Back to Journals » Drug Design, Development and Therapy » Volume 11
A comparative pharmacokinetic and tolerability analysis of the novel orotic acid salt form of tenofovir disoproxil and the fumaric acid salt form in healthy subjects
Authors Kim YK ![]() , Choi MJ, Oh TY, Yu KS
, Choi MJ, Oh TY, Yu KS ![]() , Lee S
, Lee S ![]()
Received 14 August 2017
Accepted for publication 29 September 2017
Published 6 November 2017 Volume 2017:11 Pages 3171—3177
DOI https://doi.org/10.2147/DDDT.S149125
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yu Kyong Kim,1 Mun Ju Choi,2 Tae Young Oh,2 Kyung-Sang Yu,1 SeungHwan Lee1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, 2Dong-A ST Co., Ltd., Seoul, Republic of Korea
Abstract: A novel orotic acid salt form of tenofovir disoproxil (DA-2802) was developed and is expected to replace the fumaric acid salt form. The pharmacokinetic (PK) characteristics and tolerability profiles of DA-2802 were compared to those of tenofovir disoproxil fumarate (TDF, Viread®) in healthy subjects. A randomized, open-label, single-dose study was conducted in 36 healthy subjects using a two-treatment, two-period, and two-sequence crossover design. Subjects received a single oral dose of 319 mg DA-2802 or 300 mg TDF, during each period, with a 7-day washout. Serial blood samples were collected pre-dosing and up to 72 hours post-dosing in each period, for determination of serum tenofovir concentration, which was measured by ultra-performance liquid chromatography-tandem mass spectrometry. A non-compartmental method was used to obtain PK parameters of tenofovir. For comparison between the two tenofovir disoproxil salts, the 90% confidence intervals (90% CIs) of geometric mean ratios of DA-2802 to TDF for the maximum concentration (Cmax) and the area under the concentration–time curve to the last quantifiable concentration (AUC0–t) were determined. The tolerability profiles of tenofovir were assessed by evaluation of adverse events and vital signs, physical examination, ECG, and clinical laboratory tests. The serum tenofovir concentration–time profiles of DA-2802 or TDF were comparable in 32 subjects who completed the study. In both profiles, a two-compartmental elimination with first-order elimination kinetics in the terminal phase was reported in a few subjects, showing a secondary peak in the initial phase of elimination. The geometric mean ratio (90% CI) of DA-2802 to TDF was 0.898 (0.815–0.990) for Cmax and 0.904 (0.836–0.978) for AUC0–t. There were no clinically significant findings in the tolerability assessments. DA-2802 showed comparable PK characteristics and tolerability profiles to TDF.
Keywords: phase I clinical trial, chronic hepatitis B virus infection, drug development
Introduction
Tenofovir is a nucleotide analog that exhibits an antiviral effect via inhibition of various viral DNA polymerases, resulting in termination of viral replication.1 Tenofovir exists in a very polar dianion state at physiological pH,2,3 leading to poor bioavailability. In order to overcome this hurdle, tenofovir disoproxil – as a prodrug of tenofovir – was developed.2 The prodrug form increases both the bioavailability and the stability of tenofovir at both pH 2.0 and 7.4.4 For enhancement of solubility, various salt forms can be conjugated, and the fumaric acid salt form of tenofovir disoproxil is used as the first-line monotherapy for treatment of patients with chronic hepatitis B virus infection.5
The known pharmacokinetic (PK) characteristics of tenofovir disoproxil fumarate (TDF) are that the water-soluble prodrug has a bioavailability of up to 25% in the fasted state and shows dose linearity within the dose range of 75–600 mg, without accumulation with repeated dosing. Tenofovir is barely present in plasma or the serum protein-bound state, and both tenofovir and tenofovir disoproxil are not cytochrome P450 (CYP) enzyme substrates, as shown in in vitro studies.6 The major elimination pathway of tenofovir is via the renal route, with 70%–80% of the drug excreted unchanged in urine.7 The remaining 20%–30% has been shown to be eliminated via the hepatobiliary route in an in vivo animal model.8
A novel tenofovir disoproxil orotate was developed to reduce the manufacturing cost and ultimately provide a cheaper treatment for patients. Orotic acid salt forms are widely available in conjugation with other active ingredients such as choline orotic acid.9 This novel salt form of tenofovir disoproxil is under development as an alternative treatment option to the currently available TDF, and the PK characteristics and tolerability of the two formulations were evaluated in healthy Korean men in this study.
