Back to Journals » International Journal of Nanomedicine » Volume 16
A Combined Antitumor Strategy Mediated by a New Targeted Nanosystem to Hepatocellular Carcinoma
Authors Farinha D, Migawa M, Sarmento-Ribeiro A, Faneca H ![]()
Received 16 January 2021
Accepted for publication 27 April 2021
Published 18 May 2021 Volume 2021:16 Pages 3385—3405
DOI https://doi.org/10.2147/IJN.S302288
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel Rubinstein
Dina Farinha,1,2 Michael Migawa,3 Ana Sarmento-Ribeiro,1,4,5 Henrique Faneca1,2
1CNC - Center for Neuroscience and Cell Biology (CNC), University of Coimbra, Coimbra, Portugal; 2Institute for Interdisciplinary Research (IIIUC), University of Coimbra, Coimbra, Portugal; 3Ionis Pharmaceuticals Inc, Carlsbad, USA; 4Laboratory of Oncobiology and Hematology (LOH) and University Clinic of Hematology, Faculty of Medicine, University of Coimbra, Coimbra, Portugal; 5Clinical Hematology Department, Centro Hospitalar Universitário de Coimbra (CHUC), Coimbra, Portugal
Correspondence: Henrique Faneca
CNC - Center for Neuroscience and Cell Biology (CNC), University of Coimbra, Coimbra, 3004-504, Portugal
Tel +351-239-820-190
Fax +351-239-853-607
Email [email protected]
Background: Hepatocellular carcinoma (HCC) is one of the main causes of cancer-related death. Sorafenib, which is the first-line therapy for this disease, is associated with reduced therapeutic efficacy that could potentially be overcome by combination with selumetinib. In this context, the main goal of this work was to develop a new nanosystem, composed of a polymeric core coated by a lipid bilayer containing the targeting ligand GalNAc, to specifically and efficiently co-deliver both drugs into HCC cells, in order to significantly increase their therapeutic efficacy.
Methods: The physicochemical characterization of hybrid nanosystems (HNP) and their components was performed by dynamic light scattering, zeta potential, matrix-assisted laser desorption ionization – time of flight mass spectroscopy, and transmission electron microscopy. Cellular binding, uptake and specificity of HNP were evaluated through flow cytometry and confocal microscopy. The therapeutic activity was evaluated namely through: cell viability by the Alamar Blue assay; cell death by flow cytometry using FITC-Annexin V; caspases activity by luminescence; mitochondrial membrane potential by flow cytometry; and molecular target levels by Western blot.
Results: The obtained data show that these hybrid nanosystems present high stability and loading capacity of both drugs, and suitable physicochemical properties, namely in terms of size and surface charge. Moreover, the generated formulation allows to circumvent drug resistance and presents high specificity, promoting great cell death levels in HCC cells, but not in non-tumor cells. This potentiation of the antitumor effect of co-loaded drugs was carried out by an increased programmed cell death, being associated with a strong reduction in the mitochondrial membrane potential, a significant increase in the activity of caspases 3/7 and caspase 9, and much greater number of annexin V-positive cells.
Conclusion: The developed formulation resulted in a high and synergistic antitumor effect, revealing a translational potential to improve therapeutic approaches against HCC.
Keywords: hepatocellular carcinoma, hybrid nanosystems, drug delivery, GalNAc, sorafenib, selumetinib
Introduction
Primary liver cancer is the seventh most common cancer in the world and the second leading cause of cancer mortality.1 Hepatocellular carcinoma (HCC) represents about 75–85% of primary liver cancers.2 Currently, surgical resection and liver transplantation are the most effective therapeutic approaches for HCC in early stages. However, for the majority of HCC patients, it is not possible to perform surgery at the time of diagnosis.3 In these cases, sorafenib, which is a multikinase inhibitor, represents the standard therapy for patients with advanced HCC and in intermediate-stage that are not eligible for locoregional therapies.3,4 In most studies carried out, sorafenib has shown improved survival when compared with placebo.5 Sorafenib can block tumor cell proliferation by inhibiting the activity of RAF-1, B-RAF and other kinases in the RAS/RAF/MEK/ERK signaling pathway.6 Furthermore, sorafenib can inhibit angiogenesis through targeting vascular endothelial growth factor receptor −2/-3 (VEGFR-2/-3), platelet-derived growth factor receptor (PDGFR-β) and other tyrosine kinases.7,8 Although treatment with sorafenib is potentially associated with inhibition of tumor growth and angiogenesis, exposure to sorafenib leads to up-regulation of pERK, thus promoting cellular resistance to chemotherapy and consequently treatment failure.9 In this context, the combination of sorafenib with a mitogen-activated protein kinase (MEK) inhibitor could hypothetically abrogate this effect and potentiate the antitumor activity. Selumetinib (AZD6244, ARRY142886) is a potent and selective allosteric MEK1/2 inhibitor.10,11 MEK1/2 is a critical component of the RAS/RAF/MEK/ERK pathway, and its activation is essential for cell proliferation and central to drive the growth and progression of cancer.12,13 In fact, owing to the complexity of hard-to-treat diseases such as cancer, current developments have gently shifted from a focus on monotherapy to combined or multiple therapies since the synergy of therapeutic agents or techniques could give rise to superadditive therapeutic effects.14 However, both sorafenib and selumetinib have an inherent toxicity that leads to highly undesirable side effects. In this way, unspecific toxicity and low water solubility strongly limit the use of these drugs in many clinical applications.15,16
Nanotechnology could play a fundamental role in circumventing these limitations, since it allows the development of functional nanostructures which can improve the solubility of drugs that are poorly soluble in water; target them to the specific tissue/site to minimize side effects; prolong their half-life in the systemic circulation by reducing immunogenicity; release them at a sustained rate; and deliver two or more drugs simultaneously.17,18 In this regard, liposomes and poly (lactic-co-glycolic acid) (PLGA) nanoparticles have been used as drug delivery systems. They have common characteristics, such as biocompatibility, high drug loading capacity, long circulation time and tunable size.19,20 Nevertheless, they also have unique properties. In the case of PLGA nanoparticles, they easily incorporate substances with distinct physicochemical characteristics, including proteins and anticancer drugs, and their degradation rate can be flexibly controlled, varying the molar ratio between glycolic acid and lactic acid.21 On the other hand, liposomes can load hydrophobic drugs within the lipid bilayer and hydrophilic drugs in the aqueous nucleus, and their surface chemistry can be easily changed to meet different requirements, simply by adjusting both the concentrations and the type of lipids.22 It is also possible to produce liposomes sensitive to a wide range of external stimuli, which allows for a highly controlled release of their load.23 Lipid-PLGA hybrid nanoparticles make it possible to combine the desirable characteristics of PLGA nanoparticles and liposomes.24 This type of nanoparticles exhibits great potential as delivery systems for cancer drugs, enabling the encapsulation of drugs in both the PLGA nucleus and the lipid bilayer.24 They also play an important role in overcoming the increasingly prevalent multidrug resistance (MDR), allowing a high therapeutic index with low toxicity.24,25 Moreover, the therapeutic responses are even more improved when these nanosystems are coated with targeting ligands, allowing them to specifically deliver into target cells.26,27 The asialoglycoprotein receptor is a hetero-oligomer that is expressed in the liver and overexpressed in HCC and is practically absent in the other tissues.28 This receptor is found in the hepatocyte membrane and regulates the levels of plasma glycoproteins with α2,6-galactose-linked sialic acid as terminal residue.29 In fact, it has high binding capacity to D-galactose and N-acetyl-D-galactosamine terminated carbohydrate residues, promoting its efficient cellular uptake.27 Several studies have shown that the use of specific ligands for this receptor allows the efficient and specific targeting of nanosystems to HCC models.28,30,31 In this regard, Ionis Pharmaceuticals Inc. developed a triantennary N-acetylgalactosamine (GalNAc) cluster that has a high affinity for the asialoglycoprotein receptor.32
In this study, we designed a novel delivery nanosystem, composed of PLGA nanoparticles coated with a lipid bilayer containing DSPE-PEG (2000)-GalNAc, to mediate a targeted therapeutic strategy to HCC involving the combination of selumetinib and sorafenib. This new hybrid nanosystem (HNP) allows not only a high encapsulation of selumetinib and sorafenib, but also their targeted and controlled release into hepatocellular carcinoma cells, bypassing several limitations associated with the use of free drugs, consequently promoting a great therapeutic effectiveness.
