Back to Journals » International Journal of Nanomedicine » Volume 15
A Dual Receptor Targeting- and BBB Penetrating- Peptide Functionalized Polyethyleneimine Nanocomplex for Secretory Endostatin Gene Delivery to Malignant Glioma
Authors Lu L, Chen H, Wang L, Zhao L, Cheng Y, Wang A ![]() , Wang F
, Wang F ![]() , Zhang X
, Zhang X ![]()
Received 8 July 2020
Accepted for publication 1 October 2020
Published 12 November 2020 Volume 2020:15 Pages 8875—8892
DOI https://doi.org/10.2147/IJN.S270208
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lu Lu,1 Hongyuan Chen,2 Longkun Wang,1 Lin Zhao,3 Yanna Cheng,3 Aijun Wang,4 Fengshan Wang,1 Xinke Zhang3
1Key Laboratory of Chemical Biology (Ministry of Education), Institute of Biochemical and Biotechnological Drug, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 2Department of General Surgery, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 3Key Laboratory of Chemical Biology (Ministry of Education), Department of Pharmacology, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 4Surgical Bioengineering Laboratory, Department of Surgery, UC Davis Health Medical Center, Sacramento, CA, USA
Correspondence: Fengshan Wang; Xinke Zhang
School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, No. 44 Wenhuaxi Road, Jinan 250012, People’s Republic of China
Tel/Fax +86 531 88382490
Email [email protected]; [email protected]
Purpose: Vascular endothelial growth factor receptor 2 (VEGFR-2) and neuropilin-1 (NRP-1) are two prominent synergistic receptors overexpressed on new blood vessels in glioma and may be promising targets for antiglioma therapy. The aim of this study was to design a dual receptor targeting and blood-brain barrier (BBB) penetrating peptide-modified polyethyleneimine (PEI) nanocomplex that can efficiently deliver the angiogenesis-inhibiting secretory endostatin gene (pVAXI-En) to treat glioma.
Materials and Methods: We first constructed the tandem peptide TAT-AT7 by conjugating AT7 to TAT and evaluated its binding affinity to VEGFR-2 and NRP-1, vasculature-targeting ability and BBB crossing capacity. Then, TAT-AT7-modified PEI polymer (PPTA) was synthesized, and a pVAXI-En-loaded PPTA nanocomplex (PPTA/pVAXI-En) was prepared. The physicochemical properties, cytotoxicity, transfection efficiency, capacities to cross the BBB and BTB (blood-tumor barrier) and glioma-targeting properties of PPTA/pVAXI-En were investigated. Moreover, the in vivo anti-angiogenic behaviors and anti-glioma effects of PPTA/pVAXI-En were evaluated in nude mice.
Results: The binding affinity of TAT-AT7 to VEGFR-2 and NRP-1 was approximately 3 to 10 times greater than that of AT7 or TAT. The cellular uptake of TAT-AT7 in endothelial cells was 5-fold and 119-fold greater than that of TAT and AT7 alone, respectively. TAT-AT7 also displayed remarkable efficiency in penetrating the BBB and glioma tissue in vivo. PPTA/pVAXI-En exhibited lower cytotoxicity, stronger BBB and BTB traversing abilities, higher selective glioma targeting and better gene transfection efficiency than PEI/pVAXI-En. More importantly, PPTA/pVAXI-En significantly suppressed the tube formation and migration of endothelial cells, inhibited glioma growth, and reduced the microvasculature in orthotopic U87 glioma-bearing nude mice.
Conclusion: Our study demonstrates that PPTA/pVAXI-En can be exploited as an efficient dual-targeting nanocomplex to cross the BBB and BTB, and hence it represents a feasible and promising nonviral gene delivery system for effective glioma therapy.
Keywords: VEGFR-2 and NRP-1 targeting, glioma penetration, multifunctional peptide, anti-angiogenesis, gene delivery system
Introduction
Malignant glioma is one of the most common intracranial tumors. However, it is difficult to accurately remove glioma completely by traditional surgery due to its infiltration into normal brain tissue.1 The side effects of postoperative radiotherapy and chemotherapy are serious, decreasing patient quality of life.2,3 Therefore, it is urgent to develop more effective therapeutic strategies. The neovascularization of glioma is quite rich, and high vascularity is a major physiological tissue feature of glioma. In addition, abnormal angiogenesis can cause high interstitial fluid pressure and hypoxia inside the tumor tissue, which hinder the delivery of therapeutic drugs from blood vessels to tumor tissues.4,5 Therefore, neovascularization may be an attractive therapeutic target for the treatment of malignant glioma.
Gene therapy for glioma has been extensively studied in related clinical research.6 Endostatin is a potent antiangiogenic protein that was first identified in 1997.7 Researchers have confirmed that endostatin can effectively inhibit angiogenesis, tumor growth and metastasis.8–10 In 2005, endostatin was approved by the FDA in China to treat non-small-cell lung cancer. Later, researchers found that the endostatin protein or gene could be used to treat colon carcinoma and melanoma.11,12 Importantly, some studies indicated that glioma could express a certain level of endostatin, and the increased endostatin expression suppressed glioma angiogenesis.13,14 Hence, in this study, we proposed that the endostatin gene could be used as an effective therapeutic agent to treat glioma angiogenesis. We first constructed a pVAXI-En plasmid to express endostatin. The human cytomegalovirus (CMV) immediate-early enhancer-containing promoter in the pVAXI plasmid could increase the protein expression level in eukaryotic cells. Even more importantly, the addition of a human IgGγ signal sequence to the endostatin DNA sequence could achieve secretory expression of the endostatin gene.15,16
To administer gene drugs with therapeutic potency, safe and efficient gene delivery carriers are needed.17 PEI, a cationic polymer, has been considered one of the most efficient nonviral gene vectors. PEI has excellent properties of DNA condensation, high transfection efficiency, and lysosome escape via the “proton-sponge” effect.18 However, its inherent toxicity and low targeting ability limit its potential use. Recently, many studies have revealed that PEI modified with specific ligands (such as antibodies, peptides or glycoproteins) could notably improve gene delivery efficiency and active targeting.19,20 VEGFR-2 is a tyrosine kinase receptor that is highly expressed on endothelial cells.21 NRP-1 is a cell-surface glycoprotein receptor that has been proven to be a vascular endothelial growth factor 165 (VEGF165) coreceptor that promotes VEGF165-VEGFR-2 intracellular trafficking, facilitating angiogenesis.22 In addition, NRP-1 can induce vascular permeability by interacting with VEGF165 and semaphorin 3A, contributing to blood vessel growth and tissue edema.23,24 Therefore, VEGFR-2 and NRP-1 are two ideal targets because of their synergistic effect on angiogenesis. ATWLPPR (AT7), which was identified by a phage display peptide library,25 showed high affinity to VEGFR-2 and NRP-1 simultaneously.26,27 Drug delivery systems modified with AT7 have been proven to possess efficient active targeting ability for glioma therapy.28,29 These results show that AT7 has potential as a targeting ligand for new glioma vessels.