Materials and methods
Study design, subjects, and ethics approval
The study protocol was reviewed and approved by the Seoul National University Hospital Institutional Review Board (IRB) prior to the commencement of the study procedures, and written informed consent was obtained from all subjects for this study (Trial registration number: NCT02557594, registered on September 22, 2015). The study was conducted in full accordance with the principles stipulated in the Declaration of Helsinki as amended in 2013 (Fortaleza, Brazil)10 and the International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) Guideline.11
This was a randomized, single-dose, open-label, two-treatment, two-period, two-sequence crossover study. Subjects who had a history of allergic reactions to tenofovir or had previously participated in other clinical studies within 3 months prior to the day of study drug administration were excluded from the study. Eligible 36 healthy male subjects aged between 20 and 50 (both inclusive) years were randomized to receive one tablet of 319 mg tenofovir disoproxil orotate (DA-2802; Dong-A ST Co., Ltd., Seoul, Republic of Korea) or one tablet of 300 mg tenofovir disoproxil fumarate (TDF, Viread®; Gilead Sciences, Inc., CA, USA) in Period 1 with 200 mL of water and vice versa in Period 2 after a week of washout. All subjects were hospitalized on the day before study drug administration and were required to fast up to 10 hours before dosing. Subjects were restricted from taking any concomitant medication or beverages containing xanthine or alcohol.
Serial blood samples (7 mL) were obtained for determination of serum tenofovir concentration at pre-dosing and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48, and 72 hours after dosing. Tolerability assessments were based on vital signs, physical examinations, and adverse events (AEs) throughout the study.
Determination of serum tenofovir concentration
Serum concentrations of tenofovir were determined using a validated method using ultra-performance liquid chromatography with mass spectrometry. The lower and upper limits of quantification were 5 and 2,000 ng/mL, respectively, and the calibration curve of the method was linear over this range. Where necessary, samples were diluted with normal human serum to bring the concentration within the calibration range. Calibration standards were prepared by spiking blank plasma samples with known amounts of tenofovir ranging from 5 to 2,000 ng/mL. The within-run accuracy (expressed as the percentage difference from the theoretical concentration) of the quality control samples used during the sample analysis ranged from −10.7% to −2.0%, with a within-run precision (expressed as the coefficient of variation) of ≤4.0%. The between-run accuracy ranged from −8.0% to 0.4%, with a between-run precision of ≤7.2%.
Pharmacokinetic evaluation and statistical analyses
For determination of the tenofovir PK parameters between the two salt forms, a non-compartmental analysis was conducted. By implementation of Phoenix® WinNonlin® (version 6.3; Certara USA Inc., Princeton, NJ, USA), the following PK parameters were derived: maximum concentration (Cmax) (ie, Tmax); area under the concentration-time curve to the last quantifiable concentration (AUC0–t, where t is the last time point with a quantifiable concentration) and from 0 to infinity (AUC0–inf); the terminal half-life (t1/2); and the apparent clearance (CL/F). The observed concentrations and times were used to determine the Cmax and Tmax of tenofovir. The AUCs were calculated using the linear trapezoidal method, and t1/2 was calculated as the natural logarithm of 2 divided by the elimination rate constant, which was calculated as the slope of the terminal data from the semilogarithmic plot by linear regression.
The PK parameters of log-transformed Cmax, AUC0–t, and AUC0–inf of tenofovir were compared between the two salt forms based on the two one-sided tests procedure by mixed-model analyses of variance for the crossover design – considering sequence, treatment, and period as fixed effects and the subjects nested within the sequence as a random effect. Statistical analyses were conducted using SAS (version 9.4; SAS Institute Inc., Cary, NC, USA). The 90% confidence intervals (CIs) of geometric mean ratios (GMR) of the orotic acid to fumaric acid salt forms for Cmax, AUC0–t, and AUC0–inf were calculated for statistical comparison between the two salts. The formulations were considered to be similar if the 90% CIs for these parameters were within the range of 0.8–1.25.
Safety and tolerability evaluation
Physical examinations, vital sign evaluation, laboratory tests, 12-lead ECGs, and AE monitoring were conducted throughout the study period for evaluation of safety and tolerability after single oral administrations of the two salt forms of tenofovir. For AE monitoring, the subjects were interviewed via non-leading open questions such as “how-do-you-feel.”