Experimental Section
Materials
Poly(d,l-lactic-co-glycolic acid) (PLGA) 50:50 DL (24–38kDa), polyvinyl alcohol (PVA) (MW 9000–10,000, 80% hydrolyzed), ethyl acetate (Sigma Aldrich), acetone, coumarin-6, JC-1 probe, fluorescein diacetate and propidium iodide were purchased from Sigma-Aldrich; Nile Red (TCI chemicals); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (Chol) and 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000) were purchased from Avanti PolarLipids; Selumetinib was kindly offered by AstraZeneca; sorafenib was kindly provided by Bayer HealthCare Pharmaceutical; and triantennary N‐acetylgalactosamine (GalNAc) cluster was kindly offered by Ionis Pharmaceuticals.
Production of Nanoparticles
The PLGA nanoparticles were prepared by solvent emulsification and diffusion-evaporation method.33 Briefly, 50 mg of PLGA 50:50 block copolymer (24–38 kDa) solution in 2.5 mL ethyl acetate was poured in 5 mL of aqueous phase containing 5% PVA and stirred for 30 minutes, to get a primary emulsion, using a homogenizer (T 10 basic, Ultra Turrax, IKA). The primary emulsion was then diluted with water at 50 °C and stirred overnight to remove the organic solvent. The effect of variables like PVA concentration, PLGA concentration and homogenization time, on the particle characteristics was studied. For binding/uptake studies and for studies with drugs, 50 µg of coumarin-6 or 1 mg of drugs, respectively, were dissolved in the organic phase. Nanoparticles were recovered by ultracentrifugation at 17,000 g which allowed the removal of non-encapsulated coumarin-6 or drugs in PLGA nanoparticles.
Large unilamellar liposomes (LUV) were prepared by extrusion of multilamellar liposomes (MLV) composed of 1:1 (molar ratio) mixtures of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol (Chol). Briefly, lipids dissolved in chloroform were mixed and dried under vacuum in a rotator evaporator. The dried lipid films were hydrated with deionized water and the resulting MLV were then sonicated for 3 min and extruded through two stacked polycarbonate filters of 200 nm pore diameter using a Liposofast device (Avestin, Toronto, Canada). For binding/uptake studies and for studies with drugs, 30 µg of nile red or 0.5 mg of drugs, respectively, were added in chloroform.
Liposomes containing the GalNAc ligand were prepared by the postinsertion method.34 The GalNAc PFP ester dissolved in acetone was coupled to DSPE-PEG2000-amine micelles, prepared in sodium tetraborate buffer at pH 8.5. The coupling reaction was performed overnight with stirring. GalNAc O-acetyl groups were removed by the addition of aqueous ammonia. Different organic solvent/aqueous solution ratios and reaction temperatures were tested in order to optimize the conjugation. Micelles were purified by size exclusion chromatography on a Sephadex G-25 M (GE Healthcare) column using water as running buffer to remove unreacted GalNAc. The product was analysed by MALDI-TOF-MS, by using an UltrafleXtreme mass spectrometer (Bruker, Bremen, Germany) operating in reflector positive ion detection mode with laser smartbeam 2 and under Compass flex Series 1.4 software control (Bruker, Bremen, Germany).
The insertion of DSPE-PEG-GalNAc conjugates or plain DSPE-PEG-amine onto the preformed liposomes, at 2 mol % (relative to the total lipid amount), was performed through micelles incubation with liposomes in a water bath at 40 °C for 16 hours under agitation.
Hybrid nanosystems (HNP) were prepared by adding a dispersion of PLGA nanoparticles to the preformed liposomes containing DSPE-PEG-GalNAc or DSPE-PEG-amine. The mixture was vortexed for 1 hour at 60 °C (T> Tm) and 1000 rpm. Thereafter, the dispersion was centrifuged at 9000 g for 30 min at 15 °C to separate particles from non-adsorbed lipids. The supernatant was used to determine the free lipid concentration and consequently the amount of non-adsorbed lipids.
Physicochemical Characterization of Nanoparticles
The physicochemical characterization of HNP and their components was performed in terms of average hydrodynamic diameter, polydispersity index, zeta potential and transmission electron microscopy.
The measurements of dynamic light scattering (DLS) of the nanoparticles and their components were performed in a Zetasizer Nano-ZS (Malvern Instruments Ltd., UK) and converted into average hydrodynamic diameter and polydispersity index through the software Zetasizer 7.02. Nanosystems were also characterized in relation to their zeta potential using a Zetasizer Nano-ZS (Malvern Instruments Ltd., UK) that measures electrophoretic and zeta mobility potential distribution by a method of light scattering by phase analysis. The analyses were performed at 25 °C, in water, and at a backward scattering angle of 173°.
Nanosystems were analysed in terms of their size/structure by transmission electron microscopy. The nanosystems were absorbed on copper grids covered with formvar film. To promote contrast, uranyl acetate was used. The grids were examined in a JEM 1400 Transmission Electron Microscope (Jeol, Tokyo, Japan). Images were digitally recorded using a SC1000 ORIUS CCD camera (Gatan, PA, USA) and analysed with an EDX Microanalysis System (Oxford Instruments, Abingdon, UK)
In vitro Release Studies
The in vitro release of nile red and coumarin-6 from hybrid nanosystems was studied by using dialysis membranes, with a molecular weight cut-off of 14,000 Da, against phosphate buffer solutions with two different pH values (5 and 7.4) and in the absence or presence of HCC cell extracts. Briefly, 50 mg of HNP was added to a dialysis bag which was then suspended in 50 mL of release medium at 37 °C and stirred continuously at 250 rpm. Small aliquots (1 mL) of release medium were removed over time and replaced with the same volume of fresh medium. Then, the aliquots were lyophilized, dissolved in DMSO and the fluorescence intensity was measured in a SPECTRAmax GEMINI EM fluorometer (Molecular Devices, CA, USA). The concentration of the probes was determined by comparing the fluorescence intensity of the samples to that of the standards (calibration curves).
Cell Cultures
The human hepatocellular carcinoma cell lines (HepG2 and Hep3B cells), the human glioblastoma cell line (DBTRG-05MG cells) and the human skin fibroblast cell line (HFF-1 cells) were maintained at 37 °C, under 5% CO2, in Dulbecco’s modified Eagle’s medium-high glucose (DMEM-HG) (Sigma–Aldrich, MO, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Alfagene), penicillin (100 U/mL) and streptomycin (100 mg/mL). The HepG2, Hep3B, DBTRG-05MG and HFF-1 cells were seeded at a density of 60 × 103 cells/cm2, 30 × 103 cells/cm2, 15 × 103 cells/cm2 and 20 × 103 cells/cm2, respectively. All cell lines used were purchased from ATCC.
Spheroid Cultures
To generate spheroids, cells suspended in the complete medium were seeded at a density of 6 × 103 cells/well in 96-well round-bottomed ultra-low attachment microplates (Corning® Costar®) and incubated at least for 3 days, at 37 °C, in a humidified atmosphere of 5% CO2.
Cellular Uptake and Internalization
Twenty-four hours before incubation with HNP, HepG2, Hep3B and DBTRG-05MG cells were seeded in 12-well culture plates. In the competition studies, 30 min after incubating cells with 1.7 mL of DMEM-HG medium, containing or not 1.5 mg/mL of asialofetuin, the hybrid nanosystems, prepared with coumarin-6 and nile red, were added to each well. Following 4 hours incubation at 4 °C (binding) or 37 °C (uptake), cells were washed twice with PBS, detached, and resuspended in PBS. Then, the samples were evaluated by flow cytometry (FACSCalibur flow cytometer, Becton Dickinson, NJ, USA). Live cells were gated by forward/side scattering from a total of 20,000 events and data was analysed using CellQuest software.
For confocal microscopy, HepG2, Hep3B and DBTRG-05MG cells were incubated with HNP, containing coumarin-6 and nile red, for 4 hours at 37 °C. Afterwards, cells were washed with PBS and fixed with 4% paraformaldehyde, for 15 min at room temperature. Then, cells were washed again with PBS and nuclei were stained with 1 μg/mL Hoechst 33258 dye (Molecular Probes) in PBS upon cell incubation for 5 min. Cells were finally washed with PBS and directly observed for intracellular fluorescence distribution under a confocal scanning laser microscope Zeiss LSM 510 Meta (Carl Zeiss, Oberkochen, Germany), using the 63× oil immersion objective.