However, the BBB and BTB are two obstacles that rigorously prevent therapeutic drugs from crossing from the peripheral blood circulation to the brain and glioma tissue.30,31 Cationic cell-penetrating peptides (CPPs) have been studied extensively for their efficient ability to break through various biological barriers, including cell membranes, nuclear membranes, the BBB and eye barriers. They can also facilitate the tissue penetration and internalization of modified drug carriers.32 TAT (RKKRRQRRR), one of the most promising CPPs, has been studied extensively for its efficient biological barrier penetration and high cargo (antibodies, nucleic acids and nanoparticles) delivery efficiency.33,34 Nevertheless, the disadvantages of nonselectivity and low specificity limit the further application of TAT in vivo. Interestingly, in our study, we discovered that TAT could interact with the NRP-1 in addition to penetrating the BBB. Considering the high expression of NRP-1 in brain glioma tissue and new blood vessels, we hypothesized that the new tandem peptide TAT-AT7 (constructed by connecting the TAT to AT7) might possess a stronger affinity for the NRP-1 than AT7. TAT-AT7 was expected to retain the BBB penetration property of TAT and the VEGFR-2-targeting ability of AT7 simultaneously. Accordingly, TAT-AT7 was conjugated to PEI via a heterobifunctional PEG linker to obtain PEI-PEG-TAT-AT7 (PPTA). After loading the antiangiogenic pVAXI-En plasmid, the novel nanocomplex PPTA/pVAXI-En was prepared. As illustrated in Figure 1, this multifunctional gene delivery system is expected to possess the following features: (i) high glioma angiogenesis-targeting ability because of the overexpression of VEGFR-2 and NRP-1 on vasculature endothelial cells or glioma cells; (ii) increased vascular permeability caused by the interaction of the R/KXXR/K sequence of TAT-AT7 with NRP-1; (iii) strong BBB and BTB penetration capacities mediated by TAT-AT7; and (iv) enhanced transfection efficiency induced by PPTA. From the above, the aim of our study was to investigate whether PPTA/pVAXI-En could achieve dual-targeted gene delivery to glioma neovascularization and suppress the growth of glioma.
 |
Figure 1 Schematic illustration of the TAT-AT7-modified PEI polymer delivery system encapsulating the angiogenesis-inhibiting secretory endostatin gene (PPTA/pVAXI-En) as an antiglioma therapy. PPTA/pVAXI-En could efficiently penetrate the BBB and BTB, showed high level of accumulation in glioma tissue and specifically bound VEGFR-2 and NRP-1 expressed on endothelial cells and glioma cells. |
Materials and Methods
Materials
The purified recombinant human receptor proteins VEGFR-2 and NRP-1 were purchased from R&D Systems (MN, USA). A CM5 sensor chip was obtained from GE Healthcare Life Sciences (Uppsala, Sweden). Rabbit anti-VEGFR-2 antibody was purchased from Abcam (USA). Rabbit anti-NRP-1 antibody and secondary antibody (anti-rabbit) were purchased from Cell Signaling Technology (MA, USA). MAL-PEG-NHS (MW=2 kDa) was purchased from Jenkem Technology (Beijing, China). RKKRRQRRRC (TAT), ATWLPPRC (AT7) and RKKRRQRRRCATWLPPR (TAT-AT7) were synthesized from China Peptide Co., Ltd. (Shanghai, China). The pVAXI-En and pEGFP-N1 plasmids were purified with an EndoFree Plasmid Maxi Kit (Hilden, Germany). Branched PEI (MW=25 kDa), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and indocyanine green (ICG) were purchased from Sigma-Aldrich (MO, USA). Hoechst 33342 was obtained from Aladdin (Shanghai, China). LysoTracker Red and bovine serum albumin (BSA) were purchased from Solarbio Bioscience and Technology (Shanghai, China). Matrigel was obtained from BD Biosciences (MA, USA). GoldView II was supplied by BioTeke Corporation (Beijing, China). Label IT® Nucleic Acid Labeling Kits were purchased from Mirus Bio (WI, USA). F-12K medium, heparin and endothelial cell growth supplement (ECGS) were obtained from MacGene (Beijing, China). DMEM and fetal bovine serum (FBS) were obtained from Gibco®, Life Technologies (CA, USA). All other chemicals and reagents were of the highest commercial grade available.
Cell Culture and Animal Studies
Human umbilical vein endothelial cells (HUVECs) were obtained from ATCC (Manassas, VA). The cells were cultured in F-12K medium supplemented with 100 μg/mL heparin, 50 μg/mL ECGS, and 10% FBS under a 5% CO2 atmosphere at 37°C. U87-mCherry-luc cells (human glioma cells) were obtained from Shanghai Sciencelight Biology Science & Technology Co. (Shanghai, China). bEnd.3 cells (mouse brain microvascular endothelial cells) were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). U87-mCherry-luc cells and bEnd.3 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin.
Female nude mice 4–6 weeks of age were purchased from the Jinan Pengyue Experimental Animal Breeding Co., Ltd. (Jinan, China). The mice were raised under specific pathogen-free (SPF) conditions. All animal procedures were approved by the Ethics Committee of Shandong University and performed in accordance with the Shandong Council on Animal Care.
Binding Affinity of TAT-AT7 to VEGFR-2 and NRP-1
The binding affinity of TAT-AT7 to VEGFR-2 and NRP-1 was evaluated by surface plasmon resonance (SPR) analysis using Biacore S200 instrument (GE Healthcare). Recombinant VEGFR-2 and NRP-1 proteins were immobilized on the CM5 sensor chip according to the operating instructions for amine coupling. Different peptides at a series of concentrations diluted in HBS-EP buffer were injected and flowed over the chip surface to detect resonance unit changes. The response curves were analyzed with 1:1 Langmuir binding mode with Biacore S200 evaluation software to obtain the dynamic parameter KD (equilibrium dissociation constant).
Vasculature-Targeting Ability of the TAT-AT7 in vitro
Expression of VEGFR-2 and NRP-1 in HUVECs was first evaluated by an immunostaining assay using anti-VEGFR-2 and anti-NRP-1 antibodies. Then, HUVECs were seeded in 6-well plates and cultured overnight. FITC-TAT, FITC-AT7, and FITC-TAT-AT7 (20μM) were added to HUVECs and incubated at 37°C for 1 h. Fluorescent images were captured with an inverted microscope (Olympus, Japan). After trypsinization, the cells were collected and suspended in PBS, and the fluorescence intensity was analyzed by flow cytometry (CytomicsTM FC500, Beckman Coulter, USA).
The Brain-Targeting Ability of TAT-AT7 in vivo
Nude mice were anesthetized with isoflurane and fixed on a ventricle stereotaxic instrument. Then, U87-mCherry-luc cells (2.5×105 cells in 5 µL) were injected into the right striatum of the brain. After 4 days, the bioluminescence of the glioma tissue in the brains of the mice was detected with an IVIS kinetic imaging system (Lumina II; Caliper, MA, USA) to verify successful establishment of an orthotopic U87 glioma-bearing nude mice model. The mice were randomly divided into four groups according to bioluminescence intensity (n=3). Different FITC-labeled peptides were injected via the tail vein (30 mg/kg). Mice injected with physiological saline were used as a control group. The mice were sacrificed 1 h later, and the brains were fixed with 4% paraformaldehyde and cut into slices. Then, the brain slices were further stained with DAPI and visualized using a digital fluorescent scanning microscope (Pannoramic 250 Flash III, Budapest, Hungary). The detailed fluorescence parameters were as follows. Blue channel: exposure times: 2ms; digital gain: 1; bit depth: 8. Green channel: exposure times: 80ms; digital gain: 1; bit depth: 8. Red channel: exposure times: 40ms; digital gain: 1; bit depth: 8.
Synthesis of PPTA
First, TAT-AT7 and MAL-PEG-NHS were dissolved in dimethyl sulfoxide (DMSO) at a molar ratio of 4:3, after which the mixture was stirred for 12 h at room temperature in an atmosphere of nitrogen. Second, the PEI solution was solubilized in PBS (0.1 mM; pH 7.0), after which a 4-fold molar excess of PEG-TAT-AT7 dissolved in DMSO was added to the above PEI solution. The resulting mixed solution was stirred overnight in an atmosphere of nitrogen. The resulting product, PPTA, was dialyzed against H2O (MWCO=10 kDa) and then lyophilized. PEI-PEG-TAT (PPT) and PEI-PEG-AT7 (PPA) were synthesized and obtained with the protocol described above. The chemical structures of all products in D2O were characterized by 1H NMR spectrometry (300 MHz).
