength. (A) SAHA-S-S-VE; (B) mixture of SAHA-S-S-VE and its reduced product; (C) totally reduced product of SAHA-S-S-VE. |
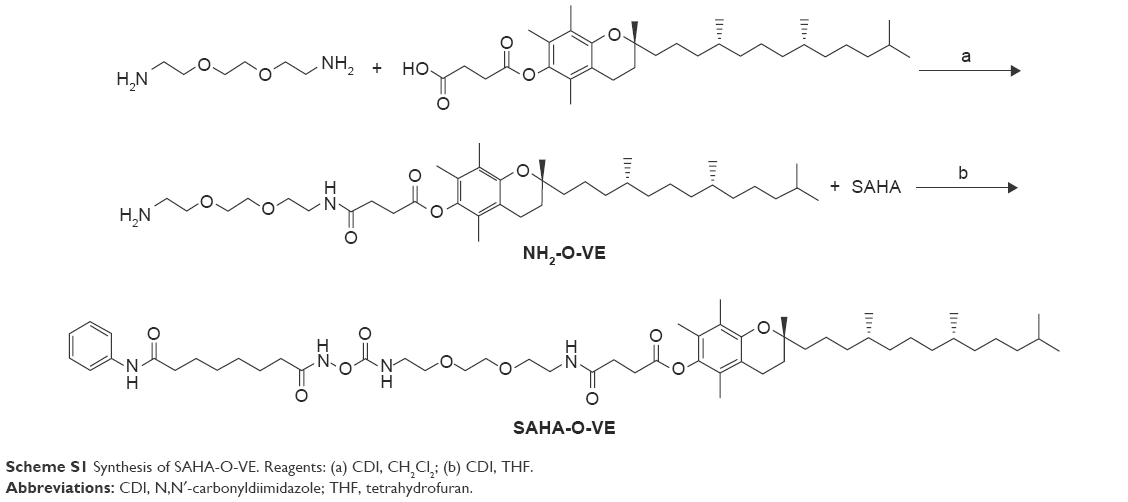 |
Scheme S1 Synthesis of SAHA-O-VE. Reagents: (a) CDI, CH2Cl2; (b) CDI, THF. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.