")
Back to Journals » Journal of Blood Medicine » Volume 12
A Deep Dive into the Pathology of Gray Platelet Syndrome: New Insights on Immune Dysregulation
Authors Glembotsky AC, De Luca G, Heller PG
Received 26 March 2021
Accepted for publication 16 June 2021
Published 11 August 2021 Volume 2021:12 Pages 719—732
DOI https://doi.org/10.2147/JBM.S270018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ana C Glembotsky,1,2 Geraldine De Luca,1,2 Paula G Heller1,2
1Departamento Hematología Investigación, Instituto de Investigaciones Médicas “Dr. A. Lanari”, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina; 2Departamento Hematología Investigación, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Universidad de Buenos Aires, Instituto de Investigaciones Médicas (IDIM), Buenos Aires, Argentina
Correspondence: Paula G Heller
Departamento Hematología Investigación,Instituto de Investigaciones Médicas “Dr. A. Lanari”, Facultad de Medicina, Universidad de Buenos Aires, Combatientes de Malvinas 3150, Buenos Aires, 1427, Argentina
Tel +54 11 5287 3872
Email [email protected]
Abstract: The gray platelet syndrome (GPS) is a rare platelet disorder, characterized by impaired alpha-granule biogenesis in megakaryocytes and platelets due to NBEAL2 mutations. Typical clinical features include macrothrombocytopenia, bleeding and elevated vitamin B12 levels, while bone marrow fibrosis and splenomegaly may develop during disease progression. Recently, the involvement of other blood lineages has been highlighted, revealing the role of NBEAL2 outside the megakaryocyte-platelet axis. Low leukocyte counts, decreased neutrophil granulation and impaired neutrophil extracellular trap formation represent prominent findings in GPS patients, reflecting deranged innate immunity and associated with an increased susceptibility to infection. In addition, low numbers and impaired degranulation of NK cells have been demonstrated in animal models. Autoimmune diseases involving different organs and a spectrum of autoantibodies are present in a substantial proportion of GPS patients, expanding the syndromic spectrum of this disorder and pointing to dysregulation of the adaptive immune response. Low-grade inflammation, as evidenced by elevation of liver-derived acute-phase reactants, is another previously unrecognized feature of GPS which may contribute to disease manifestations. This review will focus on the mechanisms underlying the pathogenesis of blood cell abnormalities in human GPS patients and NBEAL2-null animal models, providing insight into the effects of NBEAL2 in hemostasis, inflammation and immunity.
Keywords: NBEAL2, gray platelet syndrome, α-granules, immune dysregulation, neutrophils
The gray platelet syndrome (GPS) (OMIM #139090) is a rare autosomal recessive disorder characterized by moderate macrothrombocytopenia, marked decrease or absence of platelet alpha (α)-granules, which gives platelets a grayish appearance on the blood smear, and mild to moderate bleeding.1 Some patients have splenomegaly and develop bone marrow fibrosis during disease progression. First described in 1979,2 the genetic abnormality leading to GPS remained unknown for a long time. Thirty years later, using linkage analysis, the underlying molecular defect was mapped to chromosome 3p21,3,4 and shortly thereafter, mutations in NBEAL2 were recognized as the cause of GPS by three independent groups by using different approaches.5–7 NBEAL2 is involved in the biogenesis of megakaryocyte and platelet α-granules, although its role beyond hemostasis, particularly in immunity and inflammation, has been recently highlighted by Sims et al in a landmark international study.8 This review will focus on the mechanisms underlying abnormalities in the megakaryocyte-platelet axis and the immune system in GPS patients and Nbeal2-null animal models.
Spectrum of NBEAL2 Mutations
NBEAL2 is a large gene with 54 exons located on chromosome 3p21 encoding Neurobeachin-like 2, a scaffolding protein composed of 2754 amino acids.9 NBEAL2 belongs to a family of proteins known as BEACH (beige and Chediak-Higashi syndrome)–domain-containing proteins (BDCP), containing armadillo (ARM)-like, Concanavalin A-like lectin, pleckstrin homology (PH), BEACH and WD40 domains. These domains represent highly conserved regions that are crucial for protein–protein interactions, membrane dynamics, vesicle trafficking, lysosome size regulation and synaptosome formation.9 It is expressed in hematopoietic cells, including megakaryocytes, platelets, monocytes, neutrophils and NK cells.
