")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
A critical appraisal of ibrutinib in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia
Received 27 March 2015
Accepted for publication 6 May 2015
Published 23 June 2015 Volume 2015:11 Pages 979—990
DOI https://doi.org/10.2147/TCRM.S73559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Professor Garry Walsh
David L Tucker, Simon A Rule
Department of Haematology, Plymouth Hospitals NHS Trust, Plymouth, UK
Abstract: Although chemo-immunotherapy remains at the forefront of first-line treatment for mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL), small molecules, such as ibrutinib, are beginning to play a significant role, particularly in patients with multiply relapsed or chemotherapy-refractory disease and where toxicity is an overriding concern. Ibrutinib is a first-in-class, oral inhibitor of Bruton’s tyrosine kinase, which functions by irreversible inhibition of the downstream signaling pathway of the B-cell receptor, which normally promotes cell survival and proliferation. Early clinical trials have demonstrated excellent tolerability and a modest side-effect profile even in elderly and multiply pretreated patient cohorts. Although the majority of disease responses tend to be partial, efficacy data have also been encouraging with more than two-thirds of patients with CLL and MCL demonstrating a durable response, even in the high-risk disease setting. Resistance mechanisms are only partially understood and appear to be multifactorial, including the binding site mutation C481S, and escape through other common cell-signaling pathways. This article appraises the currently available data on safety and efficacy from clinical trials of ibrutinib in the management of MCL and CLL, both as a single agent and in combination with other therapies, and considers how this drug is likely to be used in future clinical practice.
Keywords: ibrutinib, mantle cell lymphoma, chronic lymphocytic leukemia, Bruton’s tyrosine kinase, lymphoproliferative disorders
Introduction
Conventional systemic therapy for non-Hodgkin lymphoma (NHL) relies upon single-agent or combination chemotherapy, often in conjunction with a monoclonal antibody for induction and subsequently maintenance of disease remission.1–5 This approach, however, is not always successful in the context of resistant or refractory disease, and is not always appropriate when treating patients with significant comorbidities where toxicity can become an overriding factor.
Recent advances in our understanding of the role of the B-cell receptor (BCR) in promoting cell survival and proliferation have generated new therapeutic opportunities for the treatment of NHL. Several non-receptor kinases downstream of the BCR such as Bruton’s tyrosine kinase (BTK), spleen tyrosine kinase, and phosphatidylinositol 3-kinase-delta (PI3Kδ) present the potential for new therapeutic targets.6 Patients with poor prognostic risk factors such as multiply relapsed/refractory (R/R) disease and high-risk chromosomal deletions appear to achieve significant and durable responses to these newer agents. Responses are often rapid and dramatic,7 and discontinuation or hospitalization, due to side effects, is an uncommon event. The most advanced of these new agents is the BTK inhibitor, ibrutinib. This review provides an overview of the data on ibrutinib focusing on the clinical trials that have supported its US Food and Drug Administration status as a breakthrough drug and discusses the common and important effects of its use in clinical practice.8
Ibrutinib (PCI-32765, trade name Imbruvica; Pharmacyclics, Sunnyvale, CA, USA) is a first-in-class, oral, irreversible inhibitor of BTK.9
Ibrutinib’s target, BTK, is a signaling molecule situated downstream of the BCR within the cytoplasm. Stimulation of the BCR triggers activation of a number of intracellular pathways, one of which results in the phosphorylation of BTK, which in turn activates cytoskeletal rearrangement and upregulation of transcriptional pathways involved in differentiation, proliferation, migration, and cell survival.10,11 Although BTK is expressed in many hematopoietic cell lines, it appears to have most importance in B-cell malignancies.12,13
BTK takes its name from an American pediatrician, who first described a condition in boys (Bruton’s disease) characterized by recurrent bacterial infections. Bruton’s disease, now known as X-linked agammaglobulinemia (XLA), is caused by an inherited or acquired defect in the BTK gene, mapping to Xq21.3-q22, resulting in very low circulating B-cell numbers with an immature phenotype and a profound hypogammaglobulinemia.14–18 Interestingly, pharmacological inhibition of BTK does not appear to have such a profound immunosuppressive effect as that seen in patients with XLA.
Pharmacology, pharmacodynamics, and mechanism of action
Ibrutinib is an off-white solid with an empirical formula of C25H24N6O2 and a molecular weight of 440.50. It is freely soluble in methanol and practically insoluble in water. The chemical name of ibrutinib is 1-[(3R)-3-[4-amin-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-y1]-1-piperidinyl]-2-propen-1-one, and its structure is shown in Figure 1.
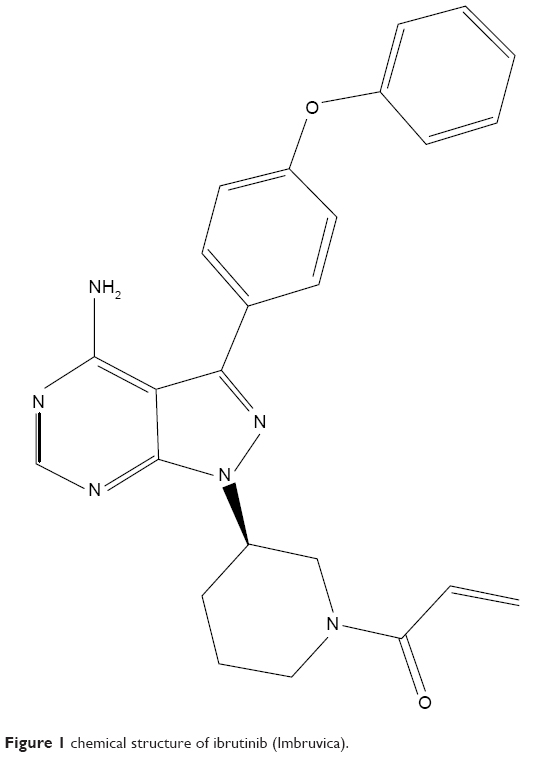 | Figure 1 chemical structure of ibrutinib (Imbruvica). |
Ibrutinib is supplied for oral administration in capsules that contain 140 mg ibrutinib as the active ingredient.19
Ibrutinib is an orally active small molecule that covalently binds to the cysteine residue C481 at the phosphorylation site of BTK, leading to irreversible inactivation.11,20 Ibrutinib actually possesses a broad kinome, which includes irreversible inhibition of several other kinases with important roles in normal and malignant B-cell signaling.9,21 Exactly why this cross-specificity does not result in a broader toxicity profile is not yet fully understood.