Cytotoxicity Evaluation of PPTA
The cytotoxicity of PPTA against endothelial cells was evaluated by MTT assay. HUVECs were seeded into 96-well plates at a density of 5×103 cells/well. After incubation overnight, the HUVECs were treated with PEI, PPA, PPT and PPTA at a series of concentrations for 24 h. Then, 20 µL of MTT solution (5 mg/mL) was added to each well, and the cells were further cultured for 4 h under dark conditions. Then, the resulting formazan crystals were dissolved in 150 µL of DMSO after the medium had been removed. After incubation for 15 min, the absorbance at 490 nm was measured using a microplate reader (Bio-Rad 680, Hercules, CA, USA).
In vivo Imaging and Distribution of PPTA in Glioma
Orthotopic U87-mCherry-luc glioma-bearing nude mice were randomly divided into 6 groups (n=3). ICG was used as a NIR dye for fluorescence imaging in vivo. Different polymers loaded with ICG (100 µL) were injected into the mouse body via the tail vein. A saline group was used as a negative control group. The mice were anesthetized with isoflurane and assessed with an IVIS kinetic imaging system at 1 h, 2 h, and 4 h after injection. At 4 h after injection, the mice were sacrificed, and the heart, liver, spleen, lung, kidney and brain were removed and observed with an IVIS kinetic imaging system.
Preparation and Characterization of PPTA/pVAXI-En
The pVAXI-En plasmid was added dropwise to an equal volume of a PPTA solution under slow vortexing, followed by incubation for 30min at room temperature to obtain PPTA/pVAXI-En. In addition, PEI/pVAXI-En, PPA/pVAXI-En and PPT/pVAXI-En were prepared using the same method. The particle size and zeta potential of different nanocomplexes were monitored with a nano particle analyzer (DelsaNano S, Beckman Coulter, USA). The morphology of nanocomplexes was observed by transmission electronic microscopy (TEM; JEM-1200EX; JEOL, Tokyo, Japan). The ability of the nanocomplexes to condense the pVAXI-En plasmid was determined by agarose gel retardation assay, in which the retardation and position of nanocomplexes were visualized with a UV transilluminator and digital imaging system (IS-2200, Alpha Innotech, USA).
Intracellular Trafficking and Lysosomal Escape of PPTA/pVAXI-En
First, the pVAXI-En plasmid was labeled with FITC using Label IT® Nucleic Acid Labeling Kits according to the instructions. HUVECs were seeded into a laser confocal dish and cultured overnight. Freshly prepared PEI/FITC-pVAXI-En, PPA/FITC-pVAXI-En, PPT/FITC-pVAXI-En and PPTA/FITC-pVAXI-En were individually added to each dish. The nanocomplexes were incubated with cells at 37°C for 2 h and 4 h in the dark, followed by incubation with LysoTracker Red for 30 min. Nuclear staining was performed by Hoechst 33342 staining. Fluorescence images of the cells were taken under a laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
In vitro BBB and BTB Penetration Assays
An in vitro BBB model was established according to previous reports.35,36 bEnd.3 cells were seeded into the apical chambers of 12-well transwell plates. After 7 days, the transendothelial electrical resistance (TEER) of the bEnd.3 cells monolayer was measured with an epithelial voltammeter (Millipore, Bedford, USA). Monolayers with a TEER over 200 Ω were used for subsequent experiments. The pVAXI-En plasmid labeled with FITC was the same as previous method. Then, freshly prepared PEI/FITC-pVAXI-En, PPA/FITC-pVAXI-En, PPT/FITC-pVAXI-En and PPTA/FITC-pVAXI-En were individually added into the apical chambers. After 4 h incubation at 37°C, the fluorescence intensities of the solutions from the lower chamber were measured with a fluorescence microplate reader.
An in vitro BTB model was established as previously reported.37 Briefly, U87 cells were seeded into the basolateral chambers, and HUVECs were seeded into the apical chambers at a 1:5 HUVEC/U87 cell ratio. After 4 days, freshly prepared PEI/FITC-pVAXI-En, PPA/FITC-pVAXI-En, PPT/FITC-pVAXI-En and PPTA/FITC-pVAXI-En were individually added to the corresponding apical chamber. After 4 h of incubation at 37°C, the fluorescence intensities of the solutions from the lower chamber were determined with a fluorescence microplate reader. The penetration ratios of the nanocomplexes are expressed as the relative fluorescence intensity compared with the total fluorescence intensity measured before the experiment.
Gene Transfection Studies
HUVECs were seeded into 6-well plates at a density of 2×105 cells/well and incubated overnight. PEI/pEGFP-N1, PPA/pEGFP-N1, PPT/pEGFP-N1 and PPTA/pEGFP-N1 (containing 4 µg of the pEGFP-N1 plasmid) were added to the HUVECs and incubated with the cells for 4 h. The cells were then incubated with freshly prepared F-12K complete medium for another 48 h at 37°C. The expression of EGFP was observed by fluorescence microscopy. The cells were digested and suspended in PBS, and the transfection efficiency was quantified by flow cytometry.
Orthotopic U87-mCherry-luc glioma-bearing nude mice were randomly divided into 4 groups (n=3). Two hundred microliters of PEI/pEGFP-N1, PPA/pEGFP-N1, PPT/pEGFP-N1 or PPTA/pEGFP-N1 (containing 40 μg of the pEGFP-N1 plasmid) or physiological saline was injected into the mice six times on days 6, 8, 10, 12, 14 and 16 via the tail vein. On day 18, the mice were sacrificed, and their glioma-bearing brains were fixed with 4.0% paraformaldehyde, embedded in paraffin and cut into slices. The brain slices were further stained with anti-EGFP antibody for the study of transfection efficiency in vivo.
Effect on Angiogenesis in vitro
The transfection process was carried out as previously described. The supernatant was harvested, and secretory endostatin protein expression was detected with a human endostatin ELISA kit according to the operating instruction. The antiangiogenic effect of PPTA/pVAXI-En was determined by tube formation assay. Briefly, Matrigel was coated onto 96-well plates at 37°C for solidification. HUVECs transfected with the different nanocomplexes were seeded into the Matrigel-coated plates (3×104 cells/well). After incubation at 37°C for 6 h, tube formation was observed with an inverted microscope, and the number of branch points was calculated with ImageJ software. A wound-healing assay was used to evaluate the effect of PPTA/pVAXI-En on endothelial cell migration. Briefly, HUVECs were seeded into 6-well culture plates (3×105 cells/well). After overnight incubation, the HUVECs monolayer was scraped with 200-µL pipette tips. Different nanocomplexes (containing 4 µg of the pVAXI-En plasmid) were added to each well. After 4 h of incubation at 37°C, the cells were then incubated with freshly prepared F-12K complete medium for another 48 h at 37°C. Images were captured after 0 h, 24 h and 48 h using an inverted microscope. The cell migration rate was measured with ImageJ software.
Pharmacodynamics in vivo
Orthotopic U87-mCherry-luc glioma-bearing nude mice were randomly divided into 6 groups (n=6). Two hundred microliters of freshly prepared PEI/pVAXI-En, PPA/pVAXI-En, PPT/pVAXI-En and PPTA/pVAXI-En (containing 40 µg of the pVAXI-En plasmid) was injected into the mice six times on days 6, 8, 10, 12, 14 and 16 via the tail vein. A saline group was used as a negative control group. Bioluminescence was measured on days 4, 9, 14 and 20. On day 21, the mice were sacrificed, and their glioma-bearing brains were fixed with 4.0% paraformaldehyde, embedded in paraffin and cut into slices. CD31 staining was conducted to detect the microvessels in glioma.