Results
Subject disposition and demographics
Of the 36 intention-to-treat subjects randomized, 34 were administered with the study drug at least once after screening, and 32 completed the study and were included for statistical evaluation. The four dropouts were due to the subjects’ withdrawal of consent. The age range of the study participants was 21–42 years, and the mean ± standard deviation (SD) of weight, height, and BMI was 70.4±7.4 kg, 175.4±5.5 cm, and 22.9±2.0 kg/m2, respectively.
PK characteristics
Tenofovir was absorbed rapidly after a single oral administration of orotic acid and fumaric acid salt form of tenofovir disoproxil to 32 healthy male subjects with the Tmax of the tenofovir ranging between 0.5 and 2.0. The mean serum tenofovir concentrations versus time profiles almost overlapped between the two salt forms (Figure 1). After reaching Tmax, tenofovir showed a two-compartmental elimination with first-order elimination kinetics in the terminal phase. During the initial phase of elimination, a few subjects showed a secondary peak in both orotic acid and fumaric acid salt forms of tenofovir disoproxil treatment groups (Figure 2).
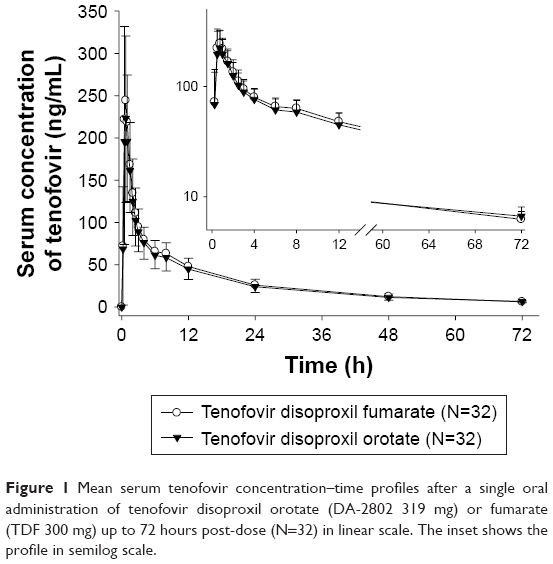 | Figure 1 Mean serum tenofovir concentration–time profiles after a single oral administration of tenofovir disoproxil orotate (DA-2802 319 mg) or fumarate (TDF 300 mg) up to 72 hours post-dose (N=32) in linear scale. The inset shows the profile in semilog scale. |
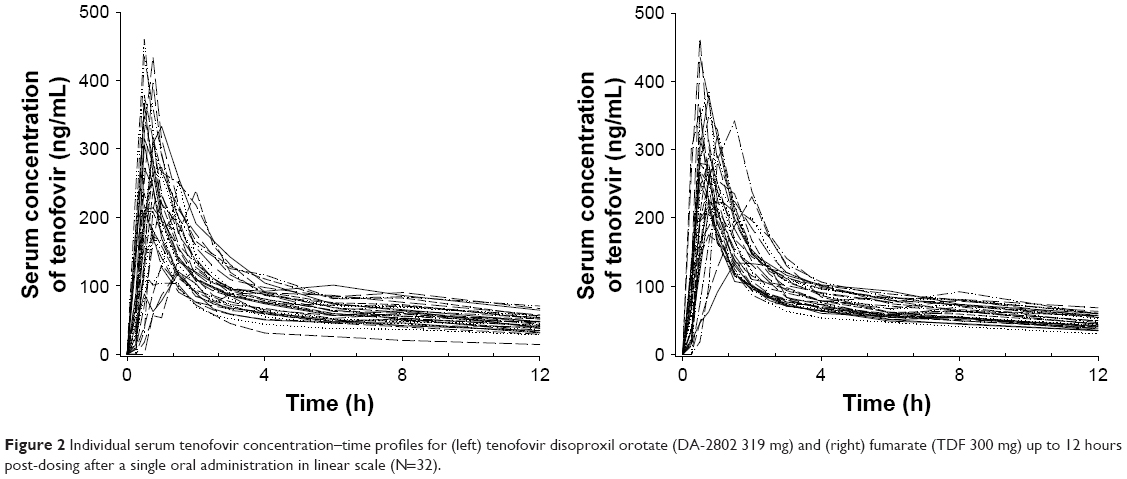 | Figure 2 Individual serum tenofovir concentration–time profiles for (left) tenofovir disoproxil orotate (DA-2802 319 mg) and (right) fumarate (TDF 300 mg) up to 12 hours post-dosing after a single oral administration in linear scale (N=32). |
As a result, the 90% CIs of GMR for Cmax and the AUCs between the two salt forms were entirely within the conventional bioequivalence range of 0.80–1.25 (Table 1 and Figure 3).