Cell Viability
The cytotoxicity of the different hybrid nanosystems or the free drugs was evaluated through the Alamar Blue assay.30 This assay measures the redox capacity of the cells due to the production of metabolites as a result of cell growth. Briefly, 0.2 mL of DMEM-HG medium containing 10% (v/v) alamar blue dye, prepared from a 0.1 mg/mL stock solution of alamar blue, was added to each well after 72 hours of incubation with HNP or free drugs. After 1 hour of incubation at 37 °C, the absorbance at 570 and 600 nm was measured in a SPECTRAmax PLUS 384 spectrophotometer (Molecular Devices, CA, USA). Cell viability (as a percentage of untreated control cells) was calculated according to the formula (A570 - A600) of treated cells x 100/(A570 - A600) of control cells.
Cell Death
To evaluate the cell death promoted by the different conditions, flow cytometry was used by using the probes FITC-Annexin V (Immunostep, Salamanca, Spain) and propidium iodide (PI, Sigma). After 48, 72 and 96 hours of incubation with hybrid nanosystems or free drugs, cell media and detached cells were harvested, washed with ice-cold PBS and resuspended in 100 μL of binding buffer (10 mM HEPES (pH 7.4), 2.5 mM CaCl2, 140 mM NaCl) to which 2 µL of FITC annexin V (0.05 mg/mL) and 1 µL of PI (0.05 mg/mL) were added. Samples were incubated for 5 min in the dark at room temperature and after that analysed in a FACSCalibur flow cytometer (Becton Dickinson, NJ, USA). FITC fluorescence was evaluated in the FL-1 channel, propidium iodide was evaluated in the FL-3 and 20,000 events were collected. The data were analysed using CellQuest software.
Caspases Activity
The activities of caspase-3/7, −8 and −9 were measured using a luminescence assay, with Caspase-Glo 3/7, Caspase-Glo 8 and Caspase-Glo 9 assay kit (Promega, USA). The assessment was performed according to the protocol given by the manufacturer. Twenty four hours after HepG2 and Hep3B cells have been plated on 96-well plates, they were incubated with the different HNP or the free drugs for 72 hours. In the case of spheroids, after 3 days of spheroid formation, they were incubated with the different HNP or the free drugs for 96 hours, under gentle agitation at 37°C. After the different treatments, 100 µL of assay reagent were added to each well and incubated for 1 hour at room temperature. Finally, the luminescence was measured in a Synergy HT luminometer (Biotek, USA).
Mitochondrial Membrane Potential
Mitochondrial membrane potential was analysed by flow cytometry using JC-1 fluorescent dye. Briefly, 24 hours after HepG2 and Hep3B cells have been plated on 12-well plates, they were incubated with the different hybrid nanosystems or the free drugs for 48 hours. Then, the cells were resuspended and incubated with fresh culture medium, containing JC-1 dye (2.5 µg/mL), for 40 minutes at 37 °C in the dark. After incubation, the cells were washed with PBS and analysed in a FACSCalibur flow cytometer (Becton Dickinson, NJ, USA). The shift from red (JC-1 oligomer) to green (JC-1 monomer) fluorescence indicates the decrease in mitochondrial membrane potential.
Western Blotting
Total protein extracts were prepared from pellets of HepG2 and Hep3B cells treated with the different HNP or the free drugs for 48 hours. The cells were lysed for 30 minutes in 1 × RIPA buffer (150 mM NaCl, 50 mM Tris-base, 5 mM EGTA, 1% Triton X-100, 0.5% DOC and 0.1% SDS) that contained a protease inhibitor cocktail (Sigma) and phosphatase inhibitors. The concentration of protein lysates was determined using the BC protein assay kit (Biorad, CA, USA). Samples containing equal amounts of protein (30 μg) were resuspended in loading buffer (20% glycerol, 10% SDS, 0.1% bromophenol blue) and incubated for 5 min at 95 °C. Then, they were resolved on SDS-PAGE in a 12% polyacrylamide gel. After that, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore). The membrane was then blocked in 5% BSA for 1 hour at room temperature and incubated, overnight at 4 °C, with anti-MEK (#9122, 1:1000), anti-pMEK (Ser217/221) (#9154, 1:1000), anti-ERK1/2 (#9107, 1:2000), anti-pERK1/2 (Thr202/Tyr204) (#4370, 1:2000) (Cell Signaling), and anti-GAPDH (#MAB1667, 1:1000) (Millipore) antibodies, and with the appropriate alkaline phosphatase labelled-secondary antibodies (1:10,000) (Thermo Fisher) for 2 hours at room temperature. After antibody incubation, the membranes were washed four times with TBS-T, and then incubated with the enzyme substrate ECF (Thermo Fisher) for 5 min at room temperature. The chemofluorescence was detected through a ChemiDoc Touch System (Bio-Rad) and the analysis of band intensity was made using the Quantity One software (Bio-Rad).
Spheroid Treatment and Growth
After the initial 3 days of formation, spheroids were incubated with different HNP or free drugs for the desired time, under gentle agitation, at 37 °C and in a humidified atmosphere of 5% CO2. Phase contrast images (Analysis imaging system CELENA® S, Logos Biosystems) were used to observe spheroid diameter and compaction over the treatment time. Live-dead staining of spheroids was done using fluorescein diacetate (FDA) and propidium iodide. Briefly, 100 μL of a solution containing 2 mg/mL propidium iodide in HBS and 2 µL of 5 mg/mL FDA in acetone was added to the spheroids and incubated for 5 minutes at room temperature. Spheroids were washed with PBS and visualized using an Axio Imager Z2 microscope (Zeiss, Munich, Germany) using an AxioCamHRc camera (Zeiss, Germany).
Statistical Analysis
Data were analysed using the Prism software (version 5.0). Statistical significance of differences between data was evaluated by Student’s t-tests and one-way ANOVA. A value of p < 0.05 was considered significant. To assess the normality of the population, the Shapiro–Wilk test was used.
Results and Discussion
Synthesis and Characterization of Hybrid Nanosystems
In this work, a new nanosystem formulation to mediate the specific co-delivery of two chemotherapeutic agents into HCC cells was developed. As illustrated in Figure 1, these nanocarriers consisted in a core-shell nanostructure constituted by a core of PLGA coated with a lipid bilayer composed of POPC, Chol, DSPE-PEG2000-amine, and DSPE-PEG2000-GalNac.
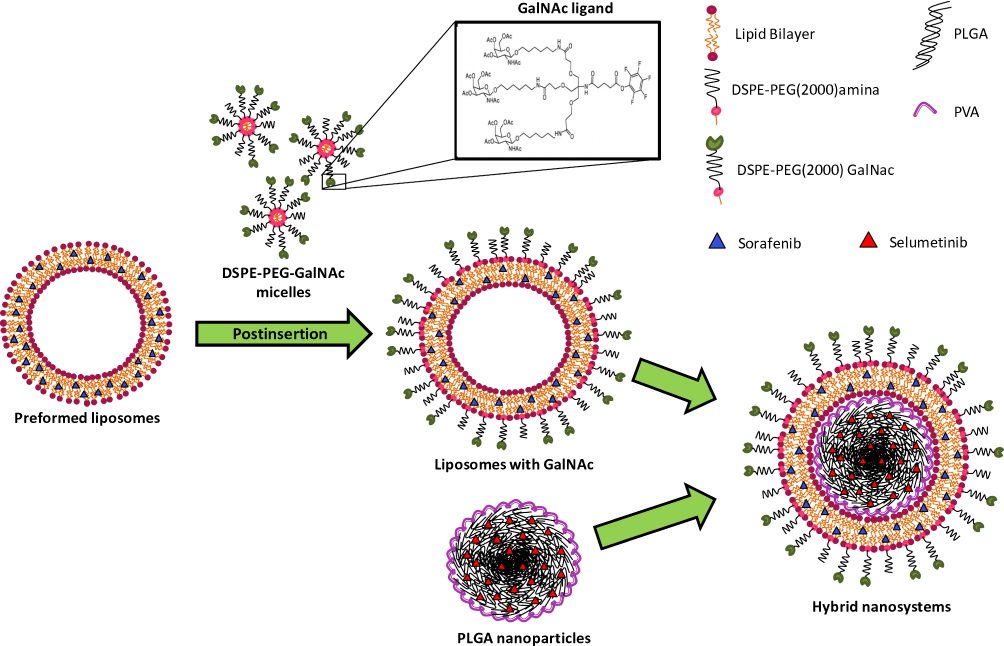 |
Figure 1 Schematic representation of the hybrid nanosystems preparation. |
The production of these hybrid nanoparticles (HNP) was performed in two stages. In the first step, the polymeric core was produced with the biodegradable and biocompatible PLGA copolymer using the solvent emulsification diffusion-evaporation method.35 Several parameters, such as polymer and surfactant concentration, and homogenization time, that are known to affect the physicochemical properties of PLGA nanoparticles, were tested.36 The obtained results (Supplementary Figure S1A–C) showed that the best formulation of PLGA nanoparticles was obtained with 2% of PLGA, 5% of PVA for 30 min of homogenization. As shown in Figure 2A, these nanoparticles presented a mean diameter of approximately 170 nm and a superficial charge near the −40 mv that remained almost unchanged during 120 days at room temperature, showing their high stability even at long-term storage, which is a critical issue in drug delivery formulation development.