Statistical Analysis
The data are presented as the mean ± standard deviation (SD). Multiple group comparisons were conducted by one-way ANOVA, followed by Tukey’s multiple comparison test using GraphPad Prism 6 software. A value of p < 0.05 was used to indicate statistical significance.
Results
Binding Affinity of TAT-AT7 to VEGFR-2 and NRP-1
SPR analysis was conducted to evaluate the binding affinity of TAT-AT7 to VEGFR-2 and NRP-1. The SPR analysis was performed using a Biacore S200 instrument. Recombinant VEGFR-2 and NRP-1 proteins were first immobilized on a CM5 chip. Different peptides, as analytes, were then flowed over the surface of the chip. The KD value calculated by SPR analysis reflects the binding level of the analytes to the ligand proteins. The smaller the KD value is, the stronger the binding ability. As shown in Figure 2A, TAT-AT7 showed the strongest binding affinity to NRP-1 with a KD value of 2.740×10−8 M compared to the TAT and AT7 with KD values of 6.052×10−8 M and 6.770×10−8 M, respectively. TAT-AT7 showed high affinity to VEGFR-2 with a KD value of 2.276×10−8 M, whereas the binding affinity of AT7 to VEGFR-2 was weaker with a KD value of 2.830×10−7 M, and the binding between TAT and VEGFR-2 was nonspecific with an unmeasurable KD value (Figure 2B). The above SPR results indicated that TAT-AT7 exhibited higher affinity to NRP-1 than to TAT or AT7 alone, and the binding ability of TAT to the NRP-1 verified our initial hypothesis.
 |
Figure 2 Binding affinity of TAT-AT7 to VEGFR-2 and NRP-1. The binding curves for AT7, TAT, and TAT-AT7 with the recombinant proteins VEGFR-2 (A) and NRP-1 (B) were assessed by SPR analysis. Different peptides at a series of concentrations (increasing from 0.0156 μM to 0.03125 μM, 0.0625 μM, 0.125 μM, and 0.25 μM) were flowed over the surface of chip-immobilized VEGFR-2 or NRP-1 proteins. The KD value reflects the level of analyte binding to ligand proteins. |
Vasculature-Targeting Ability of the TAT-AT7 in vitro
HUVECs expressing VEGFR2 and NRP-1 were used as models of tumor vascular endothelial cells. As expected, we first verified by an immunostaining assay that both VEGFR-2 and NRP-1 showed significant positive expression on the surface of HUVECs (Supplementary Figure S1). A cellular uptake experiment was conducted to explore the vascular targeting ability of the TAT-AT7 in vitro. Fluorescent micrographs of HUVECs after exposure to FITC-labeled peptides were shown in Figure 3A. The fluorescence intensity in HUVECs exposed to FITC-TAT-AT7 increased notably. Quantitative analysis by flow cytometry (Figure 3B) indicated that the mean fluorescence intensity of the FITC-TAT-AT7 group was approximately 5-fold and 119-fold that of FITC-TAT and FITC-AT7, respectively, which revealed that TAT-AT7 exhibited higher uptake efficiency and penetration capability into HUVECs than TAT or AT7 did. We supposed that the improved internalization of TAT-AT7 might depend not only on the strong penetration of TAT into HUVECs but also on the binding ability to VEGFR-2 and NRP-1 expressed on the surface of HUVECs.
 |
Figure 3 In vitro and in vivo targeting abilities of TAT-AT7. (A) Fluorescence images of HUVECs after incubation with FITC-AT7, FITC-TAT or FITC-TAT-AT7 for 1 h. Bars represent 50 μm. (B) Flow cytometric histograms and quantitative fluorescence intensities of HUVECs after incubation with FITC-AT7, FITC-TAT or FITC-TAT-AT7 for 1 h. The mean fluorescence intensity of HUVECs was measured by flow cytometry. * indicates p<0.05 when compared with the control group. # indicates p<0.05 when compared with the AT7 group and & indicates p<0.05 when compared with the TAT group. (C) Distribution of TAT-AT7 in the glioma tissue of intracranial U87 glioma-bearing mice. Frozen sections were photographed with a digital fluorescent scanning microscope. Green, different FITC-labeled peptides; blue, DAPI-stained cell nuclei; red, glioma tissue labeled with the red fluorescent protein mCherry. Bars represent 200 μm. |
The Brain-Targeting Efficacy of TAT-AT7 in vivo
To investigate the BBB penetration and glioma targeting ability of TAT-AT7 in vivo, fluorescence imaging analysis of brain tissue from orthotopic U87 glioma-bearing nude mice was conducted. As shown in a representative micrograph (Figure 3C), red fluorescence represented the location of brain glioma tissue, FITC-TAT and FITC-TAT-AT7 displayed strong green fluorescence signals in the glioma region after intravenous injection, while FITC-AT7 was unable to achieve obvious accumulation in glioma tissue due to its weak BBB penetration. Furthermore, the fluorescence intensity after injection with FITC-TAT-AT7 was much higher than that after injection with FITC-TAT. From the above results, we could conclude that TAT-AT7 retained the BBB and BTB penetration ability of TAT and showed significant targeting accumulation in VEGFR-2- and NRP-1-overexpressing glioma tissue in vivo.
Synthesis and Cytotoxicity Evaluation of PPTA
The overall synthesis of PPTA was divided into two steps, as presented in Figure 4A. Heterobifunctional MAL-PEG-NHS (PEG linker) was selected as a connecting arm, PEG-TAT-AT7 was synthesized through the reaction of the sulfhydryl groups of TAT-AT7 with the MAL groups of the PEG linker, and then PEG-TAT-AT7 was connected to the amino groups of PEI to obtain PPTA. The details of the 1H-NMR spectra were as follows (Figure 4BE): the solvent peak of D2O was found at 4.7 ppm. The characteristic peaks of PEG appeared at 3.5–3.7 ppm. The repeating units of the PEI polymer appeared at 2.5–3.2 ppm. The successful coupling of TAT-AT7 to the PEI polymer was confirmed by the appearance of the peaks for the aromatic protons of TAT-AT7 at 7.0–7.8 ppm. The PPA and PPT polymers were synthesized by the same method, and their 1H-NMR spectra were shown in Supplementary Figure S2. The major problem with PEI polymer as a nonviral gene vector is the correlation of toxicity and transfection efficiency. Consequently, current studies on PEI focus on achieving high efficiency and low toxicity.38 As shown in Figure 5C, the in vitro MTT assay revealed that as the concentrations of PEI, PPA, PPT and PPTA increased from 2 μg/mL to 32 μg/mL, the viability of HUVECs gradually decreased to varying degrees. However, PPA, PPT and PPTA showed significantly lower cell toxicity than PEI at any concentration, especially over 8 μg/mL. These results suggested that PPTA had better biocompatibility and applicability as a vehicle for gene transfection.