 | Table 1 Pharmacokinetic parameters of tenofovir and statistical comparison between tenofovir disoproxil orotate (DA-2802 319 mg) and fumarate (TDF 300 mg) after a single oral administration |
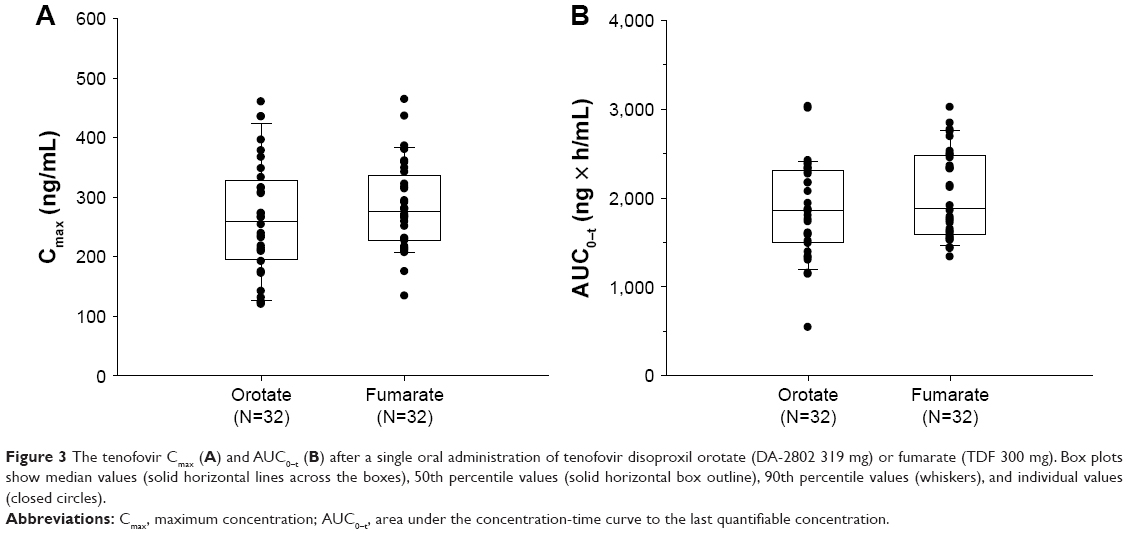 | Figure 3 The tenofovir Cmax (A) and AUC0–t (B) after a single oral administration of tenofovir disoproxil orotate (DA-2802 319 mg) or fumarate (TDF 300 mg). Box plots show median values (solid horizontal lines across the boxes), 50th percentile values (solid horizontal box outline), 90th percentile values (whiskers), and individual values (closed circles). |
Safety and tolerability
Of the 34 subjects who had received tenofovir disoproxil orotate, 2 (5.9%) reported five AEs during the study. After administration of TDF, 3 (9.4%) of the 32 subjects reported five AEs, which were considered by the investigator to be study drug-related AE. The most frequently reported study drug-related AEs after administration of TDF were fatigue, occurring in two subjects. However, the two subjects who had experienced five AEs after administration of a single oral tenofovir disoproxil orotate were considered to not be related to the study drug.
After administration of tenofovir disoproxil orotate and TDF, all reported AEs were mild in severity, and there were no deaths or serious AEs that caused withdrawals from the study. There were no clinically significant changes in the clinical laboratory results, vital sign measurements, and ECG tolerability parameters throughout the study period.