Eighty-six different NBEAL2 variants have been detected in 69 GPS pedigrees described to date, harbouring homozygous (65%) or compound heterozygous (35%) mutations.5–8,10−19 Genetic defects include frameshift, nonsense, missense, splicing and small indel variants,16 which are scattered along the gene. The spectrum of NBEAL2 variants is depicted in Figure 1 and listed in Table 1. Most of them are private mutations, while some are found in more than one pedigree. Although no mutational hotspots have been recognized, missense variants are enriched within the BEACH domain, consistent with the essential role of this region in NBEAL2 function.8 No genotype-phenotype correlation has been demonstrated, either regarding the type of mutation or its location.8 In some pedigrees, three different variants have been identified and the pathogenic role of each one of them is not clear.6,8,10,18 Current gene curation efforts for platelet disorders, such as those carried out recently for Glanzmann thrombasthenia,20 will contribute to accurate interpretation of variants. Heterozygous NBEAL2 variants may be found in unaffected carriers, who may display a mild decrease in α-granule content, although they are not thrombocytopenic.10 Genetic diagnosis may be approached by Sanger sequencing of the NBEAL2 gene, when the diagnosis of GPS is suspected based on the phenotypic features, or by high-throughput sequencing, including gene panels for inherited platelet disorders19 or whole-exome sequencing.17
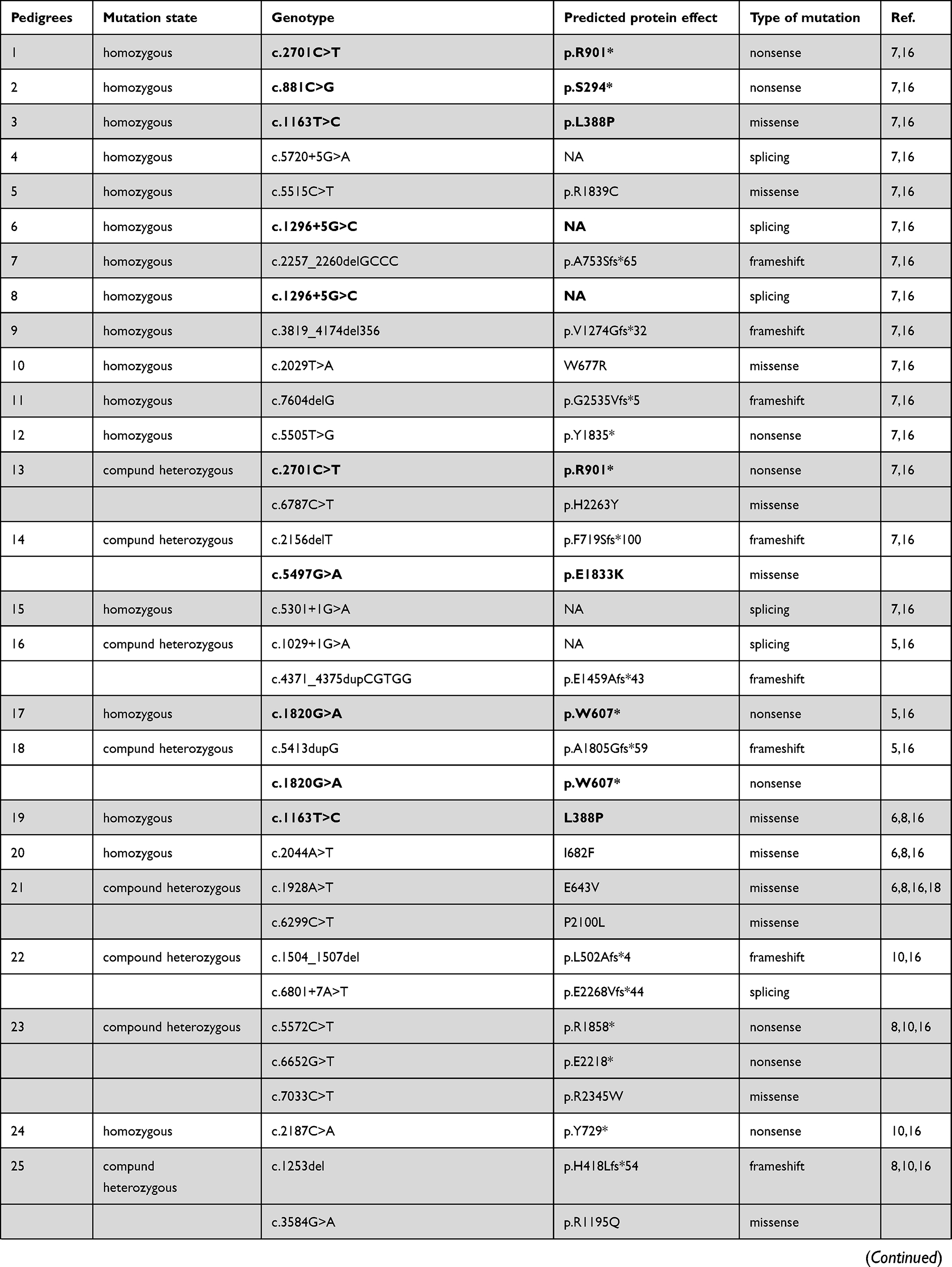 | 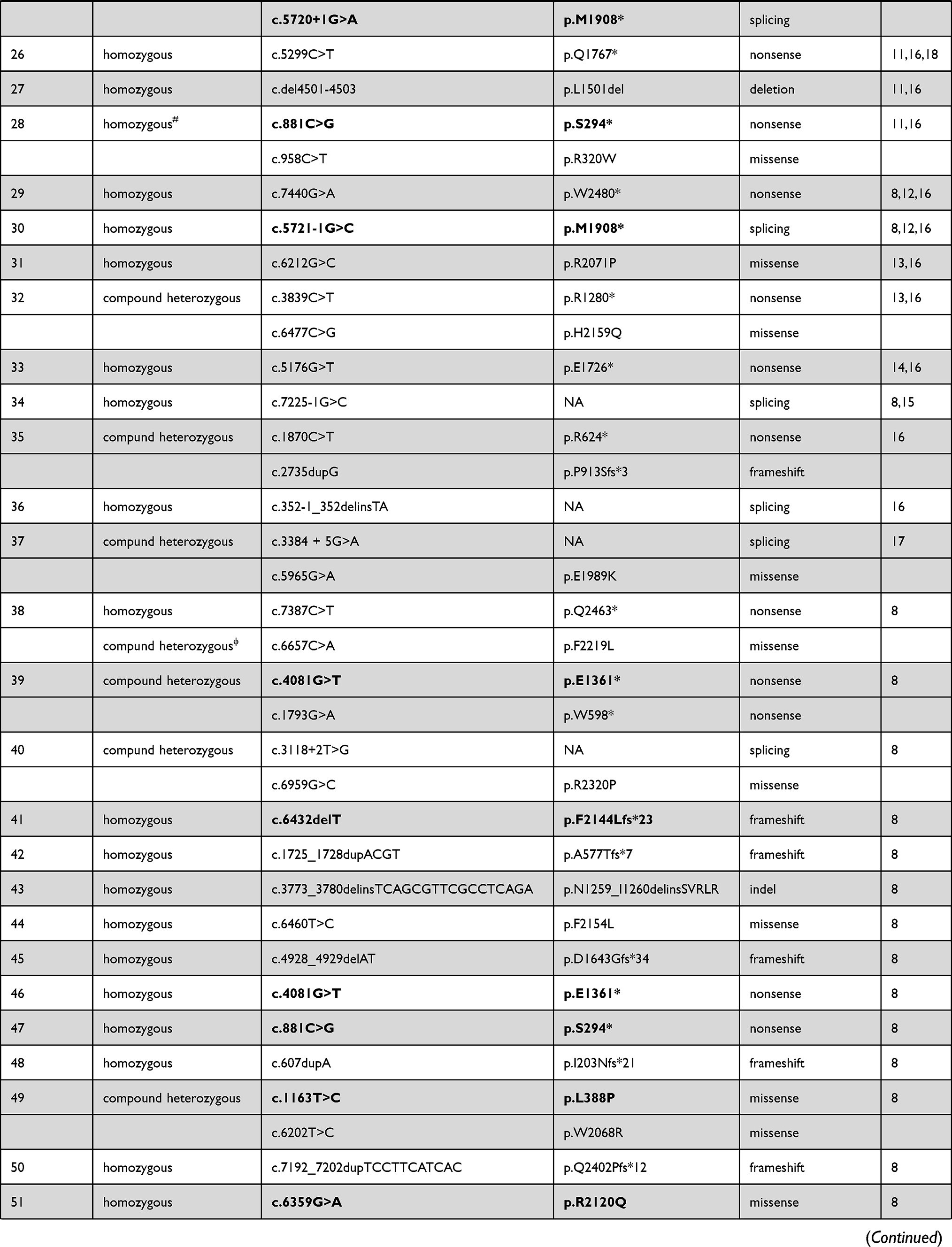 | 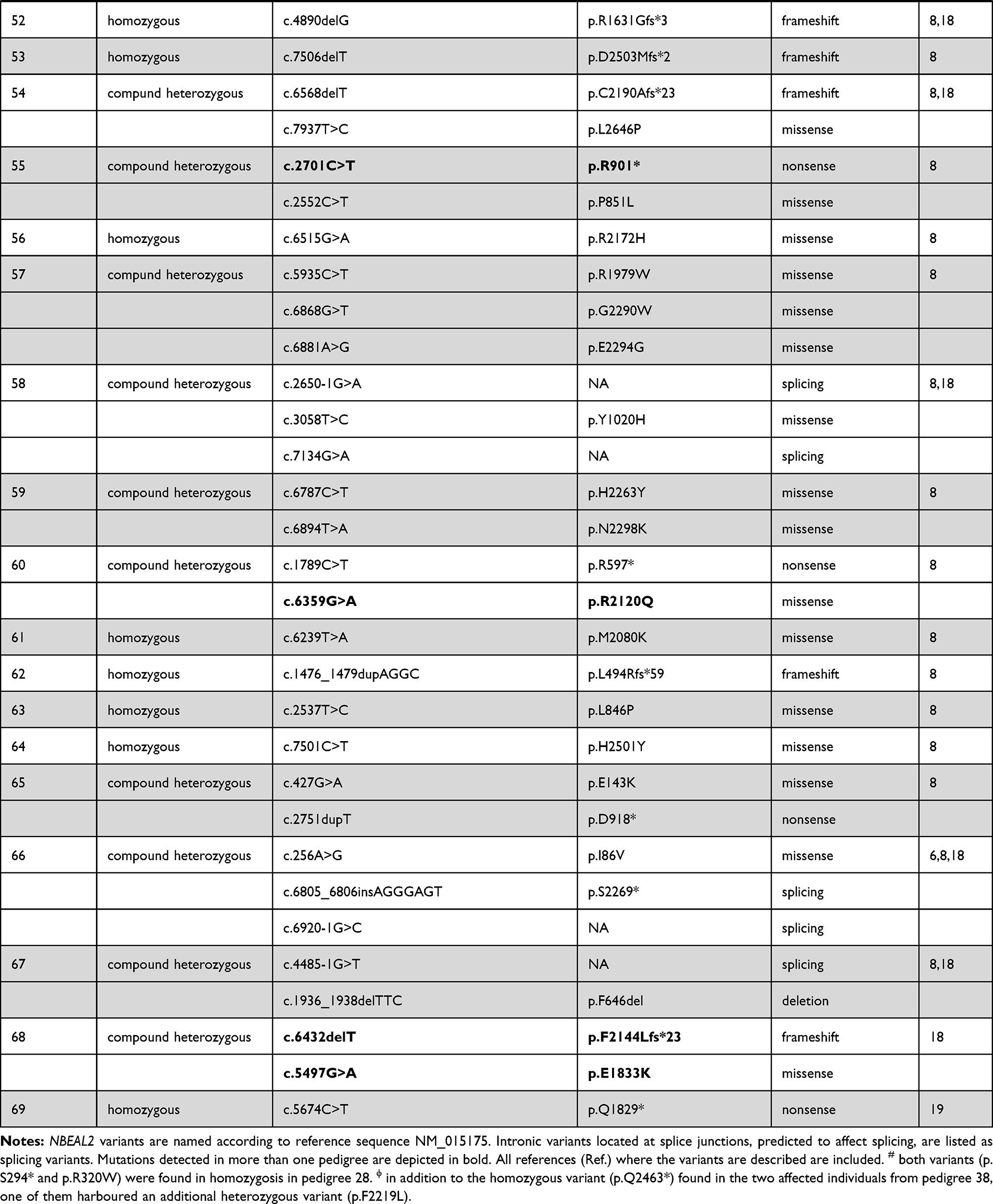 |  |
Table 1 Published NBEAL2 Variants in GPS Patients |
Alpha-Granule Deficiency in GPS Platelets and Megakaryocytes
Alpha-granule deficiency is a constant feature of GPS. Electron microscopy remains the gold standard for diagnosis. Platelets characteristically show absent or markedly reduced α-granules and frequently display prominent vacuolization of the cytoplasm, whereas the content of dense granules, lysosomes, mitochondria and peroxisomes is preserved.21 The content of α-granules may also be quantified by immunofluorescence staining of blood smears for α-granule proteins, such as thrombospondin-1 (TSP1) and platelet factor 4 (PF4), as described.10,22,23 The finding of a severe reduction in α-granules by this method correlates with the results of ultrastructural analysis.
Although GPS platelets lack typical α-granules, two types of abnormal α-granules may be recognized by immune electron microscopy, including normal-sized granules almost devoid of content with scant von Willebrand (vWF)-positive material and very small vestigious α-granule-like vesicular structures with intense vWF staining.24 In contrast to the ARC (arthrogryposis, renal dysfunction, and cholestasis) syndrome, due to mutations in VPS33B or VP16B, in which platelets lack α-granules and their membranes, the granule-limiting membranes are present in GPS platelets, as well as membrane proteins, such as P-selectin and GMP-140.25–28 These findings indicate that, whereas defects in VPS33B and VPS16B completely abrogate α-granule formation, lack of NBEAL2 seems to interfere with α-granule maturation and cargo loading and retention, reflecting that NBEAL2 acts at a later stage of α-granule development.
As recently reviewed,29 α-granules are formed by fusion of vesicles budding from the trans-Golgi network (loaded with endogenous cargo) or the cell membrane (early endosomes carrying exogenous cargo), which are directed to multivesicular bodies that represent α-granule precursors. Whereas α-granule proteins synthesized in the megakaryocyte, such as platelet factor 4 (PF4), vWF, β-thromboglobulin, thrombospondin, platelet-derived growth factor (PDGF) and fibronectin, are markedly reduced in GPS platelets, those routed to α-granules following endocytosis of plasma proteins, such as albumin, IgG and fibrinogen, are less affected,26 indicating that NBEAL2 loss of function preferentially affects the delivery of endogenously synthesized cargo to α-granules. Interestingly, analysis of the platelet proteome revealed that, in addition to downregulation of α-granule content, unexpectedly, proteins normally resident in neutrophil granules, including myeloperoxidase and elastase, were overrepresented.8 Although there is no clear explanation for this finding, the transfer of neutrophil proteins during emperipolesis, which occurs in a high proportion of GPS megakaryocytes, may represent a plausible mechanism.8