 |
Figure 4 The synthetic mechanism and 1H-NMR spectrum of PEI-PEG-TAT-AT7 (PPTA). (A) The whole process of PPTA synthesis was divided into two steps as presented. (B–E) 1H NMR spectra of PEI, TAT-AT7, NHS-PEG-MAL, and PPTA. |
 |
Figure 5 Characterization and cytological evaluation of PPTA/pVAXI-En. (A and B) SEM images and agarose gel electrophoresis of PEI/pVAXI-En, PPA/pVAXI-En, PPT/pVAXI-En and PPTA/pVAXI-En. (C) Cytotoxicity of PEI, PPA, PPT and PPTA at various concentrations against HUVECs. ** and *** Indicate p<0.01 and p<0.001 versus the PEI group, respectively. (D) Fluorescence images of the intracellular distribution of PEI/FITC-pVAXI-En, PPA/FITC-pVAXI-En, PPT/FITC-pVAXI-En and PPTA/FITC-pVAXI-En in HUVECs at 2 h (left) and 4 h (right) after the beginning of transfection. Green, FITC-labeled pVAXI-En plasmid; blue, cell nuclei stained with Hoechst 33342; red, lysosomes stained with LysoTracker Red. Bars represent 10 μm. |
Characterization of PPTA/pVAXI-En
PPTA/pVAXI-En was prepared by the electrostatic adsorption principle between electropositive PPTA and electronegative pVAXI-En plasmid. As shown in Supplementary Table S1, polymers/pVAXI-En with different N/P ratios (nitrogen in PEI to phosphates in plasmid DNA) showed different sizes (between 40 and 300 nm). It has been confirmed that nanoparticles of approximately 100 nm maintain the best passive targeted therapeutic effects, such as prolonged blood circulation and reduced mononuclear phagocyte system (MPS) capture.39 Consequently, PPTA/pVAXI-En (N/P=12), with a size of 101.6 ± 4.35 nm and ζ potential change of 17.7 ± 1.39 mV, was selected for subsequent experiments. The TEM image revealed that PPTA/pVAXI-En (N/P=12) had a homogeneous spheroid morphology (Figure 5A). The results of the agarose gel retardation assay (Figure 5B) also showed that PPTA/pVAXI-En (N/P=12) had good DNA condensing and neutralizing properties.
Cellular Uptake and Lysosomal Escape of PPTA/pVAXI-En
The process of gene transfection includes cell uptake and the successful expression of DNA. The important steps in the process and indispensable factors for transfection efficiency are DNA internalization and escape from lysosomes. pVAXI-En plasmid was first labeled by FITC to track the intracellular location of PPTA/FITC-pVAXI-En. As shown in Figure 5D, PPTA/FITC-pVAXI-En (green fluorescence) and the other groups all colocalized with lysosomes (red fluorescence) after 2 hours (left). When the incubation time reached 4 hours, the green and red fluorescence were separated to different degrees (right). These results clearly demonstrated that PPTA/pVAXI-En had good lysosomal uptake and escape abilities at a comparable level to PEI/pVAXI-En.
Ability of PPTA/pVAXI-En to Penetrate the BTB and BTB in vitro
The BBB is the main physiological barrier for drug delivery into the brain. To determine the ability of PPTA/pVAXI-En to penetrate the BBB, an in vitro BBB model was established. A monolayer of bEnd3 cells seeded in the apical chamber was used as a biomimetic BBB (Figure 6A). Fluorescence intensity passing through the apical chamber was detected to evaluate the ability of PPTA/pVAXI-En to penetrate the BBB. As shown in Figure 6B, the penetration ratios of PPTA/pVAXI-En (1.78 ± 0.11%) and PPT/pVAXI-En (1.70 ± 0.06%) were clearly increased in comparison to those of PEI/pVAXI-En (0.95 ± 0.07%) and PPA/pVAXI-En (1.14 ± 0.11%). The BTB is another major physiological barrier that restricts the transport of drugs to brain glioma tissue. An in vitro HUVEC/U87 cell coculture model was established (Figure 6A). In this coculture system, a monolayer of HUVECs seeded in the apical chamber can be endowed with an angiogenic phenotype after stimulation with U87 cells, which can imitate the BTB in vivo under physiological conditions.40 The results in Figure 6C showed that the penetration ratio of PPTA/pVAXI-En (2.72 ± 0.13%) was significantly higher than those of PEI/pVAXI-En (1.28 ± 0.08%), PPA/pVAXI-En (1.47 ± 0.17%) and PPT/pVAXI-En (2.12 ± 0.19%). The above results indicated that both PPTA/pVAXI-En and PPT/pVAXI-En displayed a superior ability to traverse the BBB and BTB compared with that of PEI/pVAXI-En and PPA/pVAXI-En. Furthermore, PPTA/pVAXI-En exhibited a greater ability to penetrate the BTB than PPT/pVAXI-En.
 |
Figure 6 Ability of PPTA/pVAXI-En to penetrate the BBB and BTB in vitro. (A) Schematic illustration of the in vitro BBB and BTB models. The penetration ratios of PEI/pVAXI-En, PPA/pVAXI-En, PPT/pVAXI-En and PPTA/pVAXI-En in the in vitro BBB model (B) and BTB model (C). * Indicates p<0.05 when compared with PEI/pVAXI-En, # indicates p<0.05 when compared with PPA/pVAXI-En, and & indicates p<0.05 when compared with PPT/pVAXI-En. |
In vivo Glioma Targeting Property of PPTA
To assess the in vivo brain targeting ability of PPTA, we chose ICG as the imaging agent. PEI/ICG, PPA/ICG, PPT/ICG and PPTA/ICG complexes were intravenously injected into orthotopic U87 glioma-bearing nude mice. Free ICG was used as the control group. Fluorescence imaging in nude mice (Figure 7A) showed that PPTA significantly prolonged the retention time of ICG in the brain. PPTA/ICG showed a stronger fluorescence signal than the other groups at 1 h, 2 h and 4 h. Ex vivo fluorescence imaging of the heart, liver, spleen, lung, kidney and brain was further conducted after 4 hours and indicated that the fluorescence intensity of PPTA/ICG in isolated brain tissue was much higher than that of PEI/ICG, PPA/ICG or PPT/ICG (Figure 7B), which provided compelling evidence that PPTA had remarkable BBB penetration and brain targeting abilities.
 |
Figure 7 The brain-targeting property of PPTA. (A) In vivo fluorescence images of intracranial U87 glioma-bearing nude mice at different time points after intravenous injection with the PEI/ICG, PPA/ICG, PPT/ICG or PPTA/ICG complexes. (B) Ex vivo fluorescence images of the heart, liver, spleen, lung, kidney and brain organs from nude mice after 4 h. |
The Gene Transfection Efficiency of the PPTA/DNA Nanocomplex in vitro and in vivo
We demonstrated the low cytotoxicity of PPTA and favorable lysosomal escape ability of PPTA/pVAXI-En, which were closely related to DNA transfection efficiency. Next, we chose the pEGFP-N1 plasmid as the reporter gene to investigate the transfection efficiency in vitro and in vivo. HUVECs were transfected with PEI/pEGFP-N1, PPA/pEGFP-N1, PPT/pEGFP-N1 and PPTA/pEGFP-N1, and the expression of enhanced green fluorescent protein (EGFP) was observed by fluorescence microscopy and quantified by flow cytometry. On the basis of the expression of EGFP (Figure 8AC), we could see that the transfection efficiency of PPTA/pEGFP-N1 (33.02 ± 3.66%) in HUVECs was significantly higher than that of PEI/pEGFP-N1 (21.82 ± 3.60%) and PPA/pEGFP-N1 (26.08 ± 2.23%). PPT/pEGFP-N1 (29.90 ± 3.14%) also showed higher transfection efficiency than PEI/pEGFP-N1. We confirmed the outstanding BBB penetration and brain targeting abilities of PPTA in vivo. Next, we further evaluated the transfection efficiency of PPTA/pEGFP-N1 in orthotopic U87 glioma-bearing nude mice. As shown in Figure 8D, the expression of EGFP in glioma tissue transfected with PPTA/pEGFP-N1 was much higher than that in other groups, which further indicated the glioma targeting potency and gene delivery capacity of PPTA in vivo.