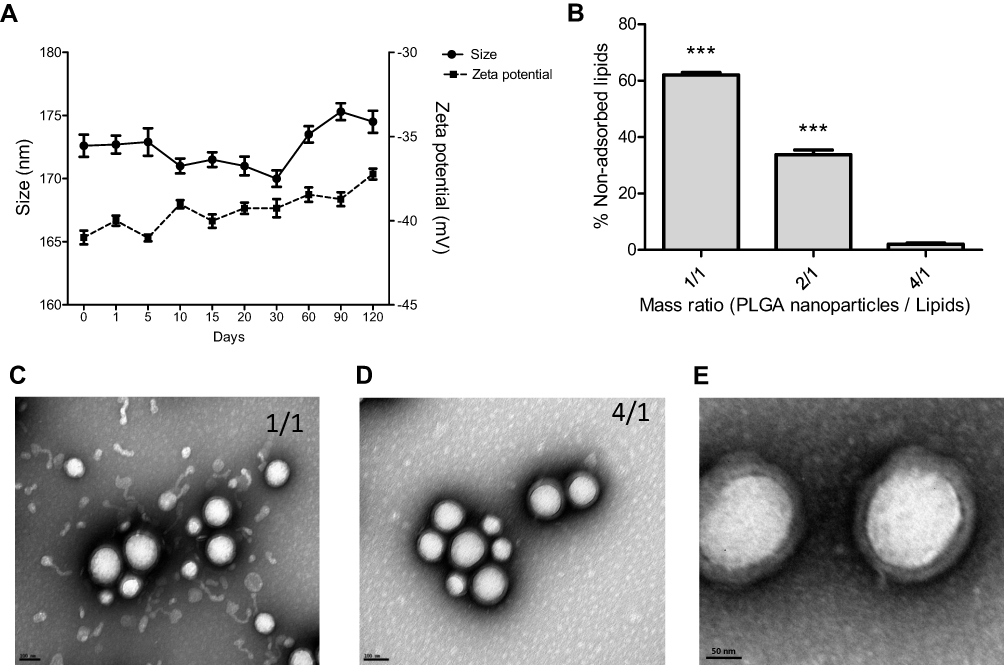 |
Figure 2 Development of the hybrid nanosystems. (A) Stability and physicochemical properties of PLGA nanoparticles, size and zeta potential, over 120 days at room temperature. (B) Percentage of non-adsorbed lipids for the different PLGA/lipid mass ratios tested in the HNP development. Data are expressed as mean ± SD obtained from four in dependent samples (n= 4). (C) and (D) Transmission electron microscopy representative images of HNP with different PLGA/lipid mass ratios ((C) 1/1 mass ratio and (D) 4/1 mass ratio) (scale bar = 100 nm). (E) Transmission electron microscopy representative image of HNP (PLGA/lipid 4/1 mass ratio), showing the lipid-coated PLGA nanoparticles (scale bar = 50 nm). (***P< 0.001) Denotes a statistically significant difference in the percentage of non-adsorbed lipids when compared to that obtained in the production of the hybrid nanoparticles in the 4/1 mass ratio. |
Then, in the second stage, the PLGA nanoparticles were coated with the lipid bilayer, through their mixture with the preformed liposomes, as previously described.37 Different PLGA/lipid mass ratios were tested in the production of the hybrid nanosystems (HNP), in order to determine the lowest amount of liposomes needed to cover all PLGA nanoparticles. As illustrated in Figure 2C–E, the TEM images revealed that for the PLGA/lipid mass ratios tested the PLGA nanoparticles were entirely coated, which is denoted by the darker outline, having been observed an excess of free liposomes in the case of the 1/1 mass ratio (Figure 2C) that was not detected for the 4/1 mass ratio (Figure 2D). These results were corroborated through the quantification of the non-adsorbed lipids (Figure 2B), which demonstrated that for the 1/1 mass ratio it was observed 62% of free liposomes, while in the case of the 4/1 ratio, only 2% of free lipids was registered. Moreover, as shown in Table 1, the differences between the physicochemical properties of the HNP (4/1 mass ratio) and those of the PLGA nanoparticles also confirmed the coating of the later with the lipid bilayer. In this context, to prepare the hybrid nanosystems for the subsequent studies the 4/1 PLGA/lipid mass ratio was selected.
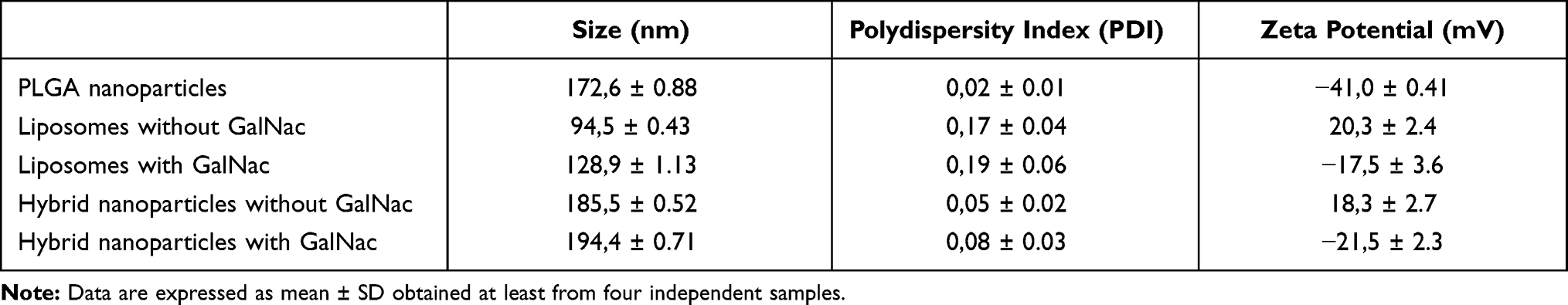 |
Table 1 Physicochemical Characterization of Hybrid Nanosystems and Their Components |
Since the triantennary N-acetylgalactosamine (GalNAc) cluster was recently described as having high affinity to the asialoglycoprotein receptor (ASGP-R), which is overexpressed in HCC cells, we selected this cluster as the targeting moiety of our HNP to confer specificity to HCC cells.32,38–40 To this purpose, we conjugated the GalNAc cluster to the DSPE-PEG2000-amine, testing different conjugation conditions as described in the material and methods section, and evaluated it by MALDI-TOF-MS.41–43 As presented in Supplementary Table S1, for the best conditions of reaction it was obtained a conjugation efficiency of 93%. This high yield was confirmed by MALDI-TOF-MS since, as illustrated in Figure 3A, the profile of the detected molecular weights of DSPE-PEG2000 suffered a shift to the right (higher molecular weights) after GalNAc cluster conjugation. In fact, the peak of the DSPE-PEG2000-amine was observed between 2300 and 3360 m/z, while the peak of the DSPE-PEG2000-GalNAc was registered between 3970 and 4660 m/z, demonstrating the successful conjugation of the GalNAc cluster to DSPE-PEG2000-amine.41
 |
Figure 3 Coating nanosystems with triantennary N-acetylgalactosamine (GalNAc) cluster. (A) Successful synthesis of DSPE-PEG2000-GalNAc demonstrated by MALDI-TOF-MS analysis; Ai) DSPE-PEG2000; Aii) DSPE-PEG2000-GalNAc. (B) Stability and physicochemical characteristics, size and zeta potential, of HNP with (blue) and without (green) DSPE-PEG2000-GalNAc, over 30 days at room temperature. |
Additionally, the physicochemical properties of the nanosystems (Table 1), both the liposomes and the hybrid nanosystems, also confirm the effective GalNAc cluster conjugation to DSPE-PEG2000-amine, since this conjugation induced an increase in their mean diameter and a strong decrease in their zeta potential, most probably due to the suppression of the positive charge of the DSPE-PEG2000 terminal end amine group by the conjugation of the GalNAc cluster, which exposes the N-acetylgalactosamine groups at the surface of the nanosystems conferring them a negative surface charge.44 The surface charge of nanosystems, such as this negative zeta potential of HNP, favours the electrostatic repulsions between them, contributing to their stability even over long storage periods at room temperature (Figure 3B).45
Loading and Release Capacity of the Hybrid Nanosystems
In order to evaluate the ability of the HNP to transport more than one chemotherapeutic agent, we loaded the nanosystems with two hydrophobic fluorescent dyes, coumarin-6 and nile red.46 The coumarin-6 was encapsulated in the polymeric core, while the nile red was loaded in the lipid bilayer of the hybrid nanosystems. The loading efficiency was 76% for coumarin-6 and 44% for nile red, most probably due to the higher hydrophobic area of the PLGA core when compared with that of the lipid bilayer. Regarding the release kinetics of the fluorescent dyes, as illustrated in Figure 4, it was observed a greater and faster release for nile red than for coumarin-6, possibly justified by the fact that the later needs to cross both the polymeric core and the lipid bilayer.47 Moreover, it was registered a faster release of the dyes in acid conditions (pH 5), than at pH 7.4, and in the presence of HCC cell lysates, indicating that the developed hybrid nanosystems are responsive to these factors, most probably being sensitive to the endosome acidic pH as well as to the interaction with cells, which could result in a more efficient release of the drugs in target cells.48,49