Similar to platelet findings, ultrastructural analysis of bone marrow or cultured GPS megakaryocytes disclosed the absence of typical α-granules, although very small granules, consistent with immature α-granule precursors, were evident.24,28 In vitro differentiation of GPS primary megakaryocytes further revealed that vWF is abundantly synthesized but fails to be correctly packaged into granules and is misdirected or discharged into the lumen of the demarcation membrane system. On the contrary, P-selectin was immunodetected in multivesicular bodies in the maturing megakaryocyte and in the membrane of vacuolar structures deprived of soluble content resembling empty α-granules.25 Altogether, these findings support the notion that NBEAL2 deficiency leads to a defect in the transfer of protein cargo into the lumen of developing α-granules or in the retention of granule content, rather than a primary impairment in α-granule biosynthesis.
Nbeal2 knockout mouse models have been generated by three different groups and all reveal a platelet phenotype which resembles human GPS.30–32 Ultrastructural analysis of megakaryocytes disclosed greatly reduced numbers of α-granules in two of these studies.30,32 Intracellular tracking via fluorescence microscopy showed that initial trafficking of endocytosed proteins was preserved, but they were subsequently delivered to recycling endosomes and released, and the same fate was demonstrated for endogenous proteins, reinforcing that, as shown in humans, the main function NBEAL2 in murine cells relies on the retention of molecular cargo, possibly through its interaction with P-selectin.33 In contrast to the absence of megakaryocyte α-granules found in the two former mouse strains, Guerrero et al showed that mature α-granules were present in megakaryocytes in their Nbeal2-/- murine model, as shown by electron microscopy, immunostaining for vWF and Western blot analysis of α-granule proteins.31 Furthermore, normal numbers of granules were also evident in proplatelet buds, indicating preserved granule transport along the proplatelet shaft, further suggesting that α-granules are generated, but not retained within megakaryocytes.31
Role of NBEAL2 in α-Granule Biogenesis
The intrinsic mechanisms and interacting proteins underlying the role of NBEAL2 in α-granule biogenesis are beginning to be revealed.29 NBEAL2 interactome of human embryonic kidney cells, engineered to express a subdomain of NBEAL2 containing the BEACH domain and its flanking PH and WH40 regions, disclosed several interactors, including the endoplasmic reticulum-associated protein SEC16a, the phosphatidyl inositol 3,5-biphosphate regulator VAC14, and the guanidine exchange factor DOCK7.34 Considering that SEC16a participates in the transport from the endoplasmic reticulum to the Golgi and VAC14 activates the PIKfyve complex involved in the intracellular vesicle transport, the defect in both molecules could plausibly alter endosomal trafficking and cargo sorting during α-granule formation. DOCK7 is a guanine nucleotide exchange factor for small GTPases, Rac1 and Cdc42, which are major regulators of the actin cytoskeleton. Interestingly, DOCK7-deficient mice present with a bleeding phenotype.27,35 DOCK7 expression is decreased in GPS patients and mouse models, and Nbeal2-null mouse platelets show decreased actin polymerization and spreading on fibrinogen, indicating that NBEAL2 is important for cytoskeleton reorganization in platelets, most probably by modulating DOCK7.34 In addition, dysregulation of the DOCK7 signaling pathway in megakaryocytes could be a possible cause of aberrant platelet formation in GPS.34 These findings point out that NBEAL2 function may not be entirely dependent on its ability to regulate granule biogenesis, but that other cellular processes may be affected by NBEAL2 deficiency. Another NBEAL2 interacting protein was revealed in a recent study performed on human megakaryocytes and immortalized megakaryocyte progenitors, which demonstrated that NBEAL2 binds SEC22B, an endoplasmic reticulum-resident trafficking protein.36 Knockout of SEC22B in these cells resulted in the failure of α-granule synthesis, suggesting that interaction with SEC22B may facilitate the role of NBEAL2 in α-granule generation.36 SEC22B binding was mapped to a region located in an N-terminal position relative to the PH-BEACH domains and was abrogated by two GPS missense variants located within this region.36 Future research may provide further insight into the process of α-granule biogenesis and the exact place of NBEAL2 in this complex network.