 |
Figure 8 The gene transfection efficiency of PPTA in vitro and vivo. (A) Fluorescent images of HUVECs transfected with PEI/pEGFP-N1, PPA/pEGFP-N1, PPT/pEGFP-N1 or PPTA/pEGFP-N1. Bars represent 200 μm. (B) Flow cytometric histogram of the fluorescence intensity of HUVECs. (C) Quantitative evaluation of EGFP expression by flow cytometry in HUVECs. * Indicates p<0.05 when compared with the PEI group. # indicates p<0.05 when compared with the PPA group. (D) In vivo transfection efficiency. Fluorescence images of brain sections separated from intracranial U87 glioma-bearing nude mice transfected with PEI/pEGFP-N1, PPA/pEGFP-N1, PPT/pEGFP-N1 or PPTA/pEGFP-N1. Green, EGFP protein; blue, cell nuclei stained with DAPI. Bars represent 50 μm. |
The Inhibitory Effect of PPTA/pVAXI-En on Angiogenesis and the Migration of Endothelial Cells in vitro
Before we evaluated the in vitro antiangiogenic activity, the supernatant of HUVECs was collected and the endostatin protein concentration measured by ELISA. The endostatin protein level transferred with PPTA/pVAXI-En was 1.42-fold and 1.27-fold higher than that of PEI/pVAXI-En and PPA/pVAXI-En, respectively (Figure 9A), which was consistent with the trend of the EGFP transfection experiment. The tube formation assay was applied to evaluate the antiangiogenic activity of PPTA/pVAXI-En. The tubular structure was photographed after 6 hours of incubation (Figure 9B), and the numbers of branch points were counted (Figure 9C). Our results showed that the numbers of branch points in different groups were all reduced to different degrees, but PPTA/pVAXI-En showed a smaller number of branch points than PEI/pVAXI-En and PPA/pVAXI-En. To explore the effect of PPTA/pVAXI-En on endothelial cell migration, a wound healing assay was adopted. The region between the two vertical lines reflected the migration of cells from both sides to the middle. The migration rates at 24 hours and 48 hours were calculated to show the degree of endothelial cell migration. As shown in Figure 9D and E, the migration rates of HUVECs treated with different nanocomplexes were all lower than those of the control group. The above results indicated that the secretory expression of the endostatin gene had prominent antiangiogenesis and antimigration effects on HUVECs in vitro.
 |
Figure 9 The inhibitory effect of PPTA/pVAXI-En on angiogenesis and the migration of HUVECs in vitro. (A) Expression level of the endostatin protein in the supernatant of HUVECs transfected with different nanocomplexes. * indicates p<0.05 when compared with PEI/pVAXI-En. # indicates p<0.05 when compared with PPA/pVAXI-En. (B and C) Photographs of tube formation and the quantification of branch points upon treatment with different nanocomplexes. The arrows indicate representative new blood vessel branch points. * indicates p<0.05 when compared with PEI/pVAXI-En. # Indicates p<0.05 when compared with PPA/pVAXI-En. Bars represent 50 μm. (D and E) The statistical migration rates and photographs showing the migration of HUVECs treated with different nanocomplexes. *, **, *** and **** Represent p<0.05, p<0.01, p<0.001 and p<0.0001 versus the control group, respectively. Bars represent 200 μm. |
In vivo Antiglioma Efficacy
Nude mice bearing an orthotopic U87-mCherry-luc glioma model were used for the antiglioma studies. U87-mCherry-luc cells contained the mCherry and luciferase genes. Luciferase was able to react with the D-Luciferin potassium salt and generate bioluminescence, which could be imaged by an IVIS spectrum imaging system. The growth of the glioma could be visualized by the bioluminescence intensity of U87-mCherry-luc cells at different time points. As illustrated in Figure 10A, the growth rate of bioluminescence intensity resulting from PPTA/pVAXI-En was lower than that observed for PEI/pVAXI-En, PPA/pVAXI-En and PPT/pVAXI-En. The bioluminescence intensity statistics on day 20 showed that PPTA/pVAXI-En inhibited glioma growth more effectively (Figure 10B). CD31 is a typical microvascular marker that can be used to assess microvessel density (MVD) and the degree of angiogenesis. CD31 staining results showed that PPTA/pVAXI-En significantly reduced CD31 expression and MVD in glioma tissue (Figure 10C).
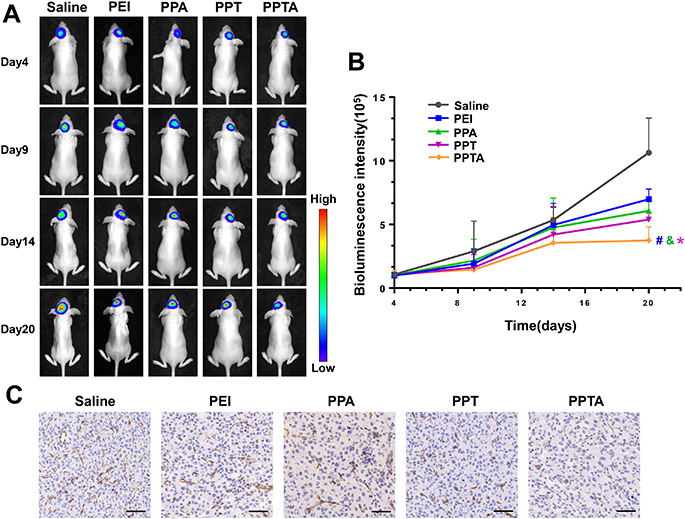 |
Figure 10 The in vivo antitumor efficacy of PPTA/pVAXI-En was evaluated in orthotopic U87-mCherry-luc glioma-bearing nude mice. (A) The growth of glioma was visualized by bioluminescence imaging on days 4, 9, 14 and 20. (B) Changes of glioma bioluminescence intensity in the brains of nude mice. * Indicates p<0.05 when compared with PEI/pVAXI-En, # Indicates p<0.05 when compared with PPA/pVAXI-En, and & indicates p<0.05 when compared with PPT/pVAXI-En. (C) Immunohistochemical staining for CD31. Bars represent 50 μm. |
Discussion
High vascularity is one of the characteristic features of malignant glioma.4 In our study, the endostatin gene was chosen to treat glioma because of its outstanding antiangiogenic ability. However, the BBB and BTB impede the transport of exogenous therapeutic genes to glioma tissue.30,31 How to enhance the specificity of the endostatin gene for new vessels in glioma and improve gene transfection efficiency are key points. Active targeted delivery systems modified by functional ligands have attracted increasing attention owing to their advantages of improved targeting ability, increased bioavailability and reduced side effects.41 Many researchers have designed a variety of dual-ligand-decorated delivery systems using a CPP and a targeting ligand for brain glioma targeting. The CPP ligand plays a role in carrying drugs across the BBB, and the targeting ligand shows affinity to specific receptors.42,43 However, the conjugation of two different functional ligands may weaken the penetrability of the CPP, the targeting ability of the ligand, or both. Therefore, it is advisable to choose two ligands with synergistic effects to maintain their respective activity.
Recently, researchers proposed and demonstrated that peptides containing a C-terminal arginine residue or internal R/KXXR/K sequence could interact with the NRP-1 and then lead to vascular internalization and tissue penetration.44 The analysis of amino acid composition indicated that there were two sequences in TAT, “RKKR” and “RQRR”, that conformed to the R/KXXR/K rules. Motivated by the above findings, we hypothesized that TAT might be able to bind to the NRP-1. The KD value calculated by SPR analysis validated the similar binding properties of TAT and AT7 with the NRP-1. The higher affinity of TAT-AT7 to the NRP-1 revealed that the connection of TAT to AT7 increased the receptor binding capacity. In addition, TAT-AT7 and AT7 showed similar affinity to VEGFR-2, which indicated that the TAT sequence did not affect the targeting ability of TAT-AT7 to VEGFR-2. Moreover, cellular uptake experiments showed that the uptake and permeability of TAT-AT7 into HUVECs overexpressing VEGFR-2 and NRP-1 were higher than those of either TAT or AT7. Most importantly, a more prominent distribution of TAT-AT7 than of TAT or AT7 was clearly observed in the brain glioma tissue of nude mice after intravenous injection. All of the above results indicated that TAT-AT7 could be a multifunctional ligand with high affinity to VEGFR-2 and NRP-1, strong BBB and BTB penetration capabilities, and glioma targeting ability due to the synergistic roles of TAT and AT7.