Discussion
In this study, we first evaluated the PK of the novel salt form of tenofovir disoproxil after a single oral administration in healthy male subjects. In comparison with those of the currently available fumaric acid salt form (TDF 300 mg), there were no significant differences in the Tmax as well as the Cmax and AUC values (Table 1), which were within the bioequivalence criteria stated by the Korea Ministry of Food and Drug Safety12 and the US Food and Drug Safety.13 This is significant because such data support the purpose of incrementally modified drug development, which ultimately improves the patient’s quality of life by 1) lowering the cost involved in manufacturing process for salt conjugation, which would ultimately be advantageous for the consumers, and 2) improving stability over that of the currently marketed fumarate form by prolonging its shelf life.14 It is thus expected that the new orotic acid salt form of tenofovir disoproxil may be used as an alternative therapy to the fumaric acid salt form.
In line with the previously described tenofovir PK,15 some individuals showed secondary peaks of various sizes at 6–10 hours in both or only in one of the treatment groups (Figure 2). The secondary peaks in both the treatment groups were larger than those in one of the treatment groups, suggesting that such phenomenon is caused by the characteristics of the compound rather than the differences in the salt forms. Such compound characteristics may have possibly occurred due to the involvement of enterohepatic circulation. As previously shown, up to 80% of tenofovir is excreted unchanged in urine within 72 hours after dosing.7 Therefore, the remaining 20% of tenofovir may be subjected to reabsorption via enterohepatic circulation, leading to the secondary peak in some individuals. In addition, due to interindividual variabilities and the relatively small portion of drug eliminated via the enterohepatic route, only a few subjects may have shown this kind of profile.
When comparing the tenofovir Cmax and AUC0–t of the two treatment groups, there were approximately three- to fivefold differences between the subjects who showed the highest and the lowest values (Figure 3B). Such variations in the tenofovir Cmax and AUC0–t were observed for each individual, regardless of treatment-group differences (Figure S1), and individual factors may be the cause of the observed variations. Because there were no particularities in either the renal functions or the demographic characteristics among all subjects, and presuming for possibilities in other interindividual variabilities, differences in transporter abundance between the subjects may be suspected. The multidrug resistance-associated protein 2 (MRP2), present on the apical side of the membrane with the role of non-bile salt organic anion excretion, contributes toward hepatobiliary elimination of tenofovir16 without affecting the intestinal absorption of the total tenofovir equivalents.8 Such variations in MRP2 expression might have, therefore, resulted in the interindividual variabilities in exposure. For this speculation to be confirmed, MRP2 genotype information of individual subjects would be supportive, which was not determined in the present study.
Other limitations are that the study was carried out only in healthy subjects. The PK characteristics of tenofovir disoproxil orotate are still unknown in the patient population with chronic hepatitis B; however, there have been reports that similar PK was observed in the HIV-1-infected patient population when compared to that in healthy subjects.7 Therefore, the new orotic acid salt form of tenofovir disoproxil is expected to have similar PK in the patient population.
Conclusion
The novel orotic acid salt form of tenofovir disoproxil oral formulation (DA-2802 319 mg) showed similar PK characteristics with comparable tolerability profiles as that of the fumaric acid salt form (TDF 300 mg). These similarities of the new salt formulation provide evidence for it being a suitable candidate as an alternative to the existing tenofovir disoproxil for patients with chronic hepatitis B.
Acknowledgments
The first author (Yu Kyong Kim) received a scholarship from the BK21-plus education program provided by the National Research Foundation of Korea. This study was sponsored by Dong-A ST Co., Ltd.
Disclosure
Ms Mun Ju Choi and Mr Tae Young Oh are employees of Dong-A ST Co., Ltd. None of the authors of this study are involved in financial or personal relationships with agencies, institutions, or organizations that could inappropriately influence the statistical analysis plan or interpretation of this work. The authors report no other conflicts of interest in this work.