 |
Figure 4 Release profiles of probes in the hybrid nanosystems. (A) Effect of pH and HCC cell extracts on the release kinetics of nile red encapsulated in the lipid bilayer of HNP. (B) Effect of pH and HCC cell extracts on the release kinetics of coumarin-6 encapsulated in the PLGA nucleus of HNP. (***P< 0.001) Denotes a statistically significant difference in the release kinetics at 72 hours when compared to the release kinetics occurred at pH 7.4, for each probe. |
Cell Binding, Uptake and Specificity of Hybrid Nanosystems
The effect of GalNAc cluster association on the cell binding and internalization of HNP as well as on their specificity to target cells was evaluated in different cell lines, both ASGP-R-expressing and non-expressing cells. For this purpose, the HNP, loaded with the fluorescent dyes coumarin-6 and nile red, and prepared with or without the GalNAc cluster, were incubated with two HCC cell lines (HepG2 and Hep3B) and one non-liver cancer cell line (DBTRG-05MG glioblastoma cell line) at 4°C (for cell binding) or at 37°C (for cell uptake).31,50 After incubation, cells were analysed by flow cytometry (Figure 5A, B and E) and confocal microscopy (Figure 5C and D).
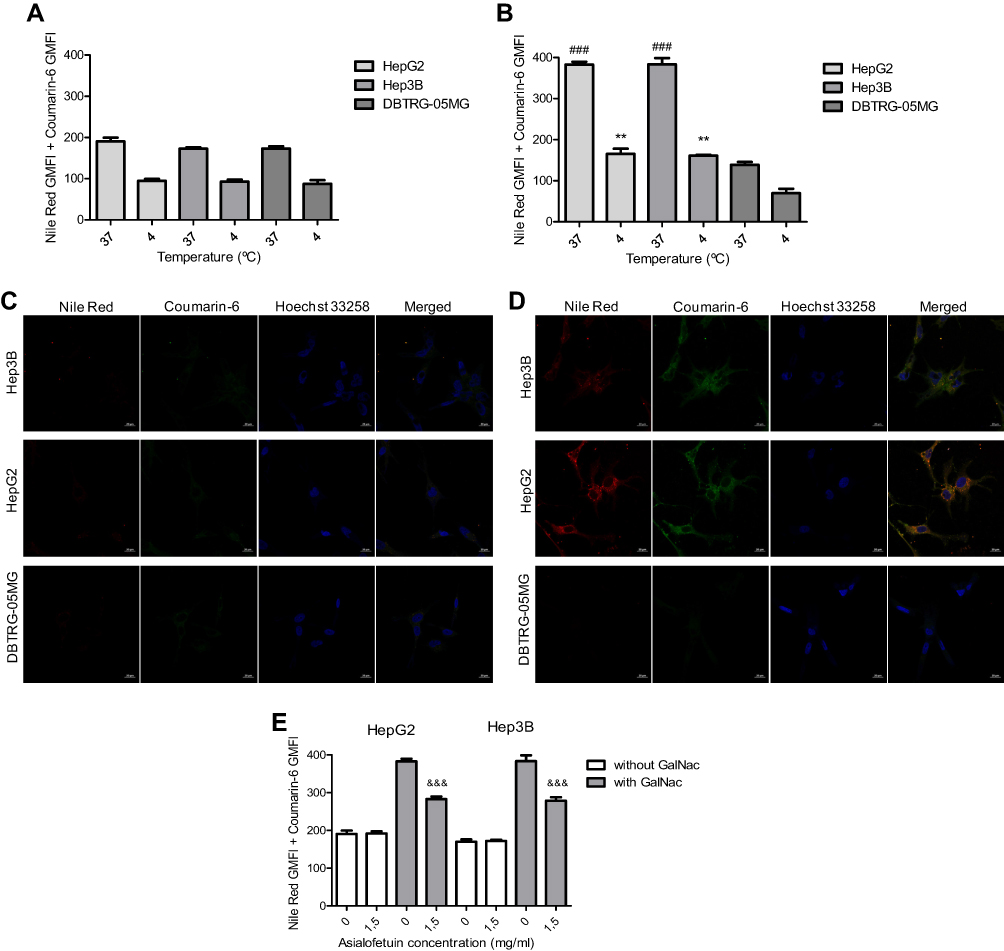 |
Figure 5 Evaluation of cell binding, uptake and specificity of hybrid nanosystems. (A and B) For cell binding and uptake, 0.5 μg/mL of HNP, loaded with coumarin-6 and nile red and prepared with (B) or without (A) the GalNAc cluster, were incubated with HepG2 and Hep3B HCC cells and the DBTRG-05MG glioblastoma cell line at 4 °C (for cell binding) or at 37 °C (for cell uptake), for 4 hours. (C and D) Representative confocal laser scanning microscopic images showing the uptake of the HNP, loaded with coumarin-6 and nile red and prepared with (D) or without (C) the GalNAc cluster, in the HepG2, Hep3B and DBTRG-05MG cells (scale bar = 20 µm). The Hoechst 33258 was used to mark the nuclei in blue. The green colour corresponds to coumarin-6 and the red one to nile red encapsulated in the HNP. (E) To further assess the specificity of the HNP, they were incubated with the HCC cell lines in the presence or absence of 1.5 mg/mL asialofetuin. The flow cytometry results were expressed through the sum of the Geometric mean fluorescence intensity in FL1 (coumarin-6) with the Geometric mean fluorescence intensity in FL4 (Nile Red). Data are expressed as mean ± SD obtained from triplicates of three independent experiments (n=3). (**P< 0.01) Denotes statistically significant difference in cell binding when compared to DBTRG-05MG cells. (###P < 0.001) Denotes statistically significant difference in cell uptake when compared to DBTRG-05MG cells. (&&&P < 0.001) Denotes statistically significant difference when compared to cells in the absence of 1.5 mg/mL asialofetuin. |
The obtained results showed that the extent of cell binding and uptake of HNP prepared without GalNAc did not present significant differences between the three cell lines, as observed both by flow cytometry (Figure 5A) and confocal microscopy (Figure 5C). However, the comparison between the results presented in Figure 5A with those presented in Figure 5B showed that the conjugation of the GalNAc cluster to nanosystems promoted a substantial increase in their levels of cell binding and uptake in both HCC cell lines but not in the glioblastoma cells. In fact, with DBTRG-05MG cells, it was even observed a slight decrease in those levels when HNP prepared with GalNAc were used. These observations, which were corroborated by confocal microscopy data (Figure 5C and D), were most probably due to the fact that HNP containing the GalNAc cluster have the ability to interact with the ASGP-R, resulting in a higher cell binding and internalization in HCC cells than their counterpart without GalNAc.30,32,39 In the case of glioblastoma cells, the slight decrease in cell binding and uptake of nanosystems prepared with GalNAc is justified by the absence of the ASGP-R in these cells and by the negative surface charge of these nanosystems (Table 1) that hampers their interaction with the negatively charged cell membrane, which most possibly do not happen with the positively charged HNP prepared without GalNAc.35
In order to clarify if the potentiation of cell binding and internalization of HNP in HCC cells, promoted by the association of GalNAc to nanosystems, is due to a specific interaction with the ASGP-R, a competitive inhibition study with asialofetuin (a specific ligand for the ASGP-R30,51) was performed. As illustrated in Figure 5E, the presence of 1.5 mg/mL of free asialofetuin promoted a significant decrease in the cellular uptake of HNP produced with GalNAc but not in those prepared without the cluster. This effect was not associated with any cytotoxicity (Supplementary Figure S2). These results (Figure 5) indicate that binding and internalization of these HNP by HCC cells are due to the GalNAc cluster interaction with the ASGP-R.