In our study, TAT-AT7 was conjugated to PEI through a heterobifunctional PEG crosslinker to form the dual-targeting gene delivery vector PPTA. Then, we used the secretory endostatin gene as a therapeutic gene to form a novel gene delivery system, PPTA/pVAXI-En, for the treatment of glioma. PPTA showed lower cytotoxicity than PEI to HUVECs in the MTT assay, largely because PEGylation and peptide modification could shield the partial positive charge of PEI.45,46 The intracerebral delivery of gene therapeutic agents is significantly impaired by shielding from the BBB and BTB. In vitro BBB and BTB penetration assays revealed that TAT-AT7 modification clearly enhanced the in vitro BBB and BTB penetration efficiencies of PPTA/pVAXI-En. In accordance with the above in vitro results, in vivo fluorescence distribution experiments further confirmed that TAT-AT7-modified PPTA could cross the BBB and more efficiently accumulated in the intracranial glioma tissue of nude mice. Therefore, it was deduced that PPTA/pVAXI-En penetrated the BBB and BTB mainly through the biological membrane permeability of TAT and adsorption-mediated transcytosis via electrostatic interaction of the cationic polymer with the negatively charged BBB and BTB.47,48 Futhermore, PPTA/pVAXI-En penetrated the BTB more efficiently via the specific binding of TAT-AT7 to VEGFR-2 and NRP-1, both of which were overexpressed on the endothelial cells of BTB. The interaction of R/KXXR/K sequence of TAT-AT7 with the NRP-1 also increased vascular permeability and tissue internalization.44
Successful transfection of PPTA/pVAXI-En depends on many factors, mainly including particle size, zeta potential, cell uptake and the intracellular stability of DNA. First, we explored the particle size and zeta potential of PPTA/pVAXI-En at different N/P ratios of the gene delivery system. When N/P was 12, pVAXI-En plasmid could be completely compressed by PPTA into nanoparticles, and PPTA/pVAXI-En presented the best particle size and positive charge for gene transfection. After internalization into cells, nanoparticles are usually ingested by acidic lysosomes. Therefore, the protection of plasmid DNA from lysosomal degradation is a very important factor affecting transfection efficiency. The unique “proton-sponge effect” of PEI can cause osmotic swelling of lysosomes, leading to the release nanoparticles from the lysosomes, and ultimately protect DNA from degradation.20 Our study showed that PEI/pVAXI-En, PPA/pVAXI-En, PPT/pVAXI-En and PPTA/pVAXI-En all could escape from lysosomes at 4 hours, which confirmed the protective effect of PEI on DNA.
We used the pEGFP-N1 plasmid as a reporter gene to investigate the transfection efficiency of PPTA in vitro and in vivo. The PPT and PPTA groups showed higher transfection efficiency than the PEI and PPA groups in HUVECs, which suggested that the modification of PEI with TAT-AT7 or TAT could improve membrane penetration ability and enhance gene transfection efficiency. EGFP expression in the glioma tissue of nude mice in the PPTA group was significantly higher than that in the PPT group, which further proved the heightened BBB and BTB penetrability and glioma targeting abilities of PPTA. In combination with the gene transfection and distribution experiments of PPTA in vivo, modification with TAT-AT7 was believed to be the main reason for the high transfection efficiency and active glioma targeting of PPTA.
Next, we analyzed the therapeutic effects of PPTA/pVAXI-En in vitro. Endostatin proteins in the supernatant of HUVECs were tested by ELISA, which revealed that our secretory pVAXI-En plasmid could be successfully transfected by PPTA and secreted outside HUVECs. The tube formation assay and wound healing assay demonstrated that PPTA/pVAXI-En could inhibit the tube formation and migration of HUVECs. The antiangiogenic activity of both PPT/pVAXI-En and PPTA/pVAXI-En was remarkable. All groups exhibited obvious antimigration activity at 48 hours. In principle, the higher the transfection efficiency is, the stronger the antimigration activity. There was no significant difference in the antimigration activity among the PEI, PPA, PPT and PPTA groups. We suspected the reason might be that the antimigration effects on HUVECs could be exhibited clearly over a certain concentration expression level. In vivo pharmacodynamics analysis revealed that the secretory endostatin gene pVAXI-En plasmid could be a promising therapeutic agent for the treatment of glioma, and PPTA/pVAXI-En achieved more effective inhibition of glioma growth and angiogenesis than the other groups, including saline, PEI/pVAXI-En, PPA/pVAXI-En and PPT/pVAXI-En, in orthotopic U87 glioma-bearing nude mice.
Conclusion
In summary, we successfully developed a novel dual targeting TAT-AT7-modified PPTA/pVAXI-En nanocomplex for secretory endostatin gene delivery to treat glioma. PPTA/pVAXI-En exhibited enhanced gene transfection efficiency, increased BBB and BTB penetration, and better glioma targeting ability, as well as superior inhibitory effects on angiogenesis and glioma growth in an orthotopic U87-bearing nude mice model. Our results not only demonstrate that TAT-AT7 is expected to be a promising multifunctional ligand to mediate nanocomplex to overcome multiple biological barriers and precisely target glioma tissue but also suggest PPTA/pVAXI-En as a potential gene delivery nanocomplex in glioma therapy. The current findings can provide insight into the application of a nonviral targeted drug delivery system for noninvasive gene therapy of glioma.
Funding
This work was supported by the National Natural Science Foundation of China grant [grant numbers 81302686]; the Primary Research & Development Plan of Shandong Province [grant number 2016GSF201083]; and the Major Project of Science and Technology of Shandong Province [grant number 2015ZDJS04001].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Prim. 2015;1:15017.
2. Stupp R, Brada M, Van Den Bent MJ, et al. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25:iii93iii101. doi:10.1093/annonc/mdu050
3. Alifieris C, Trafalis DT. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. doi:10.1016/j.pharmthera.2015.05.005
4. Da Ponte KF, Berro DH, Collet S, et al. Guillamo, In Vivo Relationship between Hypoxia and Angiogenesis in Human Glioblastoma: A Multimodal Imaging Study. J Nucl Med. 2017;58:1574–1579.
5. Overchuk M, Zheng G. Overcoming obstacles in the tumor microenvironment: recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials. 2018;156:217–237. doi:10.1016/j.biomaterials.2017.10.024
6. Kane JR, Miska J, Young JS, et al. Sui generis: gene therapy and delivery systems for the treatment of glioblastoma. Neuro Oncol. 2015;17:ii24ii36. doi:10.1093/neuonc/nou355
7. O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi:10.1016/S0092-8674(00)81848-6
8. Abdollahi A, Hlatky L, Huber PE. Endostatin: the logic of antiangiogenic therapy. Drug Resist Updat. 2005;8:59–74.
9. Poluzzi C, Iozzo RV, Schaefer L. Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev. 2016;97:156–173. doi:10.1016/j.addr.2015.10.012
10. Blezinger P, Wang J, Gondo M, et al. Systemic inhibition of tumor growth and tumor metastases by intramuscular administration of the endostatin gene. Nat Biotechnol. 1999;17:343. doi:10.1038/7895
11. Zhou Y, Gu H, Xu Y, et al. Targeted antiangiogenesis gene therapy using targeted cationic microbubbles conjugated with cd105 antibody compared with untargeted cationic and neutral microbubbles. Theranostics. 2015;5:399. doi:10.7150/thno.10351
12. Cui C, Mao L, Chi Z, et al. A Phase II, randomized, double-blind, placebo-controlled multicenter trial of endostar in patients with metastatic melanoma. Mol Ther. 2013;21:1456–1463. doi:10.1038/mt.2013.79
13. Morimoto T, Aoyagi M, Tamaki M, et al. Increased levels of tissue endostatin in human malignant gliomas. Clin Cancer Res. 2002;8:2933–2938.