Originally, ibrutinib or PCI-32765 was developed by Celera, and later by Pharmacyclics, as a targeted therapy for rheumatoid arthritis, and subsequently, lymphoma.20,22 Preclinical studies demonstrated that ibrutinib inhibited phosphorylation of BTK at very low concentrations (IC50, 0.5 nM) in B-cell lymphoma cell lines as well as inhibiting activation of B-cells after direct stimulation of the BCR.22 An objective response rate of 38% was observed in the first animal studies of ibrutinib in canines with B-cell lymphoma.20
In vivo, ibrutinib blocks BCR-induced activation, thus inhibiting proliferation and attenuating cell survival as well as impairing the ability of lymphoma cells to home to their protective niche in lymphoid tissue or bone marrow.20,23–26 Clinically, this translates into a rapid reduction in tissue burden, often in association with a peripheral lymphocytosis, which appears to be dependent on continuous administration of the drug.
Ibrutinib is rapidly absorbed and eliminated after oral administration. Mean peak plasma concentrations occur between 1 hour and 2 hours after dosing, and drug exposure occurs in a dose-proportional manner. The mean half-life is 2–3 hours, and there is no accumulation of ibrutinib after repeated daily oral dosing.27
Ibrutinib is metabolized in the liver by cytochrome CYP3A, and metabolism is the major route of elimination. The elimination half-life is 4–6 hours, and clearance is not altered by age (37–84 years). Dose adjustments should be made for patients with mild-to-moderate hepatic impairment, and ibrutinib is not recommended for patients with severe liver impairment. Ibrutinib is not cleared significantly by the kidneys, and no dose adjustment is required for severe renal impairment (creatinine clearance <30 mL/min); however, patients should be monitored more closely for signs of toxicity.
Systemic exposure is increased significantly when dosed concomitantly with a strong CYP3A inhibitor such as ketoconazole and reduced with a strong CYP3A inducer, such as rifampicin. Therefore, patients taking ibrutinib are advised to avoid grapefruit juice or Seville oranges.27,28
Ibrutinib in chronic lymphocytic leukemia and mantle cell lymphoma
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults with an incidence rate of five per 100,000 per year in the US and can exhibit an extremely heterogenous clinical course.29 Some patients are diagnosed incidentally, live for decades, and never require treatment, whereas others require immediate therapy at a young age.30 Chemo-immunotherapy, most commonly in the form of rituximab with fludarabine and cyclophosphamide, is the gold-standard treatment for fit, younger patients with symptomatic disease.31 However, for patients with high-risk prognostic features such as del17p13.1 or del11q22.3 and those unfit for, or refractory to, fludarabine-based therapy, treatment options are often unsatisfactory.31
Mantle cell lymphoma (MCL) is characterized by the t(11;14) chromosomal translocation resulting in overexpression of cyclin D1 and deregulation of the cell cycle.32 It accounts for 4%–10% of NHL with an incidence of approximately 0.5 per 100,000 in Europe and the USA.29,33 It often has an aggressive clinical course and a poor prognosis.34 Current front-line therapy comprises chemoimmunotherapy followed by autologous stem-cell transplantation for those fit enough to receive it, and although these regimens have high initial response rates, most patients eventually relapse and die from the disease.2,35
Therefore, there is an unmet need for new treatments for both CLL and MCL.
Cell survival pathways dependent on BTK as a downstream mediator of the BCR are constitutively activated in both CLL and MCL contributing to tumor proliferation and cell survival.12,36 Ibrutinib has therefore been tested as a single agent and in combination with chemo-immunotherapy in patients with MCL and CLL, including patients with adverse cytogenetics, patients with disease which is refractory to purine analog, and treatment-naive elderly patients for whom aggressive chemo-immunotherapy is not appropriate.
Phase I studies
Phase I study of ibrutinib in R/R B-cell NHL
In the initial Phase I clinical trial of ibrutinib, 56 patients with R/R B-cell NHL, including MCL and CLL/small lymphocytic lymphoma (SLL), received single-agent ibrutinib once daily in a classical, Phase I, dose-escalation schedule.27 Two schedules were evaluated, one in which patients were treated daily for 28 days followed by a 7-day rest period, and another in which patients were dosed once daily continuously. The median patient age was 65 years. The median number of previous therapies was three, and 93% of patients had previously received the anti-CD20 monoclonal antibody rituximab.
Drug tolerability was very good. The most common adverse events (AEs) were typically grade 1 or 2 in severity with more severe events being infrequent and independent of dose. Diarrhea was a common side effect (42%), the majority being mild, usually self-limiting and resolving without dose interruption. Forty-one percent of patients experienced mild nausea, and 37% of patients experienced mild fatigue. Other common, non-severe side effects included muscle spasm/myalgia (37.5%) and “other pain” (62%). Grade 3–4 hematological toxicities were uncommon but included neutropenia (12.5%), thrombocytopenia (7.2%), and anemia (7.1%). In patients with CLL, a transient increase in lymphocyte count was typically seen at the same time as a reduction in nodal disease. This was understood to reflect the disruption of BCR-mediated stromal chemotaxis and adhesion resulting in an egress of tissue-resident malignant lymphocytes.23,37,38 This was sometimes very dramatic but did not result in any clinical sequelae.
The disease response rates were also encouraging, with an overall response rate (ORR) of 54% in the intention-to-treat population. Although responses were observed across all histologies, the best activity was seen in MCL (77%), Waldenströms macroglobulinemia (75%), and CLL (69%). Also, responses appeared durable with a median progression-free survival (PFS) of 13.6 months at the time of data cut-off. At the time of publication, 20 patients remained on treatment in an extension trial due to ongoing clinical benefit.
Although the maximum tolerated dose of ibrutinib was not reached, pharmacodynamic studies revealed >95% BTK active site occupancy and similar response rates at all but the lowest dose at a range of individual body weights. A fixed dose of 560 mg was chosen as the dose for future ibrutinib studies in MCL because it was well tolerated and led to full target occupancy. Also of note, in patients with CLL, the intermittent dosing schedule was associated with a transient reversal of the treatment-related lymphocytosis during the 7-day-off drug period, suggesting a reversal of the biological effect. On this basis, continuous dosing was selected for Phase II studies.
Phase I study of ibrutinib in R/R CLL
The first Phase I trial to focus specifically on patients with CLL was a multicenter single-arm study in which 85 patients with R/R CLL were treated with single-agent ibrutinib. The median age was 66 years with a median of four previous therapies. The vast majority had received a nucleoside analog, an alkylator, and rituximab. Sixty-nine percent of patients had adverse cytogenetic markers (17p13.1 deletion or del11q22.3).