Drug Loading and Extent of Cytotoxicity
The developed hybrid nanosystems have the ability to simultaneously transport different chemotherapeutic agents and to specifically deliver them into HCC cells, potentially resulting in an increase of their therapeutic efficiency and reduction of side effects, strongly associated with antineoplastic drugs such as selumetinib and sorafenib. In order to maximize the transport of these drugs in our nanosystems several loading strategies were tested, from individual to simultaneous drug loading in the polymeric core and/or in the lipid bilayer. The results presented in Table 2 show that the highest loading capacity for the two drugs was obtained for the encapsulation of the selumetinib in the polymeric core (7,76 µg/mg HNP) and the sorafenib in the lipid bilayer (6,58 µg/mg HNP) of HNP. On the other hand, the smallest loading capacity was registered for the simultaneous encapsulation of selumetinib (2,62 µg/mg HNP) and sorafenib (2,53 µg/mg HNP) in the lipid bilayer (Table 2).
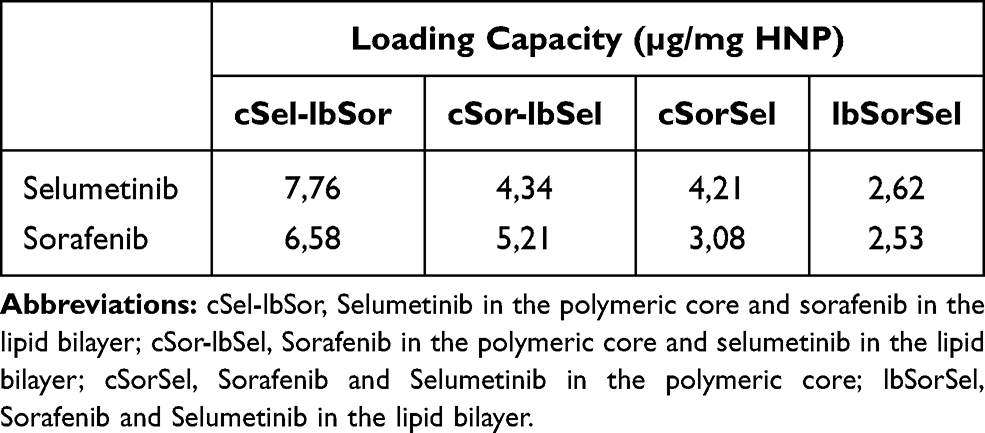 |
Table 2 Loading Capacity for Sorafenib and Selumetinib in the Hybrid Nanosystems |
In order to correlate the several loading strategies with the therapeutic potential of the obtained formulations, the cytotoxicity of different amounts of HNP, prepared with the GalNAc cluster, was evaluated in two HCC cell lines (HepG2 and Hep3B). As illustrated in Figure 6A and B, the cell death effect was dose-dependent for all formulations and the highest outcome was obtained, for both cell lines, with HNP encapsulating selumetinib in the polymeric core and sorafenib in the lipid bilayer, which corresponds to the formulation with maximum loading capacity. This formulation was consequently selected to the subsequent studies.
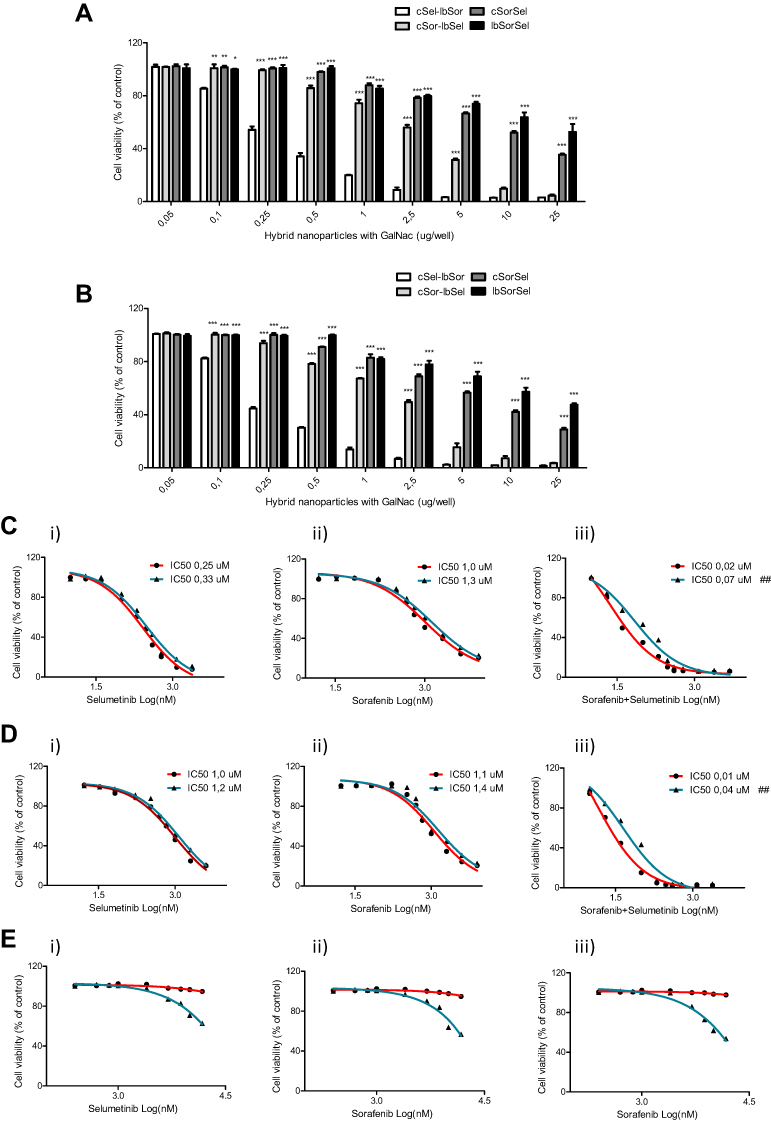 |
Figure 6 Therapeutic potential of the different formulations and of the distinct strategies mediated or not by the new hybrid nanosystems. (A) HepG2 and (B) Hep3B cells were treated with different amounts (0–25 μg/well) of distinct formulations of HNP, for 72 hours, and then the viability was analysed using the Alamar blue assay. The tested formulations were: (cSel-lbSor) Selumetinib encapsulated in the polymeric core and sorafenib in the lipid bilayer of the HNP; (cSor-lbSel) Sorafenib encapsulated in the polymeric core and selumetinib in the lipid bilayer of the HNP; (cSorSel) Sorafenib and Selumetinib encapsulated in the polymeric core of the HNP; (lbSorSel) Sorafenib and Selumetinib encapsulated in the lipid bilayer of the HNP. Dose-response curves for (C) HepG2, (D) Hep3B and (E) HFF-1 cells after treatment for 72 hours with: (i) HNP containing only selumetinib (red) or free selumetinib (blue); (ii) HNP containing only sorafenib (red) or free sorafenib (blue); and (iii) HNP containing selumetinib and sorafenib (red) or the combination of both free drugs (blue). Drug concentrations from 0 to 14μM were tested. Cell viability was analysed using the alamar blue assay and is expressed as a percentage of untreated control cells. Curves represent the average values from at least three independent experiments (n=3). (***P< 0.001); (**P< 0.01); (*P< 0.05) Denotes a statistically significant difference in cell viability when compared with cells treated with hybrid nanosystems containing selumetinib encapsulated in the polymeric core and sorafenib in the lipid bilayer (cSel-lbSor), for the same µg/well of hybrid nanoparticles. (##P< 0.01) Denotes a statistically significant difference in the IC50 values when compared to the IC50 of the drugs encapsulated in the hybrid nanoparticles, in the same cell line. |
The cytotoxicity of HNP by themselves (unloaded nanosystems) was also evaluated to clarify if the registered cell death effect was exclusively due to the chemotherapeutic agents. For this purpose, HepG2 and Hep3B HCC cell lines and one non-cancer cell line (HFF-1) were incubated with increasing doses of unloaded HNP. The obtained data showed a complete absence of cytotoxicity until 100µg of unloaded HNP per well (100µg/well) (Supplementary Figure S3), which corresponds to a large excess of nanosystems, indicating that the observed cell death effect registered with loaded HNP (Figure 6) was only due to the selumetinib and sorafenib.