14. Yang L, Lin Z, Lin J, et al. Antitumor effect of endostatin overexpressed in C6 glioma cells is associated with the down-regulation of VEGF. Int J Oncol. 2011;38:465–471.
15. Zhang X, Chen H, Yang Y, et al. Development and evaluation of chitosan gene nanoparticles loaded with recombinant plasmid PVAX1/Tat PTD-Endostatin. Shandong Univenity. 2017;55:49–54.
16. Zhang Y, Ma C, Sun W. Construction of recombinant plasmid with human endostatin CDNA in high-biosafety. Shandong Univenity. 2004;5.
17. Zhou Z, Liu X, Zhu D, et al. Nonviral cancer gene therapy: delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev. 2017;115:115–154. doi:10.1016/j.addr.2017.07.021
18. Hall A, Lächelt U, Bartek J, et al. Polyplex evolution: understanding biology, optimizing performance. Mol Ther. 2017;25:1476–1490. doi:10.1016/j.ymthe.2017.01.024
19. Choi E, Oh J, Lee D, et al. A ternary-complex of a suicide gene, a RAGE-binding peptide, and polyethylenimine as a gene delivery system with anti-tumor and anti-angiogenic dual effects in glioblastoma. J Control Release. 2018;279:40–52. doi:10.1016/j.jconrel.2018.04.021
20. Zhang Y, Tan X, Ren T, et al. Folate-modified carboxymethyl-chitosan/polyethylenimine/bovine serum albumin based complexes for tumor site-specific drug delivery. Carbohyd Polym. 2018;198:76–85. doi:10.1016/j.carbpol.2018.06.055
21. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611.
22. Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov Today. 2013;18:447–455. doi:10.1016/j.drudis.2012.11.013
23. Roth L, Prahst C, Ruckdeschel T, et al. Neuropilin-1 mediates vascular permeability independently of vascular endothelial growth factor receptor-2 activation. Sci Signal. 2016;9:ra42. doi:10.1126/scisignal.aad3812
24. Fantin A, Lampropoulou A, Senatore V, et al. VEGF165-induced vascular permeability requires NRP1 for ABL-mediated SRC family kinase activation. J Exp Med. 2017;214:1049–1064. doi:10.1084/jem.20160311
25. Binétruy-Tournaire R, Demangel C, Malavaud B, et al. Identification of a peptide blocking vascular endothelial growth factor (VEGF)‐mediated Angiogenesis. EMBO J. 2000;19:1525–1533.
26. Tirand L, Frochot C, Vanderesse R, et al. A peptide competing with VEGF165 binding on neuropilin-1 mediates targeting of a chlorin-type photosensitizer and potentiates its photodynamic activity in human endothelial cells. J Control Release. 2006;111:153–164. doi:10.1016/j.jconrel.2005.11.017
27. Ying M, Shen Q, Zhan C, et al. A stabilized peptide ligand for multifunctional glioma targeted drug delivery. J Control Release. 2016;243:86–98. doi:10.1016/j.jconrel.2016.09.035
28. Hu Q, Kang T, Feng J, et al. Tumor microenvironment and angiogenic blood vessels dual-targeting for enhanced anti-glioma therapy. ACS Appl Mater Interfaces. 2016;8:23568–23579. doi:10.1021/acsami.6b08239
29. Zhang Y, Zhai M, Chen Z, et al. Dual-modified liposome codelivery of doxorubicin and vincristine improve targeting and therapeutic efficacy of glioma. Drug Deliv. 2017;24:1045–1055. doi:10.1080/10717544.2017.1344334
30. Xie J, Shen Z, Anraku Y, et al. Nanomaterial-Based Blood-Brain-Barrier (BBB) crossing strategies. Biomaterials. 2019;224:119491. doi:10.1016/j.biomaterials.2019.119491
31. Van Tellingen O, Yetkin-Arik B, De Gooijer MC, et al. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1–12. doi:10.1016/j.drup.2015.02.002
32. Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406–424. doi:10.1016/j.tips.2017.01.003
33. Kim T, Braun GB, She Z, et al. Composite porous silicon-silver nanoparticles as theranostic antibacterial agents. ACS Appl Mater Interfaces. 2016;8:30449–30457. doi:10.1021/acsami.6b09518
34. Liu L, Guo K, Lu J, et al. Biologically active core/shell nanoparticles self-assembled from cholesterol-terminated PEG-TAT for drug delivery across the blood-brain barrier. Biomaterials. 2008;29:1509–1517. doi:10.1016/j.biomaterials.2007.11.014
35. Han B, Xie W, Zhang Y, et al. The influx/efflux mechanisms of d-peptide ligand of nAChRs across the blood-brain barrier and its therapeutic value in treating glioma. J Control Release. 2020;327:384–396. doi:10.1016/j.jconrel.2020.08.010
36. Wang X, Yin S, Li M, et al. Autophagy inhibition changes the disposition of non-viral gene carriers during blood-brain barrier penetration and enhances TRAIL-induced apoptosis in brain metastatic tumor. J Control Release. 2020;321:497–508. doi:10.1016/j.jconrel.2020.02.042
37. Ran D, Mao J, Zhan C, et al. d-retroenantiomer of quorum-sensing peptide-modified polymeric micelles for brain tumor-targeted drug delivery. ACS Appl Mater Interfaces. 2017;9:25672–25682. doi:10.1021/acsami.7b03518
38. Evans CW, Fitzgerald M, Clemons TD, et al. Multimodal analysis of pei-mediated endocytosis of nanoparticles in neural cells. ACS Nano. 2011;5:8640–8648.
39. Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496–504. doi:10.1021/mp800049w
40. Khodarev NN, Yu J, Labay E, et al. Tumour-endothelium interactions in co-culture: coordinated changes of gene expression profiles and phenotypic properties of endothelial cells. J Cell Sci. 2003;116:1013–1022. doi:10.1242/jcs.00281
41. Srinivasarao M, Low PS. Ligand-targeted drug delivery. Chem Rev. 2017;117:12133–12164. doi:10.1021/acs.chemrev.7b00013
42. Liu Y, Ran R, Chen J, et al. Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics. 2016;6:177. doi:10.7150/thno.13532
43. Zhu Y, Jiang Y, Meng F, et al. Highly efficacious and specific anti-glioma chemotherapy by tandem nanomicelles co-functionalized with brain tumor-targeting and cell-penetrating peptides. J Control Release. 2018;278:1–8. doi:10.1016/j.jconrel.2018.03.025
44. Teesalu T, Sugahara KN, Kotamraju VR, et al. C-End rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci. 2009;106:16157–16162. doi:10.1073/pnas.0908201106
45. Kievit FM, Veiseh O, Bhattarai N, et al. PEI-PEG-chitosan-copolymer-coated iron oxide nanoparticles for safe gene delivery: synthesis, complexation, and transfection. Adv Funct Mater. 2009;19:2244–2251. doi:10.1002/adfm.200801844
46. Guan X, Guo Z, Lin L, et al. Ultrasensitive ph triggered charge/size dual-rebound gene delivery system. Nano Lett. 2016;16:6823–6831. doi:10.1021/acs.nanolett.6b02536
47. Broadwell RD. Transcytosis of macromolecules through the blood-brain barrier: a cell biological perspective and critical appraisal. Acta Neuropathol. 1989;79:117–128. doi:10.1007/BF00294368
48. Qin Y, Chen H, Yuan W, et al. Liposome formulated with TAT-modified cholesterol for enhancing the brain delivery. Int J Pharm. 2011;419:85–95. doi:10.1016/j.ijpharm.2011.07.021
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.