Patients were allocated to a fixed daily dose of either 420 mg or 840 mg on a continuous schedule until onset of disease progression or unacceptable toxicity.
Drug tolerability was good with 64% of patients remaining on treatment with a median follow-up of 20.9 months. Most AEs were mild and resolved without the need to interrupt treatment, the most common being diarrhea (49%), fatigue (32%), and upper respiratory tract infection (33%). Moderate-to-severe hematological toxic effects were infrequent (anemia 6%, neutropenia 15%, and thrombocytopenia 6%), and bleeding events of grade 3 or higher occurred in only four (5%) patients. Perhaps unsurprisingly in such a heavily pretreated group, eight (9%) patients died within 30 days of the last dose of ibrutinib – three from pneumonia, one from systemic inflammatory response syndrome, one from sarcoma, and three due to disease progression.
At a median follow-up of 20.9 months, an ORR of 71% was observed with the vast majority being partial responses (PRs). There was no significant difference in response between the 420 mg and 840 mg dose, which led to a 420 mg dose being selected for subsequent trials in CLL. An additional 15 patients (18%) had a PR with lymphocytosis. Interestingly, response did not vary much according to high-risk features such as 17p deletion (ORR 68%). The 26-month estimated PFS and overall survival (OS) were remarkably good (75% and 83%, respectively). In the 17p13.1-deleted patients, the OS at 20.9 months was 70%.39
Phase I study of ibrutinib in treatment-naive patients with CLL
O’Brien et al subsequently demonstrated excellent tolerability and encouraging efficacy in previously untreated, elderly patients with CLL. Thirty-one patients received oral ibrutinib once daily in 28-day cycles.40 The median treatment duration was 21 months with a relative dose intensity of 98%.
In line with the previous Phase I studies, the most common AE was diarrhea (68%) which was predominantly mild, self-limiting and resolving without discontinuation of the study drug. Other common AEs reported included mild nausea, fatigue, and hypertension. Hematological toxicities were mild and uncommon. Only two patients discontinued treatment due to AEs (fatigue and viral infection), and only nine (29%) required interruption of treatment due to grade 3 or greater toxicity. The majority of patients continued treatment in the long-term extension study, and no patients died from causes related to treatment or within 30 days of the last dose of study drug.
Response rates were also consistent with previous studies with an ORR of 71% and a 24-month PFS of 96.3%. Four (13%) patients achieved a complete response (CR) with the remainder achieving PR. An additional four patients (13%) achieved a PR with lymphocytosis, of whom two eventually achieved a CR and seven achieved a PR with resolution of the lymphocytosis. In fact, ibrutinib led to lymphocytosis in 55% of patients, peaking at 1 week and then slowly declining. This occurred together with a significant reduction in tissue bulk that was sustained over the course of treatment. The observation of a sustained reduction in tissue burden with lymphocytosis that slowly diminishes over time is now considered to be a class effect in all BCR antagonists and has led to a change in the response criteria for CLL. Encouragingly, this trial also demonstrated no significant variation in response in the presence of high-risk prognostic factors such as 17p deletion, although the number of patients with high-risk factors was relatively low.
Phase II studies of ibrutinib in CLL
In a Phase II, single-arm study, 51 patients with previously untreated (n=35) and R/R (n=16) high-risk CLL with TP53 aberrations were treated with single-agent ibrutinib.41 The primary end point was overall response to treatment after six cycles of therapy at 24 weeks. With a median follow-up of 24 months, 41 (82%) patients are still on treatment without disease progression. Nine patients discontinued due to disease progression (10%) and death (6%) with one patient found to have Hodgkin lymphoma. The median time to progression was 7.5 months. At 24 weeks, 92% of patients achieved an objective response, the vast majority being PRs. Similar response rates were seen when subgroup analysis was performed according to IGHV mutation status, age, β2 microglobulin, or the presence of bulky disease. The estimated PFS rate at 24 months was 82%, and OS at 24 months was 80% for all patients. In keeping with other studies of ibrutinib, treatment was well tolerated.
This study demonstrates durable responses in patients with CLL with TP53 aberrations, especially patients with previously untreated disease. This study also demonstrates that the TP53 aberrant CLL clone is as equally sensitive to ibrutinib as other TP53 wild-type cells, which supports a P53-independent mechanism of action. Furthermore, histological transformation rates appear to be lower than those seen with conventional chemotherapy when used first line, emphasizing the role of up-front ibrutinib in patients with TP53 aberrations.
Phase III and long-term extension studies in CLL
The first randomized trial of ibrutinib against a licensed drug for CLL was a multicenter, Phase III, open-label study in which 391 patients with R/R CLL/SLL received daily oral ibrutinib (420 mg) or the anti-CD20 monoclonal antibody, ofatumumab (Arzerra®). This was given once weekly for up to 24 weeks on a dose-escalation protocol.42 Patients had received at least one previous therapy and were considered inappropriate candidates for purine analog treatment because of a short progression-free interval after chemo-immunotherapy, coexisting illness, an age of greater than 70 years, or chromosome 17p13.1 deletion.
Ibrutinib’s AE profile was consistent with that from previous trials. Diarrhea (48%), fatigue (28%), and nausea (26%) were the most common, and very few of these were moderate or severe. Bleeding-related AEs (most commonly petechiae and ecchymosis) were more common in the ibrutinib group (44% vs 12%), but major hemorrhage requiring transfusion was rare (1% ibrutinib, 2% ofatumumab). Changes in creatinine levels were similar in both groups (16% ibrutinib, 17% ofatumumab). Interestingly, there appeared to be a higher rate of development of atrial fibrillation in the ibrutinib group (10% compared to <1%), and this led to discontinuation of ibrutinib in one patient. However, discontinuation rates due to AEs were low (4% in both groups).
Response rates were significantly better in the ibrutinib arm. There was a 78% risk reduction of progression or death in the ibrutinib arm compared with ofatumumab. At 6 months, 88% of the ibrutinib group were still alive with no disease progression compared to 65% in the ofatumumab arm. This was independent of baseline characteristics or molecular features. OS was also significantly prolonged in the ibrutinib group (hazard ratio 0.43, 95% confidence interval 0.24–0.79; P=0.005). The OS at 12 months was 90% in the ibrutinib arm and 81% in the ofatumumab group. This effect was maintained despite the crossover of 57 patients from the ofatumumab arm after disease progression.
Therefore, for older patients with R/R or high-risk CLL, ibrutinib has compared favorably with established therapies in terms of tolerance and response both as a single agent and in combination with other drugs.