Platelet Features
Thrombocytopenia is a universal feature of GPS, being of moderate degree in most patients. In a recent study performed by Sims et al, which included the largest GPS cohort gathered to date, comprising 47 patients belonging to 38 pedigrees recruited worldwide, median platelet count was 57 (28–105) x 109/L.8 Notably, hemoglobin, total leukocyte counts, as well as neutrophils, eosinophils, basophils, monocytes and lymphocyte counts, were all shown to be lower in this GPS cohort compared to more than 45,000 healthy controls recruited to the INTERVAL trial,8 indicating that abnormal blood parameters are not limited to platelets, as previously acknowledged, but involve multiple hematopoietic lineages. Thrombocytopenia may worsen with increasing age,3 reflecting the progressive nature of this disorder. The sequential drop in platelet counts may be due to the development of bone marrow fibrosis or, alternatively to splenomegaly and splenic sequestration. The finding of an inverse relationship between platelet counts and bone marrow fibrosis but not with splenomegaly supports the former possibility.8 Consistent with this notion, splenectomy may lead to mild increase but not complete restoration of platelet counts.
The finding of pale large and giant platelets in the peripheral blood smear is a hallmark feature of GPS, as shown in Figure 2, and often the clue to diagnosis, although other platelet disorders associated with α-granule deficiency, such as those associated with mutations in transcription factors GATA-1 or GFI1B or the ARC syndrome, which is caused by VPS33B or VPS16B mutations, must also be considered.37 Mean platelet volume is usually increased, although this parameter may be underestimated or may not be informed by electronic cell counters, as described for other large platelet disorders. In such cases, measurement of the platelet diameter by image analysis of blood smears may provide useful information, showing increased mean values and/or increased proportion of large platelets in GPS patients.23,38 The bleeding diathesis is usually mild to moderate but may range from absent to severe intracranial hemorrhage.1,16
Significantly impaired response to different agonists such as thrombin and collagen, variable response to ADP and ristocetin and normal response to arachidonic acid were demonstrated in platelet aggregation studies,1,14,39 although aggregation responses may be variable among different patients.1 The expression of platelet glycoproteins was normal in most of the patients described,39 although higher fluorescence intensity for GPIb and GPIIbIIIa due to the large size of the platelets may also be found,14 whereas loss of GPVI was described in one patient.40 Interestingly, impaired platelet response to thrombin, protease-activated receptor 1 (PAR1)- and PAR4-AP (activating peptide) was a consistent finding in several studies1,22,39 and seems to be a common defect in GPS patients, which may be paralleled by a reduction in PAR1 and PAR4 expression in megakaryocytes and platelets.22,39 Although the total P-selectin pool may be normal or mildly reduced in GPS platelets,1 P-selectin was found to be constitutively expressed on the platelet surface in one study,18 whereas upregulation of P-selectin exposure in response to agonists, was markedly impaired, both in this work18 and in a separate study.39
Mice lacking NBEAL2 display platelet functional abnormalities,30–32 delayed arterial thrombus formation30 and protection from thrombo-inflammatory brain injury following focal cerebral ischemia.32 In addition, impaired dermal healing due to reduced transforming growth factor (TGF)-β release from mutant platelets32 and protection against cancer metastasis31 were other remarkable findings, pointing to the relevance of platelet α-granule content beyond hemostasis, specifically in wound healing, tumorigenesis and metastatic spread.
Megakaryocyte Abnormalities
The pathogenesis of low platelet counts in GPS has been attributed to impaired platelet production, as platelet survival is largely normal or only mildly decreased. To address the underlying mechanisms, megakaryocytes were cultured from peripheral blood and/or bone marrow hematopoietic progenitors from four GPS patients.41 Both megakaryocyte output and maturation were normal but proplatelet formation was markedly impaired and proplatelets showed aberrant architecture with decreased branching leading to lower number of tips, indicating that thrombocytopenia is due to defective thrombopoiesis and platelet release. Interestingly, proplatelet tips were larger than control, which is consistent with the finding of large platelets in circulation. In addition, abnormal megakaryocyte interaction with extracellular matrix also shown in this work may also lead to reduced platelet production.41 As mentioned before, bone marrow fibrosis, and, to a lesser extent, splenic sequestration may also contribute to low platelet counts. The study of megakaryocyte biology in Nbeal2 knockout mouse models has revealed discrepant findings. Kahr et al showed altered development, polyploidization and proplatelet formation,30 whereas Depperman et al and Guerrero et al showed normal megakaryocyte development and proplatelet formation.31,32 Different megakaryocyte source (bone marrow vs fetal liver) or culture techniques might account for the differences found between these animal models.