Therapeutic Potential of the Combined Strategy Mediated by Hybrid Nanosystems
After selection of the formulation and verification of the non-cytotoxicity of the unloaded HNP, the therapeutic potential of the combined strategy, mediated by the nanosystems, was compared with the combination of free drugs and with the individual approaches (selumetinib alone or sorafenib alone), mediated or not by the hybrid nanosystems, in the above-mentioned cell lines (HepG2, Hep3B and HFF-1). The results presented in Figure 6C and D demonstrated that the therapeutic potential of the combined and individual strategies, mediated by the developed hybrid nanosystems, was higher, in both HCC cell lines, than that of the corresponding free drugs, since the IC50 values of these HNP-mediated strategies were always smaller than those of the corresponding free drugs. This difference was particularly evident in the case of the combined approach, as the IC50 values obtained after HCC cell treatment with HNP containing both selumetinib and sorafenib were approximately 4-fold smaller than those registered with the combination of these free drugs (Figure 6C and D). The lower IC50s of chemotherapeutic agents delivered by HNP, when compared with the corresponding free drugs, was most probably due to the higher cellular internalization of the drugs mediated by the targeted HNP, through their interaction with the ASGP-Rs overexpressed in HCC cells, such as HepG2 and Hep3B, that promoted their cellular uptake. This higher internalization of the drugs resulted in a better therapeutic efficiency and consequently in the reduction of the IC50s. Moreover, this effect could also involve the ability of HNP to avoid the drug efflux process of cancer cells, which could also contribute to lower IC50s.52 Our data also revealed that the combined strategies were always much more toxic to HCC cells than individual approaches, showing a synergistic effect.
On the other hand, with the non-cancer HFF-1 cell line the IC50s of free drugs, both combined and individually used, were much lower than that observed with the HNP-mediated strategies (Figure 6E), most probably due to the lack of the ASGP-R in HFF-1 cells and to the negative surface charge of the generated HNP, which impaired the interaction and internalization of the targeted nanosystems by HFF-1 cells, consequently reducing the cellular internalization of the drugs and the subsequent cell death effect on non-cancer cells.28,35 These observations constitute an evidence that the combined strategy mediated by HNP presents higher therapeutic potential and less side effects that the free drugs.
Cell Death Mechanisms
In order to clarify the mechanisms involved in the antitumor activity of the individual and combined strategies, mediated by the targeted HNP, the extent of apoptosis and necrosis/late apoptosis was evaluated by cell staining with annexin V and PI. As illustrated in Figure 7A and C, for both HCC cell lines, cell treatment for 72h with HNP containing both selumetinib and sorafenib resulted in almost all cells stained with PI and/or annexin V, which was not observed in no other experimental condition, neither the individual strategies mediated by the HNP nor the free drug combination (at the same concentration), validating the higher cell death effect observed with the combined strategy mediated by HNP (Figure 6).
 |
Figure 7 Apoptosis levels promoted by the therapeutic strategies in HCC cells. HepG2 (A (72h) and B) and Hep3B (C (72h) and D) cells lines were stained with Annexin V and PI double-staining for 15 min after being treated, for 48, 72 and 96 hours, with empty HNP; HNP containing only selumetinib or sorafenib; HNP containing sorafenib and selumetinib; or with both free drugs. 4μg of HNP or the corresponding free drug concentrations were added to each well of 12-well plates. The apoptosis levels of cells were analysed by flow cytometry. The percentages of live cells, cells in early apoptosis and cells in late apoptosis/necrosis were presented in bar graphs (B and D). Data are expressed as mean ± SD (n=3) obtained from three independent experiments. (***, ### and &&&P < 0.001) Denotes a statistically significant difference in terms of living cells when compared with cells treated with HNP containing sorafenib and selumetinib, for 48, 72 and 96 hours of treatment, respectively. |
Moreover, this observation was reinforced by the strong synergistic effect, in terms of the amount of cells with double staining, registered with the combined strategy mediated by HNP, but not with the combined free drugs, when compared to the individual approaches.
Nevertheless, the high staining with annexin V and PI (Figure 7A and C) did not allow the elucidation between cells in late apoptosis or in necrosis.53 However, looking for the obtained data at 48 h (Figure 7B and D; and Supplementary Figure S4) it was possible to observe that treatment with the combined strategy mediated by HNP resulted in a small percentage of HCC cells with double staining (annexin V and PI), while still promoted a high percentage of HCC cells stained with annexin V. On the other hand, at 96 h (Figure 7B and D; and Supplementary Figure S4) it was registered an increase in the percentage of HCC cells with double staining (annexin V and PI), when compared to that observed at 72 h, indicating that most probably this combined therapeutic strategy promoted an early apoptotic process that evolve to late apoptosis.53 These studies demonstrated that the simultaneous treatment of HCC cells with these two drugs mediated by our HNP was more efficient than the combination of the free drugs. As previously referred, this fact is most probably due to the higher cellular internalization of the drugs, when delivered by the targeted nanosystems, and to the ability of these HNP to overcome at least partially the chemoresistance. In fact, HCC cells are known to overexpress the ABC (ATP-binding cassette) transporters, including P-glycoprotein (P-gp/MDR1/ABCB1), multidrug resistance-associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/MXR/ABCG2), that are responsible for their high resistance to chemotherapy.51,54 Therefore, the encapsulation of selumetinib and sorafenib in HNP hinder the drugs access to the ABC transporters, reducing their efflux and consequently increasing the cell death effect.
Caspases activation is a crucial event in the apoptosis induction. Initiator caspases, caspase-9 and caspase-8, are activated by intrinsic and extrinsic apoptotic pathways, respectively, leading to the activation of effector caspases (caspase-3/7) that executed the apoptosis through the cleavage of cellular components.55 In order to quantify the caspases activation and to determine the involved apoptotic pathway in HCC cells death induced by the different treatments, the activity of caspases-3/7, −8 and −9 were evaluated. As shown in Figure 8A and B, the obtained results revealed a strong activation of caspases-3/7 and −9, in both HCC cell lines, after treatment with the combined strategy (selumetinib and sorafenib) mediated by the developed HNP, this activation being much higher than that observed with the individual drugs and better than the combination of free drugs. On the other hand, the activation of caspase-8 was residual for both HCC cells, not presenting significant differences between the different experimental conditions.
 |
Figure 8 Levels of caspase activation and mitochondrial membrane potential of treated cells. (A and B) Relative luminescence produced by the activity of caspase −3/7, −8 and −9 in (A) HepG2 and (B) Hep3B cells treated, for 72 hours, with empty HNP; HNP containing only selumetinib or sorafenib; HNP containing sorafenib and selumetinib; or with both free drugs. 0.5 μg of HNP or the corresponding free drug concentrations were added to each well of 96-well plates. (C and D) The plot of JC-1 red verse green emission from flow cytometry on mitochondrial membrane potential in HepG2 (C) and Hep3B (D) cells. (E and F) Percentage of HepG2 (E) and Hep3B (F) cells with JC-1 green or red fluorescence quantified by flow cytometry. HepG2 and Hep3B cells were stained with JC-1, for 40 min at 37 °C, after being treated with the different therapeutic approaches. The percentage of green and red cells are expressed as mean ± SD obtained at least from three independent experiments (n=3). Asterisks represent significant differences in caspase 3/7 activity. Cardinals represent significant differences regarding the activity of caspase 9. Ampersands represent significant differences regarding the percentage of cells with JC-1 green fluorescence. (***, ### and &&&P < 0.001); (** and ##P< 0.01)); (#P< 0.05) Denotes a statistically significant difference when compared with cells treated with hybrid nanosystems containing sorafenib and selumetinib. |
The parallel up-regulation of caspase-3/7 and caspase-9 demonstrated the involvement of the intrinsic apoptotic pathway, indicating that most probably occurred mitochondrial changes, such as the collapse of mitochondrial membrane potential, alterations in mitochondrial permeability, and release of cytochrome c to cytosol.56,57 In this context, we analyzed the mitochondrial membrane potential through the JC-1 assay. This fluorescent cationic probe accumulates and aggregates in the mitochondria in the case of high mitochondrial membrane potential, yielding a red fluorescence, and remain as monomers in the cytosol at low concentration in the case of collapse of mitochondrial membrane potential, exhibiting green fluorescence.58–60 As illustrated in Figure 8C–F, the treatment of both HepG2 and Hep3B cells with the combination of selumetinib and sorafenib, delivered by the targeted HNP, resulted in a much higher percentage of green cells than that obtained with the other experimental conditions, including the simultaneous treatment with the two free drugs.