Ibrutinib in combination with other agents in CLL
Ibrutinib with rituximab
In a single-arm Phase Ib/II study, 40 adult patients (36 previously treated, four previously untreated) with high-risk CLL were treated with ibrutinib (420 mg once daily, continuously) in combination with rituximab (375 mg/m2 intravenously each week during cycle 1, then once per cycle until cycle 6). Fifty percent of patients had del17p or TP53 mutation, and the average age was 63 years.
At a median follow-up of 16.8 months, 31 (78%) patients remained on therapy (including 14 of the 20 with del17p or TP53 mutation). Nine patients discontinued (two died during study from pneumonia and central nervous system infection, and one patient died in remission) and six died after discontinuation (infectious complications or disease progression). Two patients (5%) discontinued due to treatment toxicity (grade 3 mucositis and grade 3 recurrent ear and pulmonary infections).
The combination of ibrutinib and rituximab achieved a higher ORR, 95% (87% being PRs), than ibrutinib alone, with a median duration of response of 15.4 months. The PFS of all patients was 78% with an OS of 83.8% at 18 months, and these were not significantly different in patients with del17p or TP53 mutation.43
Ibrutinib with chemo-immunotherapy in CLL
In a Phase Ib/II study, 30 patients with R/R CLL/SLL received ibrutinib in combination with bendamustine and rituximab (BR) for a median of six cycles (range 2–6) with a continuous fixed-dose ibrutinib until disease progression or intolerance. The median age was 62 years. Twenty-three percent and 43% carried del17p and del11q, respectively. The observed safety profile was generally consistent with the safety profile of BR alone, and side effects were comparable to those described in other ibrutinib trials except for grade 3 or higher neutropenia which was more frequently seen (40%). Interestingly, the transient lymphocytosis observed in approximately 78% of patients with ibrutinib monotherapy occurred less frequently (27%). With a median treatment duration of 16 months, the ORR was 93% including five CRs. The estimated 12-month PFS was 90%. As with previous studies in CLL, responses appeared independent of high-risk features.44
Ibrutinib with ofatumumab in CLL
In this Phase I study, 71 patients with R/R CLL/SLL, PLL, or Richter’s transformation (RT) after greater than or equal to two prior therapies were treated in three groups in which ofatumumab was administered weekly for 8 weeks and then monthly for 4 months, and ibrutinib was given continuously at 420 mg once daily.45 Group 1 received an initial cycle of ibrutinib single agent, group 2 commenced ibrutinib and ofatumumab simultaneously, and group 3 received two cycles of ofatumumab before the addition of ibrutinib. Unsurprisingly, the most frequent AEs were diarrhea (68%), infusion-related reactions (45%), peripheral sensory neuropathy (42%), and stomatitis (37%). Sixty-one percent of patients suffered AEs of grade ≥3, the most common being neutropenia. Thirty-nine percent experienced severe AEs, and 15% of patients had to discontinue ibrutinib. Approximately 13% of this cohort died within 30 days of the last dose.
The combination appears to be highly effective, with an ORR of 100% in the CLL/SLL group, and 90% of responders remained progression-free at a median of 12 months. The three patients with RT also experienced a degree of disease control followed by progressive disease on days 471, 168, and 137, respectively.
These results, which are comparable with rituximab combination data, demonstrate acceptable tolerability and encouraging responses when ibrutinib is combined with immunotherapy.
Several larger clinical trials evaluating this and other combinations are ongoing (Table 1).
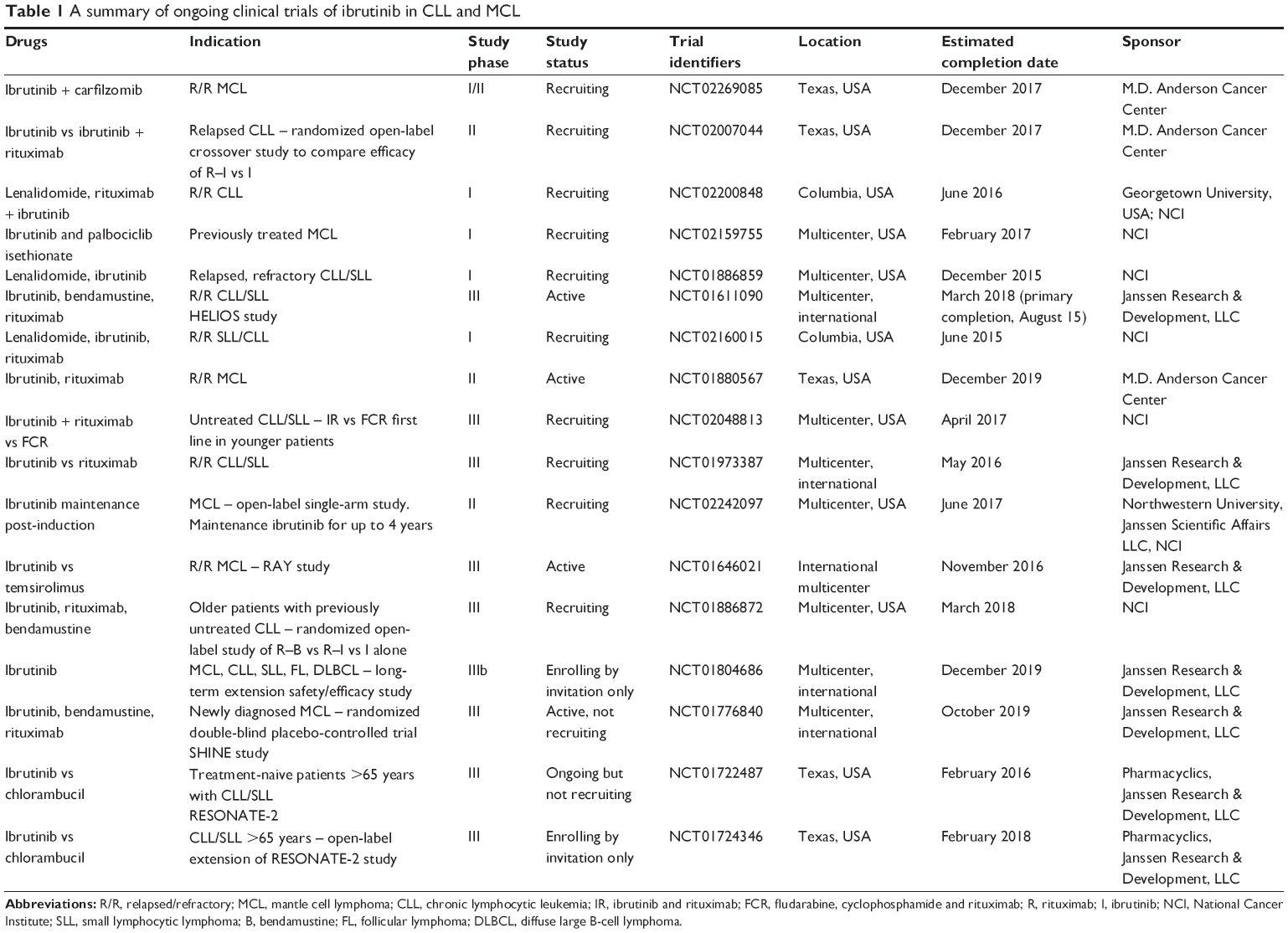 | Table 1 A summary of ongoing clinical trials of ibrutinib in CLL and MCL |
Overall, these data support the premise that ibrutinib is well tolerated and effective in patients with advanced and multiply treated CLL, both as a single agent and in combination. By way of comparison, a study of chlorambucil in combination with obinutuzumab (GA101, Gazyvaro®) on elderly patients with CLL reported a median PFS of 26.7 months with considerably higher rates of AEs due to myelotoxicity or infusion-related reactions.46 Lenalidomide single-agent therapy in the same patient group achieved a lower ORR (65%) and lower median PFS at the expense of significant cytopenias and major infections in 13% of patients.47
Mantle cell lymphoma
In the largest study of ibrutinib in MCL to date, 115 patients with R/R MCL were given single-agent ibrutinib (560 mg) on a 28-day cycle basis until progression or unacceptable AEs occurred.48 The median patient age was 68 years with a median of three prior treatments. Eleven percent had prior autologous stem-cell transplant, and 89% had prior chemo-immunotherapy.