Extensive emperipolesis is a striking feature of GPS megakaryocytes and represents a prominent finding both in GPS patients and animal models.8,30–32,39 Engulfed neutrophils are frequently found inside megakaryocytes in bone marrow biopsies. Strikingly, in vitro-differentiated megakaryocyte also showed signs of emperipolesis, indicating that this is an intrinsic cellular defect and not a consequence of altered bone marrow milieu.41 It has been proposed that mislocalization of P-selectin at the megakaryocyte surface in GPS patients fosters interaction with neutrophils, which express the PSGL counterligand, promoting emperipolesis.39 In addition, Nbeal2-/- mouse megakaryocytes show a proinflammatory gene expression profile, reflected by upregulation of several chemokines with a role in leukocyte chemotaxis. Paracrine secretion of these mediators by inflammatory megakaryocytes may attract neighbouring leukocytes favouring emperipolesis.31
GPS patients may develop bone marrow fibrosis during follow-up. In a recent study, myelofibrosis was found in 58% of patients at a median age of 28.5 years (range, 10–52 years).8 Although it may remain stable for several years, in some patients increased fiber deposition may occur during follow-up, but it seldom requires treatment. Leakage of α-granule-derived fibroblast growth factors, such as PDGF and TGFβ, to the bone marrow milieu represents a likely explanation. In addition, proinflammatory megakaryocytes may promote bone marrow fibrosis by secreting cytokines and chemokines to the surrounding environment,31 highlighting the importance of inflammation in the pathogenesis of myelofibrosis, as shown for myeloproliferative neoplasms.42 Splenomegaly may occur in patients with bone marrow fibrosis, although it may also be found in patients without fibrosis.8
Innate Immune Cell Dysfunction
Although GPS has long been considered to involve exclusively the megakaryocyte-platelet lineage, more recent evidence has revealed that the innate and adaptive immune system may also be affected by NBEAL2 loss of function, as detailed in Table 2 and depicted in Figure 3, thus broadening the phenotypic spectrum of GPS abnormalities.8,25
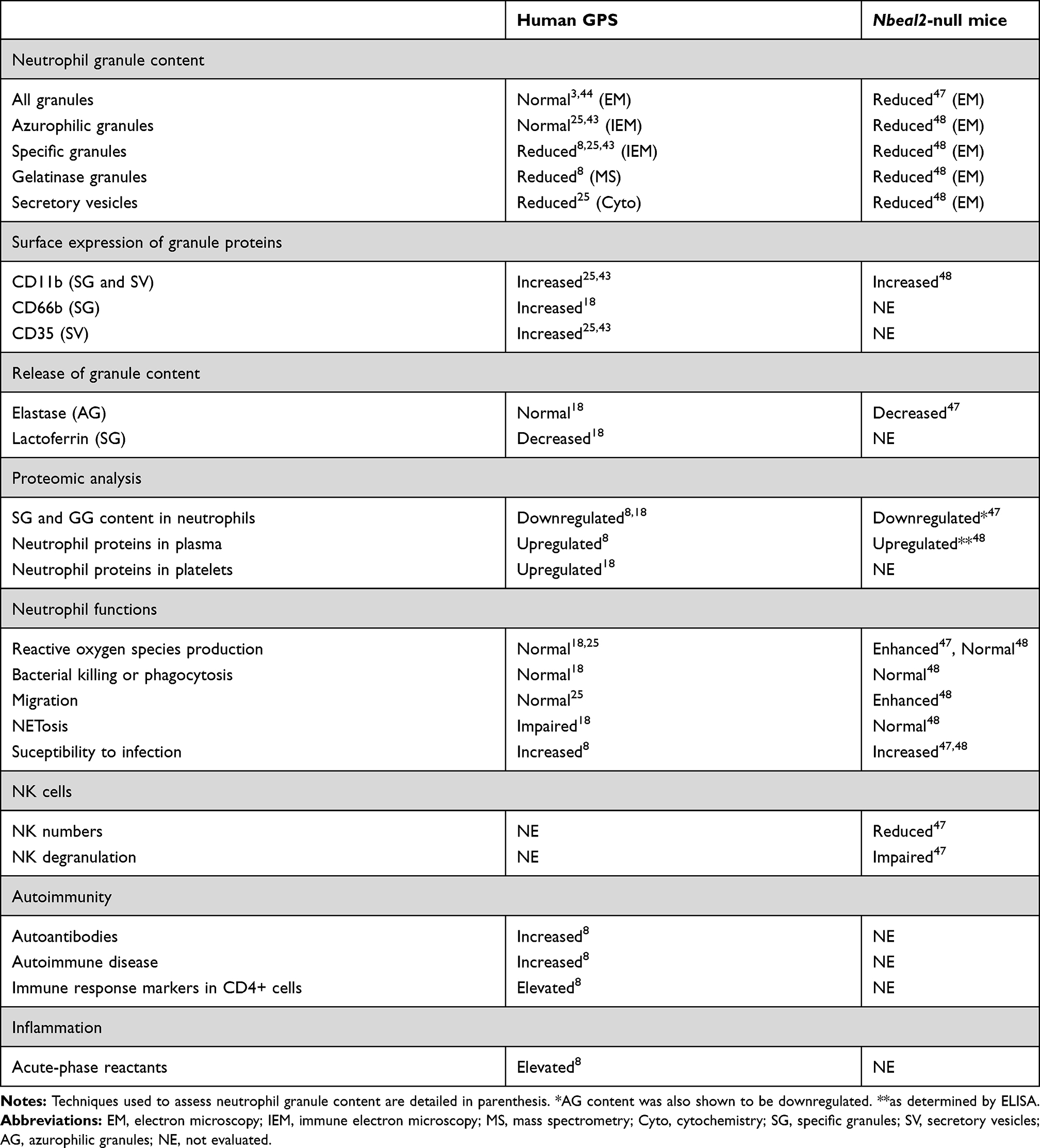 |
Table 2 Abnormalities in Innate and Acquired Immunity in GPS Patients and Animal Models |
The presence of granule defects in neutrophils, in addition to platelets, has been suggested several years ago by the group of Elisabeth Cramer,25 who described four patients form two different pedigrees displaying degranulated neutrophils with a grayish appearance on the blood smear. Reduced amounts of specific granules and secretory vesicles were shown by immune electron microscopy, whereas azurophilic granules, which contain neutrophil proteases such as elastase and myeloperoxidase, were preserved. Increased expression of granule membrane proteins, such as CD11b (specific granules) and CD35 (secretory vesicles) was shown at the cell surface of resting neutrophils, suggesting degranulation. However, despite these structural defects, no functional abnormalities were evident, as demonstrated by normal production of reactive oxygen species and neutrophil migration in response to stimuli. The same group then confirmed the presence of the same neutrophil abnormalities in a patient from an unrelated pedigree.43 In contrast to these findings, no defect in the overall number of neutrophil granules was shown by electron microscopy in GPS patients in two other studies.3,44
In the study by Sims et al, neutrophil granularity, as assessed by side scatter properties, was reduced and proteomic analysis revealed selective downregulation of proteins contained in specific granules,8 reinforcing previous findings from the French group,25 and disclosed a concomitant decrease in the content of gelatinase granule constituents.8 Reduced protein abundance was not due to decreased gene expression, as molecules underrepresented in the GPS proteome were expressed normally at the transcript level. The reduction in neutrophil content of specific granules was paralleled by increased levels of specific granule cargo, such as cathelicidin antimicrobial peptide, cysteine-rich secretory protein 3, and gelatinase-associated lipocalin, in circulation, as disclosed by mass spectrometry of patient plasma.8 Interestingly, inflammatory proteins synthesized in the liver, such as C reactive protein and lipopolysaccharide-binding protein, were also found to be upregulated in the plasma proteome, reflecting a systemic proinflammatory state. Raised serum vitamin B12 levels are an almost universal finding in GPS patients,3,8 although the mechanisms leading to B12 elevation remain undefined. Transcobalamin I, a B12 binding protein, is a major constituent of neutrophil specific granules. It might be hypothesized that its leakage to circulation could contribute to total B12 elevation, as reported for myeloproliferative neoplasms.45 However, transcobalamins were not listed among the proteins upregulated in the GPS plasma proteome8 and the mechanism underlying raised B12 levels remains to be determined. In addition to neutrophil abnormalities, this study also revealed decreased side scatter properties in eosinophils and reduced content of proteins which localize to granule structures in the proteome of GPS monocytes,8 hinting to a more widespread role of NBEAL2 in granule biology.