References
Balzarini J, Holy A, Jindrich J, et al. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob Agents Chemother. 1993;37(2):332–338. | ||
Fardis M, Oliyai R. Case study: tenofovir disoproxil fumarate: an oral prodrug of tenofovir. Prodrugs. New York, NY: Springer; 2007:1347–1357. | ||
Jenh AM, Thio CL, Pham PA. Tenofovir for the treatment of hepatitis B virus. Pharmacotherapy. 2009;29(10):1212–1227. | ||
Yuan LC, Dahl TC, Oliyai R. Degradation kinetics of oxycarbonyloxymethyl prodrugs of phosphonates in solution. Pharm Res. 2001;18(2):234–237. | ||
Ozaras R, Mete B, Ceylan B, et al. First-line monotherapies of tenofovir and entecavir have comparable efficacies in hepatitis B treatment. Eur J Gastroenterol Hepatol. 2014;26(7):774–780. | ||
Food And Drug Administration Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review of NDA Number 21–356, Viread. Rockville, MD US Food and Drug Administration Division of Neuropharmacological Drug Products; 2001. | ||
Viread® [drug label]. Foster City, CA: Gilead Sciences Inc; 2012. | ||
Mallants R, Van Oosterwyck K, Van Vaeck L, Mols R, De Clercq E, Augustijns P. Multidrug resistance-associated protein 2 (MRP2) affects hepatobiliary elimination but not the intestinal disposition of tenofovir disoproxil fumarate and its metabolites. Xenobiotica. 2005;35(10–11):1055–1066. | ||
Stahl PH, Wermuth CG. Handbook of Pharmaceutical Salts: Properties, Selection, and Use. Weinheim: Wiley VCH; 2002. | ||
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. | ||
Food And Drug Administration. Guidance for industry, E6 good clinical practice: consolidated guidance. Presented at International Conference on Harmonisation Good Clinical Practice. Federal Register. Rockville, MD: U.S. Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER), ICH. 1996; Vol 101997. 691–709. | ||
Korea Ministry of Food and Drug Safety. [Guidance document for bioequivalence studies updated in 2012]. 2010. Available from: http://drug.mfds.go.kr/html/CLASS/board/download.jsp?boardType=BOARD&boardSeq=104&seq=2731&fileSeq=1966. Accessed October 30, 2017. Korean. | ||
Food and Drug Administration Center for Drug Evaluation and Research. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products – general considerations. 2003. Available from: https://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf. Accessed October 30, 2017. | ||
Park SG, Lim JI, Kim YJ, et al. Novel tenofovir disoproxil salt and the preparation method thereof: Google Patents. 2015. Available from: https://www.google.com/patents/WO2015002434A1?cl=en. Accessed October 30, 2017. | ||
Yeh WW, Fraser IP, Caro L, et al. No Meaningful Pharmacokinetic Interaction between HCV Protease Inhibitor MK-5172 abd Tenofovir or Raltegravir. Presented at 2014 Conference on Retroviruses and Opportunistic Infections. March 3–6, 2014; Boston. | ||
Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126(1):322–342. |
Supplementary material
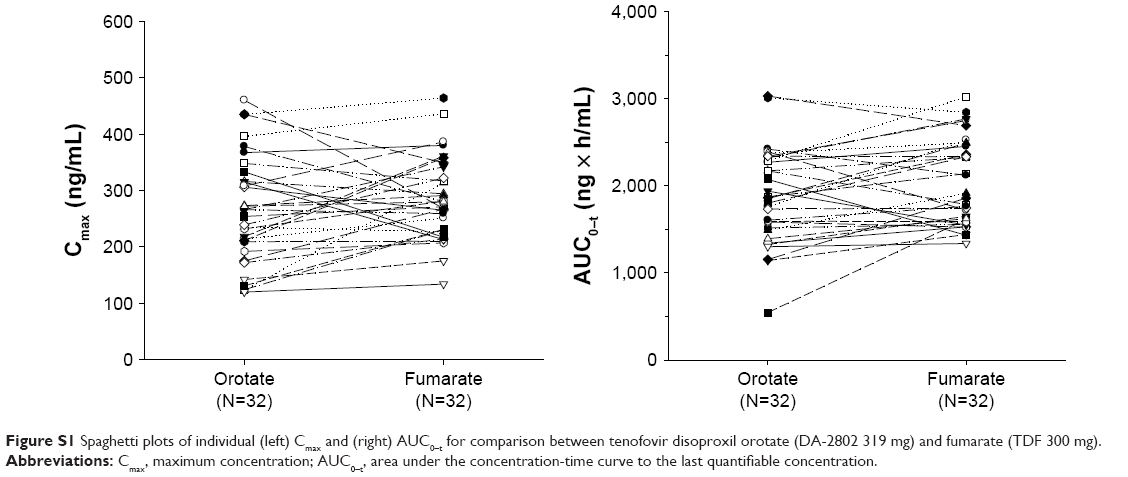 | Figure S1 Spaghetti plots of individual (left) Cmax and (right) AUC0–t for comparison between tenofovir disoproxil orotate (DA-2802 319 mg) and fumarate (TDF 300 mg). |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.