The median follow-up was 15.3 months at which 46 patients (41%) remained on therapy. Sixty-five (59%) patients discontinued. Fifty patients experienced progressive disease, and eight experienced AEs. The most common side effects were diarrhea (50% of patients), fatigue (41%), and nausea (31%), all of which were predominantly mild. Mild-to-moderate neutropenia and thrombocytopenia occurred in 18% of patients (grade 4 in 10% and 4%, respectively). Grade 3 bleeding events requiring a blood transfusion occurred in five patients (4.5%). Four patients suffered subdural hematomas, all after trauma and all while on aspirin or warfarin. This led to subsequent studies excluding the use of warfarin in clinical trials of ibrutinib; however, other anticoagulants have been permitted. Sixteen patients died during the trial, 12 due to disease progression, and four due to AEs.
Seventy-five patients (68%) achieved a response (47% PR, 21% CR), and the median response duration was 17.5 months (95% confidence interval 15.8 – not reached). The median OS was not reached, and the estimated OS was 58% at 18 months.48
Such ORRs are unprecedented for a single agent in such a heavily pretreated population with MCL. Similar rates have only previously been seen when intensive chemotherapy regimens are used, all of which are myelotoxic.49 When other single-agent regimens are used in the salvage setting, responses are far more modest with shorter remission periods.50 By means of comparison, there are three drugs licensed for use in MCL as single agents, bortezomib (Velcade™), temsirolimus (Torisel™), and lenalidomide (Revlimid™). As single agents, the ORRs for these drugs are 33% (8% CR) with bortezomib, 22% (2% CR) with temsirolimus, and 28% (8% CR) with lenalidomide.51
Therefore, ibrutinib has shown impressive single-agent responses with excellent tolerability and a modest side-effect profile in R/R MCL. An extension study is ongoing with long-term follow-up of this trial.
Ibrutinib and rituximab in MCL
A Phase II trial of ibrutinib in combination with rituximab for relapsed MCL is currently underway and reported interim results in 2014.52 Fifty patients, all of whom had received prior rituximab, were given continuous daily ibrutinib (560 mg) with rituximab (375 mg/m2) weekly for 4 weeks in cycle 1 and then on day 1 of every cycle for up to eight cycles followed by 2 years of maintenance therapy. There were no toxic deaths due to therapy, and toxicities were broadly in line with those observed in other ibrutinib studies. The ORR was 87% at a median follow-up of 6.5 months with a higher CR rate (38%) compared with historical data of ibrutinib as a single agent in this demographic. Interestingly, Ki-67 proliferation indices of <50% appear to predict better ORR and CR rates. While this trial is ongoing, the preliminary data indicate that this combination is well tolerated and efficacious, especially in patients with lower tumor proliferation indices.
With regard to currently available long-term data on ibrutinib, 3-year results from a single US center with 132 patients with CLL/SLL receiving single-agent ibrutinib have recently been published. They show that in a patient group with a median age of 68 years, 27.3% of whom have del17p, the ORR was 78% in all patients (83.9% in treatment-naive patients, 76% in RR patients, and 55.9% in R/R patients with del17p). The median duration of response has not been reached for all patients at 3 years but was 25 months for patients with del17p. Sixty-four percent of patients remain on ibrutinib at 3 years. No new safety signals were seen. Severe AEs of grade >3 were more common in the pretreated cohort and declined in frequency after 1 year on therapy.53
In summary, ibrutinib appears remarkably well tolerated and demonstrates high response rates and durable efficacy in R/R and high-risk CLL and MCL. Responses, mainly partial, are seen in approximately 70% of patients, and are higher when combined with other agents. Side effects are modest, manageable in an outpatient setting and tend to resolve with time. Lymphocytosis, which is now considered to be a class effect of all BCR antagonists, is commonly seen after a couple of weeks in patients who are responding to treatment and tends to decline with time while on treatment. The majority of the Phase III clinical trials of ibrutinib in CLL and MCL are ongoing. Table 1 summarizes the ongoing clinical studies for future reference.
Resistance and relapse
Resistance and disease relapse are recognized features of ibrutinib treatment in patients with both CLL and MCL. However, the reasons for resistance and progression are only partially understood.