A comprehensive study of neutrophil structure and function recently performed in 13 GPS patients further confirmed the reduction in specific and gelatinase granular compartments, as shown by immune electron microscopy and proteomic analysis, while azurophilic granules were preserved.18 As previously shown for CD11b,25 surface expression of CD66b, an integral membrane component of specific granules, was elevated at baseline, further pointing to degranulation and did not increase significantly upon activation. Accordingly, the release of the content of specific granules, such as lactoferrin, triggered by specific stimuli, was significantly impaired, while that of azurophilic granules was unaffected. Intriguingly, in contrast to GPS circulating neutrophils, neutrophils derived from in vitro differentiation of patient hematopoietic progenitors displayed normal abundance of specific granules, suggesting intact granule biosynthesis during granulopoiesis but lack of retention or premature exocytosis of specific granules during egress from the bone marrow or in the bloodstream. Functional studies of blood neutrophils showed preserved production of reactive oxygen species, extracellular release of neutrophil proteases, such as elastase, and killing of gram-positive and -negative bacteria and fungi. Interestingly, however, neutrophil extracellular trap (NET) formation was blunted or substantially reduced in response to classic NET inducers, such as PMA, urate crystals and Candida albicans, which engage different NET-triggering pathways.18 Considering that upstream steps of NET formation, such as ROS production and myeloperoxidase and elastase reactivity were not altered in GPS neutrophils, this finding suggests that, in addition to azurophilic granules, specific granules may play a previously unrecognized role in NET formation. This granule subset may be particularly important in the late phases of NETosis, as reflected by defective membrane rupture and chromatin web release, whereas initial steps such as chromatin decondensation and cell rounding proceeded normally.18 It remains to be explored whether a particular specific granule component is responsible for this function. Increased susceptibility to infections, particularly mild upper respiratory infections and otitis media, has been previously pointed out in isolated case reports,3,11,25,46 and was recently reported to affect 17% of GPS patients in the large international cohort mentioned above.8 However, severe or life-threatening infections do not appear to occur frequently in GPS patients.
Immune function and pathogen defense were also assessed in Nbeal2 knockout mice, by two different groups. By analyzing public repositories of gene expression data, Sowerby et al disclosed strong NBEAL2 expression in cells of the immune system, reaching the highest levels in neutrophils and NK cells, both of which rely heavily on their granule content to fulfill their role in innate immunity.47 Neutrophil content of primary, specific and gelatinase granules was found to be markedly reduced in Nbeal2 deficient mice. Proteomic analysis of Nbeal2-/- neutrophils revealed significant enrichment of granule- and vesicle-associated components among downregulated proteins and, accordingly, release of granular proteins, such as elastase, was severely dampened. Although Nbeal2-/- neutrophils showed preserved phagocytosis and enhanced respiratory burst when challenged with gram-positive bacteria (Staphylococcus aureus), mice developed uncontrolled infection and had a worse outcome. In addition to neutrophil phenotypic defects, low numbers and functional abnormalities were shown for NK cells in this animal model, mainly consisting in altered degranulation, as shown by attenuated upregulation of lysosomal protein LAMP-1 triggered by specific stimuli. Considering that perforin and granzyme released from NK lytic granules are essential for NK cell-mediated cytotoxicity, the defect in NK cell degranulation could contribute to defective immunity found in these mice.47 As shown for bacterial disease, increased susceptibility to viral (CMV) infection was demonstrated for Nbeal2-deficient mice, consistent with the important role of NK cells in antiviral immunity. In line with this study, Claushuis et al corroborated the decrease in all neutrophil granule subtypes in Nbeal2 knockout mice and extended these findings by demonstrating increased levels of neutrophil granular proteins, such as MPO and elastase, in circulation.48 Reactive oxygen species production and phagocytic capacity by Nbeal2−/− neutrophils were normal in this study, as well as bacterial (K. pneumoniae) growth, although organ damage during infection and endotoxemia was enhanced. Another study showed that the content of storage vesicles of mast cells was reduced in Nbeal2-/- mice,49 suggesting the involvement of yet another cell type in the repertoire of GPS abnormalities.
Altogether, these findings clearly demonstrate that NBEAL2 deficiency leads to neutrophil abnormalities in human GPS and animal models. However, certain differences are evident between both models, highlighting that granular biosynthesis or trafficking may differ between human and mice neutrophils. The intrinsic mechanisms underlying the effect of NBEAL2 in granule formation in leukocytes and whether this process shares the same pathways and interacting proteins operating in platelets are currently unexplored issues. Increased susceptibility to infection and worse outcome has been described in Nbeal2-/- mice, whereas, although GPS patients do not usually suffer severe infections, an increase in mild, particularly, respiratory infections, has been reported. Defective NK cell function in Nbeal2 knockout mice adds further complexity to the defect in innate immunity. It would be interesting to study whether the NK compartment is also altered in human GPS.
Autoimmune Manifestations
In addition to deranged innate immunity, there is evidence of a deregulated adaptive immune response in GPS patients. The association between GPS and autoimmune disease was first suggested by the finding of NBEAL2 variants in patients from two different pedigrees presenting with an autoimmune lymphoproliferative (ALPS)-like syndrome who concomitantly showed GPS typical platelet abnormalities.11 Recently, autoimmune features were frequently found in GPS patients recruited by Sims et al, including autoimmune disease (26%) and the presence of at least one autoantibody (59%).8 Autoimmune manifestations involved several systems, including Hashimoto’s thyroiditis, rheumatoid arthritis, skin diseases, such as alopecia, discoid lupus erythematosus and vitiligo, and atypical autoimmune lymphoproliferative syndrome. Autoantibodies were directed against different target proteins, with rheumatoid factor, perinuclear anti-neutrophil cytoplasmic, anti-thyroperoxidase and antinuclear antibodies among the most frequently detected. Markers of immune response were upregulated in GPS CD4+ T helper cells at the protein and transcript level, as shown by the analysis of the proteomic and RNA-seq profile.8 Overrepresented molecules were enriched in proteins with immunomodulatory function, such as Bruton tyrosine kinase, which is crucial for B cell development and has been suggested to be involved in autoimmunity.50 On the basis of these novel findings, this study highlights the presence of immune dysregulation in GPS patients. A more detailed phenotypic and functional characterization of B and T cells and their subsets in GPS patients would be useful to gain further insight into the mechanisms underlying autoimmunity and help explain the aberrant immune pattern.
As mentioned before, a proinflammatory signature in GPS plasma proteome, as shown by upregulation of liver-derived inflammatory markers,8 was another prominent feature of this work. This finding might reflect chronic activation of liver resident cells by myeloid-derived products leaking to the bloodstream or, alternatively, low-grade chronic infection. The potential relationship between chronic immune stimulation and the development of autoimmunity may deserve consideration. Conversely, sustained autoimmunity may, in turn, trigger further inflammation, contributing to the ongoing proinflammatory state (Figure 3).
Treatment
Most GPS patients are treated symptomatically, according to the severity of bleeding manifestations. Patients may bleed spontaneously or secondary to surgical intervention, childbirth or trauma. Non-specific hemostatic measures, such as anti-fibrinolytics, local hemostatic measures, avoidance of non-steroidal anti-inflammatory drugs and/or platelet transfusions are usually adopted to prevent or reduce excessive bleeding. Eltrombopag has been shown to increase platelet counts in certain inherited platelet conditions, such as MYH9-related disorder, ANKRD26-related thrombocytopenia, X-linked thrombocytopenia/ Wiskott-Aldrich syndrome, monoallelic Bernard-Soulier syndrome or ITGB3-related thrombocytopenia.51 The efficacy and safety of thrombopoietin receptor agonists in GPS remains unknown. Considering that these agents may promote bone marrow fibrosis, their use in GPS should be viewed with caution, especially if long-term treatment is considered. Successful hematopoietic stem cell transplantation has been achieved in a GPS patient with severe myelofibrosis and pancytopenia, with full hematopoietic recovery and resolution of bone marrow fibrosis.52 On this basis, stem cell transplantation may represent a curative treatment option for selected patients who experience a severe clinical course.