Ibrutinib does not appear to completely eliminate the malignant cell clone in CLL or MCL. Roughly 20% of patients with CLL have persistent lymphocytosis during ibrutinib therapy, and most patients have residual tumor cells which could permit the emergence of resistant subclones.54 In MCL, approximately 30% of patients exhibit primary resistance to ibrutinib, and although clinical response rates are impressive in this patient group, further resistance to treatment occurs with time on therapy.48
One explanation for resistance is a mutation of the BTK gene causing an alteration at the ibrutinib binding site. Several such mutations have been identified in patients with relapsed disease on ibrutinib, the most common being a point mutation causing a protein substitution (cysteine to serine) at the ibrutinib binding site (C481S), which causes reversible rather than irreversible inhibition of BCR signaling.55 Less frequently, mutations have been identified in the PLCγ2 gene. PLCγ2 is a gene responsible for a kinase downstream of BTK. PLCγ2 gene mutations appear to be gain-of-function mutations allowing BCR-mediated activation that is independent of BTK.55 These mutations are not secondary changes in previously mutated genes, but primary mutations in genes that are not recurrently mutated. It is noteworthy that these mutations do not appear to predate ibrutinib treatment nor are they associated with typical high-risk features of CLL, indicating that they are not relevant to the cell in the absence of the selective pressures of ibrutinib.56 Although these mutations are well described, they do not fully account for the level of resistance observed in studies and are by no means present in all patients with resistant disease. Mutations in other coding regions which then provide alternative survival signals that are not inhibited by ibrutinib, or through noncoding RNA, epigenetic activation, or gene silencing, may well play a role in resistance, and work to identify alternative mechanisms for resistance is ongoing.
Interestingly, patients with CLL on ibrutinib who are clinically responsive but with persistent lymphocytosis have not been shown to carry mutations in BTK or PLCγ2 genes.55 Therefore, the persistent lymphocytosis which is a feature of therapy is not associated with known resistance mutations.57
In MCL, in vitro studies have shown that standard, clinically achievable doses of ibrutinib elicit anti-lymphoma activity in some but not all MCL cell lines.58 The C815S mutation has not been detected in any of the cell lines unresponsive to ibrutinib. Downstream signaling events appear to better reflect resistance than phosphorylation of BTK which, when exposed to ibrutinib, is reduced in sensitive and resistant cells alike. Therefore, alternative upstream pathways or kinases appear to mediate activity of downstream events and allow a subset of MCL cells to continue to survive and grow even in the presence of BTK inhibition.58
The tumor microenvironment also plays a critical role in the survival of MCL and CLL cells and may also have a role to play in resistance to ibrutinib.59,60 This interaction, which involves a complex cell-signaling relationship between MCL cells, bone marrow, and lymphoid stromal cells, is currently under investigation.23,59,61
Relapses on ibrutinib therapy
Although ibrutinib is highly effective in MCL and CLL even in patients with high-risk disease, a substantial proportion of patients relapse on therapy or have disease resistant to treatment at initiation.
In a retrospective analysis of 42 patients with MCL treated with ibrutinib at a single US center, 31 patients discontinued ibrutinib due to progressive disease and underwent salvage therapy. For this group, the ORR and CR were 32% and 19%, respectively, with a median OS of 8.4 months, indicating that the outcome for patients experiencing MCL disease progression on ibrutinib is poor with low response rates following salvage chemotherapy.62 Elevated LDH levels at point of relapse appeared to be adversely prognostic in this group.
In a follow-up of 267 patients participating in trials of ibrutinib at a single US center, 24 (9%) patients discontinued due to progressive disease. Of these 24 patients, 16 had RT, and eight had progressive disease. All the evaluable patients with PD but only two of four patients with RT showed the C481S mutation.
In the single-arm Phase II trial of ibrutinib in high-risk CLL reported by Farooqui et al disease progression was reported in 10% of patients. All five patients had transformation manifesting as Richter’s syndrome. However, this figure is lower than that observed in another recent report of patients with similar high-risk CLL after a median of 12 months from first chemo-immunotherapy suggesting that the risk of transformation is not elevated and perhaps reduced with ibrutinib in this high-risk group.41 Furthermore, in the Phase III trial of ibrutinib vs ofatumumab, there was no difference in the incidence rate of RT between the two arms.42 Therefore, there are probably other common “escape mechanisms” through which NHL becomes resistant to ibrutinib. However, once resistance occurs, progression is often rapid, and outcomes are poor.63
In the authors’ experience, once a patient on ibrutinib begins to show evidence of progressive disease, it is important to continue ibrutinib therapy until an alternative therapeutic strategy is in place, at which point, the transition to alternative therapy should occur with as brief an interruption of therapy as possible in order to avoid the rapid disease progression that can occur once ibrutinib is discontinued.
Ibrutinib-specific safety concerns
Ibrutinib is generally very well tolerated with a very acceptable side-effect profile in all patient groups. However, early trial data raised specific concerns, which will be addressed in this section.
Eye-related abnormalities
Concern has been raised that BTK inhibitors may cause corneal opacification. In the ibrutinib vs ofatumumab for CLL study, reports of eye-related AEs were collected proactively on the basis of preclinical studies in dogs where corneal abnormalities were observed in animals receiving ibrutinib at a dose of 150 mg/kg of body weight per day (equivalent dose in humans is 81 mg/kg per day). Ocular symptoms were reported more frequently among patients in the ibrutinib group. The development of cataracts occurred in 3% of ibrutinib-treated patients compared with 1% of ofatumumab patients.42
One suggested mechanism for lens opacification was via a tyrosine kinase mechanism disrupting the EPHA2 which regulates lens clarity and organization.64 Subsequently, however, in a review of 506 patients with B-cell cancers treated with ibrutinib monotherapy, cataracts have been reported in 2.6% in a population with a median age of 66 years. This observation is consistent with the background rate in an age-matched population.65
Therefore, larger clinical trials and extension studies have failed to show an association with cataract above that of the background population, and earlier concerns may be attributable to much larger doses of ibrutinib used in preclinical studies.
Ibrutinib and bleeding
Bleeding has been reported in patients treated with ibrutinib with events of grade 3 or higher, including central nervous system hemorrhage of any grade severity, occurring in 3.4% (17 of 506 patients).64 In patients treated with ibrutinib for R/R MCL, grade 3 bleeding events occurred in five (4.5%) patients with no grade 4 or 5 hemorrhagic events. Four patients had subdural hematomas, all associated with trauma. Four of the five patients were receiving aspirin or warfarin within 2 days of occurrence.48 For this reason, the manufacturers advise caution with anticoagulant/antiplatelet therapy in conjunction with ibrutinib. However, data are lacking from randomized controlled trials to compare bleeding events in patients with BTK inhibition with other therapies in this patient group, and data on ibrutinib and platelet function studies are conflicting.66–68
The National Institutes for Health (US) conducted platelet function studies on 25 patients on ibrutinib in 2012. Three patients had abnormal platelet function tests at baseline, but these improved with time on drug, and no patients with abnormal results had spontaneous ecchymosis.66
More recent in vitro data show inhibition of collagen-induced platelet aggregation and platelet adhesion to von Willebrand factor at clinically achievable doses but only in a proportion (35%) of subjects on ibrutinib. Polymorphisms, variations in ibrutinib metabolism, redundant platelet signaling pathways, and variations in glycoprotein expression may explain why only a subset of patients treated with ibrutinib displayed spontaneous bleeding.67
In an observational study of 23 patients on ibrutinib, light transmission aggregometry (LTA) was performed on platelet-rich plasma. Seven patients (30%) had mild bleeding, only one of which was on concomitant antiplatelet/anticoagulant medication (aspirin alone). Platelet aggregation values were significantly lower in subjects with bleeding (26%) compared with those without (44%) when tested with high-dose collagen LTA. Platelet aggregation impairment was reversible 1 week after drug interruption.68
Therefore, platelet dysfunction appears to be responsible for the mild bleeding phenotype seen in about one-third of patients treated with ibrutinib, and caution must be advised over the use of antiplatelet and anticoagulant therapies.