In conclusion, recent evidence has expanded the phenotypic spectrum of GPS, providing new information which is relevant to patient care. The combined study of GPS patient samples and animal models has provided novel insight into the pathogenesis of this rare disorder. Future research will be required to further elucidate the effects of NBEAL2 in granule biogenesis and trafficking and to define its precise function in the biology of blood cells, including platelets and leukocytes, thus contributing to dissect its role in hemostasis and immunity. Increasing knowledge of the mechanisms underlying GPS will hopefully provide tools for developing appropriate therapies.
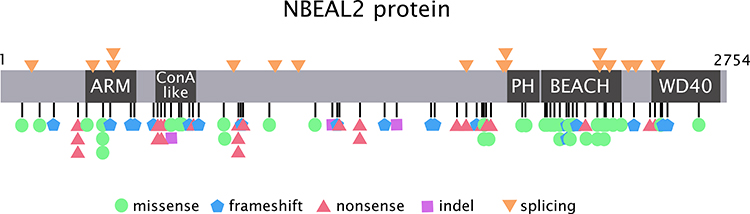 |
Figure 1 NBEAL2 variants in GPS patients. Schema of the NBEAL2 protein comprising the ARM (armadillo)-like domain, spanning amino acids (aa) 377–504, the Con (concanavalin) A-like lectin domain (aa 580–730), the PH (pleckstrin homology) domain (aa 1915–2040), the BEACH (beige and Chediak-Higashi syndrome domain (aa 2053–2345) and WD40 repeat domains (2463–2722), according to Sims et al.8 Published missense, nonsense, frameshift, indel and splicing germline NBEAL2 variants described in GPS pedigrees (Table 1) are indicated by the symbols. Missense variants are enriched within the BEACH domain. The position of splicing variants is given according to the predicted effect at the protein level. |
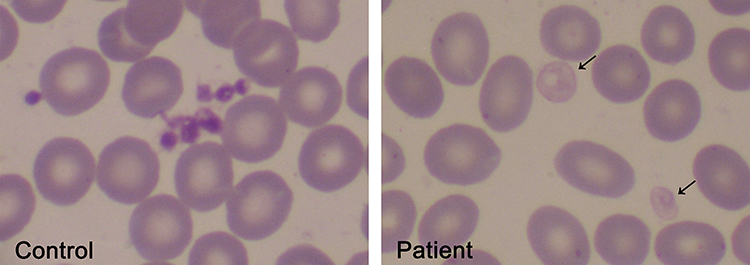 |
Figure 2 Pale platelets on the blood smear from a GPS patient. Large pale platelets due to absence or marked decrease in α-granules is a typical finding in GPS patients. A May-Grünwald Giemsa-stained blood smear from a GPS patient is shown on the right panel. Gray platelets are indicated by the arrows. A blood smear from a healthy subject stained simultaneously is shown on the left panel. Images were obtained at 1000x magnification. |
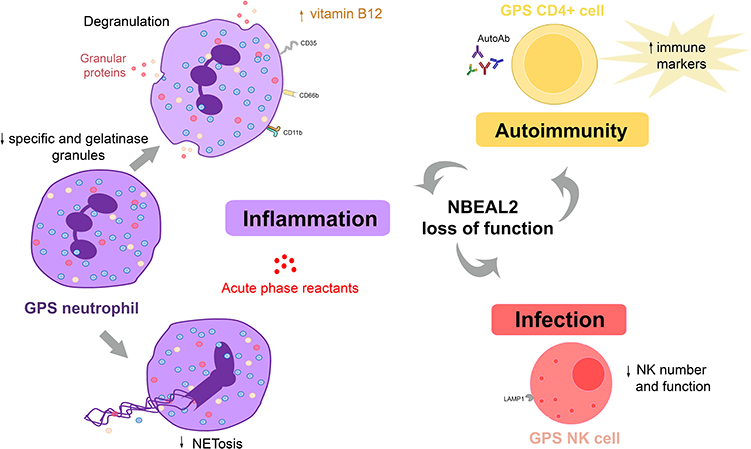 |
Figure 3 Abnormalities in the innate and adaptive immune response in GPS. GPS neutrophils display reduced numbers of specific (red circles) and gelatinase (yellow circles) granules. Whereas azurophilic (blue circles) granules are preserved in GPS patients, they are decreased in GPS animal models. Proteins resident in specific and gelatinase granules are expressed at the cell membrane, such as CD11b, CD66b and CD35, and elevated in plasma, indicating degranulation. Neutrophil extracellular trap formation (NETosis) is impaired. NK cell number and function are decreased in GPS animal models, although it is unknown whether it is also altered in patients. Altogether, these abnormalities may contribute to higher susceptibility to infections. Autoimmune manifestations and/or autoantibodies (AutoAb) are present in around half of GPS patients, coupled to upregulation of immune response markers in CD4+ cells. Liver-derived acute phase reactants are elevated in patient circulation, reflecting ongoing inflammation. |
Acknowledgments
This study was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (PICT 2018-01364) and the Fondation Nelia et Amadeo Barletta (to Paula Heller, 2020).
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Nurden AT, Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev. 2007;21(1):21–36. doi:10.1016/j.blre.2005.12.003
2. Raccuglia G. Gray platelet syndrome. A variety of qualitative platelet disorder. Am J Med. 1971;51(6):818–828.
3. Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. 2010;116(23):4990–5001.
4. Fabbro S, Kahr WH, Hinckley J, et al. Homozygosity mapping with SNP arrays confirms 3p21 as a recessive locus for gray platelet syndrome and narrows the interval significantly. Blood. 2011;117(12):3430–3434.
5. Kahr WH, Hinckley J, Li L, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;43(8):738–740.
6. Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43(8):735–737.
7. Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat Genet. 2011;43(8):732–734.
8. Sims MC, Mayer L, Collins JH, et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood. 2020;136(17):1956–1967.
9. Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic. 2013;14(7):749–766.
10. Bottega R, Pecci A, De Candia E, et al. Correlation between platelet phenotype and NBEAL2 genotype in patients with congenital thrombocytopenia and -granule deficiency. Haematologica. 2013;98(6):868–874. doi:10.3324/haematol.2012.075861
11. Rensing-Ehl A, Pannicke U, Zimmermann SY, et al. Gray platelet syndrome can mimic autoimmune lymphoproliferative syndrome. Blood. 2015;126(16):1967–1969.
12. Wijgaerts A, Wittevrongel C, Thys C, et al. The transcription factor GATA1 regulates NBEAL2 expression through a long-distance enhancer. Haematologica. 2017;102(4):695–706.
13. Bottega R, Nicchia E, Alfano C, et al. Gray platelet syndrome: novel mutations of the NBEAL2 gene. Am J Hematol. 2017;92(2):E20–E22.
14. Cao L, Su J, Jiaming J, et al. A novel nonsense NBEAL2 gene mutation causing severe bleeding in a patient with gray platelet syndrome. Platelets. 2018;29(3):288–291.
15. Steinberg-Shemer O, Tamary H. Gray platelet syndrome mimicking atypical autoimmune lymphoproliferative syndrome: the key is in the blood smear. Blood. 2018;131(24):2737.