Platelet transfusion outside the 3-hour half-life window of ibrutinib should reverse the hemostatic defect associated with ibrutinib in the event of serious bleeding. Treatment interruption for 5 days before a planned invasive procedure at high bleeding risk should be sufficient to avoid ibrutinib-related defects in hemostasis.67,68
Ibrutinib – future directions
The current requirement for continuous daily dosing coupled with the high cost of ibrutinib is likely to drive investigation of a minimal residual disease (MRD)-directed approach in which therapy may be discontinued once patients achieve molecular remission, followed by a period of regular monitoring and re-initiation at the point of MRD relapse. Two larger clinical trials currently underway have already incorporated MRD monitoring into their protocols. The SHINE study is a Phase III randomized double-blind placebo-controlled trial of rituximab plus bendamustine with or without ibrutinib for newly diagnosed MCL. The HELIOS trial is a Phase III study of ibrutinib in combination with BR in R/R CLL in which rate of MRD-negative disease remissions is a secondary end point.
Ibrutinib with other immunomodulators
The ubiquitin–proteasome pathway is an attractive target for antitumor therapy as its persistent activity is associated with tumor growth and drug resistance in many cancer types. Carfilzomib (CFZ) is a proteasome inhibitor that has an established role in multiple myeloma. In vitro studies have shown CFZ to have activity in MCL and that ibrutinib synergizes with CFZ and is toxic to both CFZ-sensitive and CFZ-resistant cell lines.69 Therefore, a Phase I single-arm safety and dose-finding trial is currently active combining ibrutinib with CFZ in patients with R/R MCL.
Lenalidomide has shown promising results in preclinical and clinical Phase I/II studies in R/R MCL, although toxicities are higher than for ibrutinib.70–72 There are several studies currently recruiting patients looking at combinations of lenalidomide and ibrutinib for MCL in the hope that this may improve response and progression rates.
Ibrutinib with other small molecules
With a greater understanding of BCR signaling pathways and potential resistance mechanisms arises the potential to combine different targeted therapies. Several studies are planned or recruiting in which ibrutinib is combined with other targeted therapies such as PI3Kδ inhibitors and the anti-BCL-2 molecule, ABT-199. These combinations may offer one solution to the problem of ibrutinib resistance, although the modest side-effect profiles of the individual drugs may not translate when used in combination.
When best to use ibrutinib? sequences and combinations
There is no doubt, therefore, that ibrutinib is highly active and well tolerated in MCL and CLL, with remarkable early- to medium-term data, even in high-risk disease. Indeed, some investigators believe that these early data may represent the dawn of a chemotherapy-free era in the treatment of some forms of NHL. Compared to conventional chemotherapy, however, the data are relatively immature, and the long-term consequences of the agent are unknown, both on disease and in terms of toxicity. Long-term treatment success will probably rely on combinations of targeted agents either with each other or with chemo-immunotherapy, and a variety of clinical studies are currently underway to establish how best to sequence and combine ibrutinib with other agents. At the writing of this review, there are 82 studies of ibrutinib currently registered at ClinicalTrials.gov for the treatment of a broad range of B-cell malignancies.73
The significant cost of ibrutinib will drive different management strategies for its use, particularly MRD-based approaches. However, this is a remarkable new therapeutic advance and heralds the very real prospect of a move away from chemotherapy for patients with CLL and possibly MCL.
Disclosure
SAR has received honoraria from Janssen-Cilag Ltd. and Pharmacyclics Inc. and research funding from Janssen-Cilag Ltd. DLT has no conflicts of interest to declare.
References
Coiffier B, Haioun C, Ketterer N, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood. 1998;92:1927–1932. | ||
Delarue R, Haioun C, Ribrag V, et al; Groupe d’Etude des Lymphomes de l’Adulte (GELA). CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood. 2013;121(1):48–53. | ||
Forstpointner R, Dreyling M, Repp R, et al; German Low-Grade Lymphoma Study Group. The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2015;104(10):3064–3072. | ||
van Oers MH, Klasa R, Marcus RE, et al. Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non-Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Baseline. 2006;108(10):3295–3301. | ||
Vose JM, Link BK, Grossbard ML, Czuczman M, Grillo-Lopez A, Fisher RI. Long-term update of a phase II study of rituximab in combination with CHOP chemotherapy in patients with previously untreated, aggressive non-Hodgkin’s lymphoma. Leuk Lymphoma. 2005;46(11):1569–1573. | ||
Byrd JC, Jones JJ, Woyach JA, Johnson AJ, Flynn JM. Entering the era of targeted therapy for chronic lymphocytic leukemia: impact on the practicing clinician. J Clin Oncol. 2014;32(27):3039–3047. | ||
European Hematology Association. 19th Congress of the European Hematology Association, Milan, Italy, June 12–15, 2014. Haematologica. 2014;99(1 suppl):249. | ||
Pazdur R. New Drug Application Approval – Imbruvica (Department of Health and Human Services, Food and Drug Admistration) Ref. ID 3395788; 2013. | ||
Pan Z, Scheerens H, Li S-J, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. Chem Med Chem. 2007;2(1): 58–61. | ||
Mohamed A, Yu L, Backesjo C, Vargas L, Berglo A, Smith CIE. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73. | ||
Buggy JJ. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;32(2):119–132. | ||
Cinar M, Hamedani F, Mo Z, Cinar B, Amin HM, Alkan S. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by ibrutinib induces apoptosis. Leuk Res. 2013;37(10):1271–1277. | ||
Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. | ||
Bruton O. Agammaglobulinemia. Pediatrics. 1952;9:722–728. | ||
Tsukada S, Rawlings DJ, Witte ON. Role of Bruton’s tyrosine kinase in immunodeficiency. Curr Opin Immunol. 1994;6:623–630. | ||
Satterthwaite AB, Witte ON, Witte O. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;6:120–127. | ||
Maas A, Hendriks RW. Role of Bruton’s tyrosine kinase in B cell development. Dev Immunol. 2001;8:171–181. | ||
Ponader S, Burger JA. Bruton’s tyrosine kinase: from X-linked agammaglobulinemia toward targeted therapy for B-cell malignancies. J Clin Oncol. 2014;32(17):1830–1839. | ||
Indications and usage Imbruvica is indicated for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. This indication is based on overall response rate. An improvement in survival or disease-related s; 2013. | ||
Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–13080. | ||
Hutchinson CV, Dyer MJS. Breaking good: the inexorable rise of BTK inhibitors in the treatment of chronic lymphocytic leukaemia. Br J Haematol. 2014;166:12–22. | ||
Burger JA, Buggy JJ. Emerging drug profiles: Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765). Leuk Lymphoma. 2013;54(11):2385–2391. | ||
Chang BY, Francesco M, De Rooij MF, et al. Egress of CD19(+) CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122(14):2412–2424. | ||
Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-κB. Cell Signal. 2013;25:106–112. | ||
Herman SE, Mustafa RZ, Gyamfi JA, et al. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286–3295. | ||
Zheng X, Ding N, Song Y, Feng L, Zhu J. Different sensitivity of germinal center B cell-like diffuse large B cell lymphoma cells towards ibrutinib treatment. Cancer Cell Int. 2014;14(1):32. | ||
Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. | ||
Indications and Usage Imbruvica. Full Prescribing Information. Pharmacyclics, Inc. Ref. ID 3395788; 2013. | ||
Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107(1):265–276. | ||
Gribben JG. How I treat CLL up front. Blood. 2010;115(2):187–197. | ||
Gribben JG, O’Brien S. Update on therapy of chronic lymphocytic leukemia. J Clin Oncol. 2011;29(5):544–550. | ||
Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361–1392. | ||
Zhou Y, Wang H, Fang W. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer. 2008;113(4):791–798. | ||
Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with rituximab-hyperCVAD alternating with rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol. 2010;150(1995):200–208. | ||
Chen Y, Wang M, Romaguera J. Current regimens and novel agents for mantle cell lymphoma. Br J Haematol. 2014;167(1):3–18. | ||
Packham G, Stevenson F. The role of the B-cell receptor in the pathogenesis of chronic lymphocytic leukaemia. Semin Cancer Biol. 2010;20(6):391–399. | ||
de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–2594. | ||
Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. | ||
Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. | ||
O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48–58. | ||
Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2):169–176. | ||
Byrd JC, Brown JR, O’Brien S, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. | ||
Burger JA, Keating MJ, Wierda WG, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15(10):1090–1099. | ||
Brown JC, Barr PM, Flinn I, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study [Abstract 525]. In: 55th ASH Annual Meeting and Exposition; 2013. | ||
Jaglowski SM, Jones JA, Flynn JM, et al. A phase 1b/2 study evaluating activity and tolerability of the BTK inhibitor ibrutinib in combination with ofatumumab in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and related diseases. J Clin Oncol. 2014;32(suppl; abstr 7009):5s. | ||
Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–1110. | ||
Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood. 2015;118(13):3489–3499. | ||
Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. | ||
Wang M, Fayad L, Cabanillas F. Phase 2 trial of rituximab plus hyper-CVAD alternating with rituximab plus methotrexate-cytarabine for relapsed or refractory aggressive mantle cell lymphoma. Cancer. 2008;113(10):2734–2741. | ||
Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. | ||
Campo E, Rule S. Mantle cell lymphoma – evolving management strategies. Blood. 2015;125(1). | ||
Wang ML, Hagemeister F, Westin JR, et al. Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. J Clin Oncol. 2014. | ||
O’Brien SM, Furman RR, Coutre SE, et al. Independent evaluation of ibrutinib efficacy 3 years post-initiation of monotherapy in patients with chronic lymphocytic leukemia/small lymphocytic leukemia including deletion 17p disease. ASCO Meet Abstr. 2014;32(15_suppl):7014. | ||
Young RM, Staudt LM. Ibrutinib treatment of CLL: the cancer fights back. Cancer Cell. 2014;26(1):11–13. | ||
Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. | ||
European Hematology Association. XIII Congress of the Italian Society of Experimental Hematology Rimini, Italy, October 15–17, 2014. Haematology. 2014;99(suppl 2):S1–S113. | ||
Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123:1810–1817. | ||
Ma J, Lu P, Guo A, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166(6):849–861. | ||
Medina DJ, Goodell L, Glod J, Gélinas C, Rabson AB, Strair RK. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor κB pathways. Haematologica. 2012;97(8):1255–1263. | ||
Zhang L, Yang J, Qian J, et al. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood. 2012;120(18):3783–3792. | ||
Medina DJ, Abass-Shereef J, Walton K, et al. Cobblestone-area forming cells derived from patients with mantle cell lymphoma are enriched for CD133+ tumor-initiating cells. PLoS One. 2014;9(4):e91042. | ||
Cheah CY, Chihara D, Romaguera JE, et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol. 2015. | ||
Woyach JA, Ruppert AS, Lozanski G, et al. Association of disease progression on ibrutinib therapy with the acquisition of resistance mutations: a single-center experience of 267 patients. ASCO Meet Abstr. 2014;32(15_suppl):7010. | ||
Byrd JC, O’Brien S, James DF. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:1278–1279. | ||
Klein BEK, Klein R, Lee KE, Gangnon RE. Incidence of age-related cataract over a 15-year interval. The Beaver Dam Eye Study. Ophthalmology. 2008;115:477–482. | ||
Farooqui M, Lozier J, Valdez J, et al. Ibrutinib (PCI 32765) rapidly improves platelet counts in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) patients and has minimal effects on platelet aggregation. Blood. 2012;120:Abstract1789. | ||
Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2015;124(26):3991–3996. | ||
Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2014:1–5. | ||
Ou Z, Zhang L, Newberry K, et al. Abstract 2432: the Bruton’s tyrosine kinase inhibitor ibrutinib synergized with the proteasome inhibitor carfilzomib and overcame immunoproteasome-mediated carfilzomib resistance in mantle cell lymphoma. Cancer Res. 2013;73(8 suppl):2432. | ||
Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol. 2012;13(7):716–723. | ||
Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol. 2013;31(29):3688–3695. | ||
Eve HE, Carey S, Richardson SJ, et al. Single-agent lenalidomide in relapsed/refractory mantle cell lymphoma: results from a UK phase II study suggest activity and possible gender differences. Br J Haematol. 2012;159(2):154–163. | ||
Burger JA. Bruton’s tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep. 2014;9:44–49. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.