16. Pluthero FG, Di Paola J, Carcao MD, Kahr WHA. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets. 2018;29(6):632–635.
17. Saliba AN, Ferrer A, Gangat N, et al. Aetiology and outcomes of secondary myelofibrosis occurring in the context of inherited platelet disorders: a single institutional study of four patients. Br J Haematol. 2020;190(5):e316–e320.
18. Aarts CEM, Downes K, Hoogendijk AJ, et al. Neutrophil specific granule and NETosis defects in gray platelet syndrome. Blood Adv. 2021;5(2):549–564.
19. Tariq H, Perez Botero J, Higgins RA, Medina EA. Gray Platelet Syndrome presenting with pancytopenia, splenomegaly, and bone marrow fibrosis. Am J Clin Pathol. 2021;aqaa229. doi:10.1093/ajcp/aqaa229.
20. Ross JE, Zhang BM, Lee K, et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: clinGen Platelet Disorder Variant Curation Panel. Blood Adv. 2021;5(2):414–431.
21. White JG. Ultrastructural studies of the gray platelet syndrome. Am J Pathol. 1979;95(2):445–462.
22. De Candia E, Pecci A, Ciabattoni G, et al. Defective platelet responsiveness to thrombin and protease-activated receptors agonists in a novel case of gray platelet syndrome: correlation between the platelet defect and the alpha-granule content in the patient and four relatives. J Thromb Haemost. 2007;5(3):551–559.
23. Glembotsky AC, Marta RF, Pecci A, et al. International collaboration as a tool for diagnosis of patients with inherited thrombocytopenia in the setting of a developing country. J Thromb Haemost. 2012;10(8):1653–1661.
24. Cramer EM, Vainchenker W, Vinci G, Guichard J, Breton-Gorius J. Gray platelet syndrome: immunoelectron microscopic localization of fibrinogen and von Willebrand factor in platelets and megakaryocytes. Blood. 1985;66(6):1309–1316.
25. Drouin A, Favier R, Massé JM, et al. Newly recognized cellular abnormalities in the gray platelet syndrome. Blood. 2001;98(5):1382–1391.
26. Rosa JP, George JN, Bainton DF, Nurden AT, Caen JP, McEver RP. Gray platelet syndrome: demonstration of a-granule membranes that can fuse with the cell surface. J Clin Invest. 1987;80:1138–1146.
27. Pluthero FG, Kahr WHA. Gray platelet syndrome: NBEAL2 mutations are associated with pathology beyond megakaryocyte and platelet function defects. J Thromb Haemost. 2021;19(2):318–322.
28. Breton-Gorius J, Vainchenker W, Nurden A, Levy-Toledano S, Caen J. Defective alpha-granule production in megakaryocytes from gray platelet syndrome: ultrastructural studies of bone marrow cells and megakaryocytes growing in culture from blood precursors. Am J Pathol. 1981;102(1):10–19.
29. Chen CH, Lo RW, Urban D, Pluthero FG, Kahr WH. α-granule biogenesis: from disease to discovery. Platelets. 2017;28(2):147–154.
30. Kahr WH, Lo RW, Li L, et al. Abnormal megakaryocyte development and platelet function in Nbeal2(-/-) mice. Blood. 2013;122(19):3349–3358.
31. Guerrero JA, Bennett C. van der Weyden L et al. Gray platelet syndrome: proinflammatory megakaryocytes and α-granule loss cause myelofibrosis and confer metastasis resistance in mice. Blood. 2014;124(24):3624–3635.
32. Deppermann C, Cherpokova D, Nurden P, et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest. 2013;123(8):3331–3342.
33. Lo RW, Li L, Leung R, Pluthero FG, Kahr W. NBEAL2 (Neurobeachin-Like 2) is required for retention of cargo proteins by α-granules during their production by megakaryocytes. Arterioscler Thromb Vasc Biol. 2018;38(10):2435–2447.
34. Mayer L, Jasztal M, Pardo M, et al. The BEACH-domain containing protein, Nbeal2, interacts with Dock7, Sec16a and Vac14. Blood. 2018;131:1000–1011.
35. Sviderskaya EV, Novak EK, Swank RT, Bennett DC. The murine misty mutation: phenotypic effects on melanocytes, platelets and brown fat. Genetics. 1998;148(1):381–390.
36. Lo RW, Li L, Pluthero FG, Leung R, Eto K, Kahr WHA. The endoplasmic reticulum protein SEC22B interacts with NBEAL2 and is required for megakaryocyte α-granule biogenesis. Blood. 2020;136(6):715–725.
37. Nurden AT, Nurden P. Should any genetic defect affecting α-granules in platelets be classified as gray platelet syndrome? Am J Hematol. 2016;91(7):714–718.
38. Noris P, Biino G, Pecci A, et al. Platelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disorders. Blood. 2014;124(6):e4–e10.
39. Larocca LM, Heller PG, Podda G, et al. Megakaryocytic emperipolesis and platelet function abnormalities in five patients with gray platelet syndrome. Platelets. 2015;26(8):751–757.
40. Nurden P, Jandrot-Perrus M, Combrié R, et al. Severe deficiency of glycoprotein VI in a patient with gray platelet syndrome. Blood. 2004;104(1):107–114.
41. Di Buduo CA, Alberelli MA, Glembotsky AC, et al. Abnormal proplatelet formation and emperipolesis in cultured human megakaryocytes from gray platelet syndrome patients. Sci Rep. 2016;6:25027.
42. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356–1363.
43. Chedani H, Dupuy E, Massé JM, Cramer EM. Neutrophil secretory defect in the gray platelet syndrome: a new case. Platelets. 2006;17(1):14–19.
44. White JG, Brunning RD. Neutrophils in the gray platelet syndrome. Platelets. 2004;15(5):333–340.
45. Zittoun J, Zittoun R, Marquet J, Sultan C. The three transcobalamins in myeloproliferative disorders and acute leukemia. Br J Haematol. 1975;31(3):287–298.
46. Kahr WH, Dror Y. Gray platelet syndrome: macrothrombocytopenia with deficient α-granules. Blood. 2012;120(13):2543.
47. Sowerby JM, Thomas DC, Clare S, et al. NBEAL2 is required for neutrophil and NK cell function and pathogen defense. J Clin Invest. 2017;127(9):3521–3526.
48. Claushuis TAM, de Stoppelaar SF, de Vos AF, et al. Nbeal2 Deficiency Increases Organ Damage but Does Not Affect Host Defense During Gram-Negative Pneumonia-Derived Sepsis. Arterioscler Thromb Vasc Biol. 2018;38(8):1772–1784.
49. Drube S, Grimlowski R, Deppermann C, et al. The Neurobeachin-like 2 Protein Regulates Mast Cell Homeostasis. J Immunol. 2017;199(8):2948–2957.
50. Rip J, Van Der Ploeg EK, Hendriks RW, Corneth OBJ. The role of Bruton’s Tyrosine Kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol. 2018;38(1):17–62.
51. Zaninetti C, Gresele P, Bertomoro A, et al. Eltrombopag for the treatment of inherited thrombocytopenias: a Phase II clinical trial. Haematologica. 2020;105(3):820–828.
52. Favier R, Roussel X, Audia S, et al. Correction of severe myelofibrosis, impaired platelet functions and abnormalities in a patient with Gray Platelet Syndrome successfully treated by stem cell transplantation. Platelets. 2020;31(4):536–540.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.