These data clearly revealed that the combined strategy mediated by the developed nanosystems promoted a strong reduction in the mitochondrial membrane potential, characteristic from the apoptotic processes, that was not observed with the other therapeutic approaches. This fact corroborates the results presented above demonstrating that this strategy (combined treatment mediated by the HNP) has much more therapeutic potential than individual approaches or treatment with the combined free drugs.
Then, we evaluated the levels of pERK and pMEK in the two HCC cell lines through immunoblotting, after treatment with HNP containing the two drugs in combination or separately and the free drugs under the same conditions (Figure 9). As expected, in cells treated with free sorafenib, we identified an up-regulation of phospho-ERK (Figure 9A–C and E).8 This up-regulation was significantly attenuated when sorafenib was encapsulated in HNP (Figure 9A–C and E). This is possibly due to the fact that hybrid nanoparticles allow to circumvent the cell resistance mechanisms associated with sorafenib. As expected, we found that in the presence of selumetinib, ERK phosphorylation was reduced (Figure 9A–C and E).9,10 This observation becomes more evident when selumetinib is encapsulated in HNP, as well as when combined with sorafenib. The exposure to the MEK inhibitor selumetinib resulted in an increase of p-MEK (Figure 9A, B, D and F). This is possibly due to the fact that the aromatic fluorine of allosteric MEK inhibitor selumetinib interacts with MEK residue S212, which results in exposure of the phosphorylation sites S218/S222 that are then susceptible to RAF mediated phosphorylation due to the feedback loop of MAPK pathway inhibition.61,62 Although MEK phosphorylation in presence of selumetinib can still occur, the transduction of the signal by MEK-mediated phosphorylation of ERK is no longer possible. These results, as well as those presented above, demonstrate once again that the combined strategy mediated by HNP has much more therapeutic potential than individual approaches or treatment with combined free drugs.
 |
Figure 9 Effect of therapeutic strategies in ERK and MEK protein levels. Western blot analysis of phosphorylated (p) and total (t) ERK, and phosphorylated (p) and total (t) MEK in HepG2 (A, C and D) and Hep3B (B, E and F) cells after being treated, with empty HNP; HNP containing only selumetinib or sorafenib; HNP containing sorafenib and selumetinib; or with free drugs (A–F); 8 μg of HNP or the corresponding free drug concentrations were added to each well of 6-well plates. Representative Western blot images displaying the levels of p/t-ERK and p/t-MEK in HepG2 (A) and Hep3B (B) cells. Graphical representation of the p-ERK/t-ERK (C and E) and p-MEK/t-MEK (D and F) ratios in HepG2 and Hep3B cells. Data are expressed as mean ± SD obtained from three independent experiments (n=3). (***P < 0.001); (**P< 0.01); (*P< 0.05) Denotes a statistically significant difference when compared with cells treated with hybrid nanosystems containing sorafenib and selumetinib. |
Antitumor Activity in Three-Dimensional Cell Culture Systems
The three-dimensional (3D) cell culture systems have been used to overcome some limitations associated with 2D cell cultures, allowing to obtain data physiologically more relevant and more extrapolative. In this regard, we evaluated the different therapeutic approaches in spheroids obtained with HepG2 and Hep3B cells, with a mean diameter of approximately 300 µm. The results presented in Figure 10A–D demonstrated that both HepG2 and Hep3B spheroids suffered a substantial reduction in their sizes/volumes only in the cases they were treated with the combination of both chemotherapeutic agents, this being particularly evident when mediated by the developed HNP, not only in terms of the spheroid size (Figure 10A and B) but also in terms of their structure (Figure 10C) and amount of live cells (Figure 10D). In fact, at the end of treatment (8 days), only spheroids incubated with the combined strategy mediated by HNP did not present a spheroid structure but rather a structure of small aggregates (Figure 10Cii), which were composed in its majority by dead cells (Figure 10Dii).
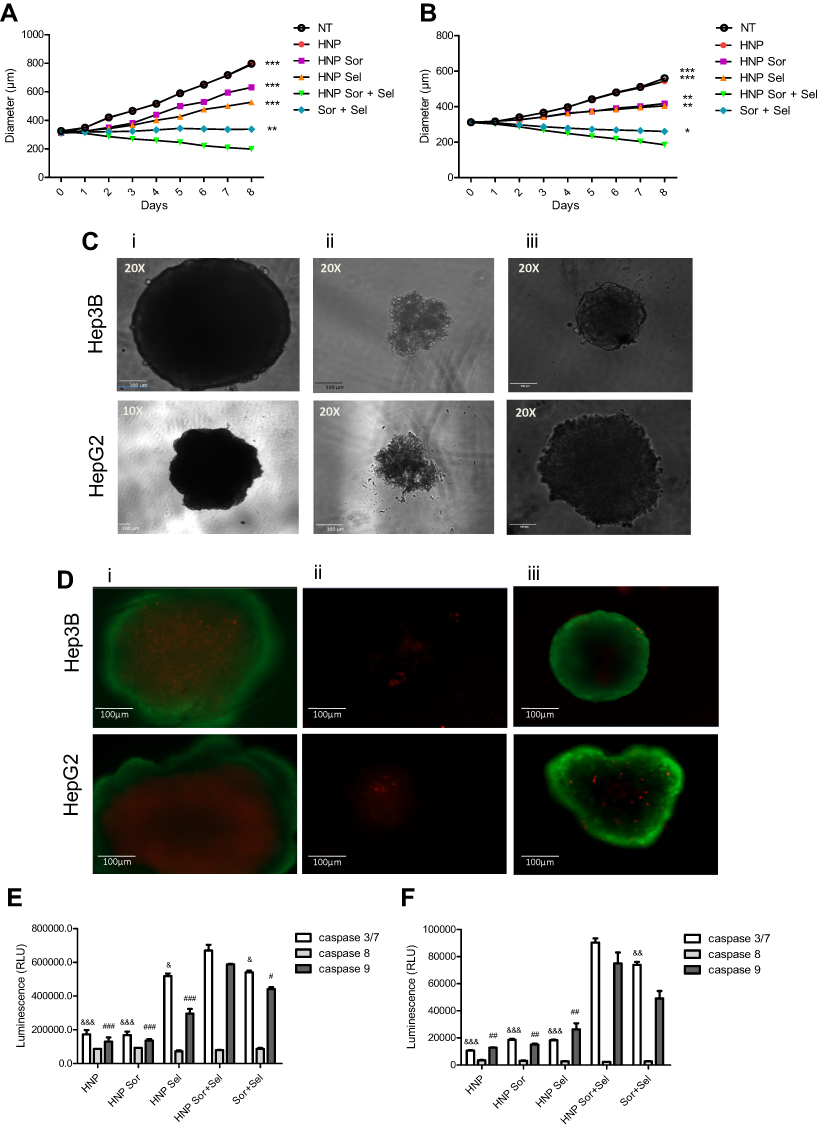 |
Figure 10 Impact of the therapeutic approaches mediated by the hybrid nanosystems in 3D cultures of HCC cells. Growth curves of (A) HepG2- and (B) Hep3B-derived spheroids after treatment, over 8 days, with empty HNP; HNP containing only selumetinib or sorafenib; HNP containing sorafenib and selumetinib; or both free drugs. 5 μg of HNP or the corresponding free drug concentrations were added to each well of 96-well plates. Representative phase contrast images of spheroids at the end of 8 days of treatment with (Ci) empty HNP; (Cii) HNP containing sorafenib and selumetinib; and (Ciii) both free drugs (scale bar = 100 µm). Representative fluorescence images of spheroids at the end of 8 days of treatment with (Di) empty HNP; (Dii) HNP containing sorafenib and selumetinib; and (Diii) both free drugs; using FDA-PIas live-dead staining (scale bar = 100 µm). Relative luminescence produced by the activity of caspase-3/7, −8 and −9 in (E) HepG2- and (F) Hep3B-derived spheroids treated, for 4 days, with empty HNP; HNP containing only selumetinib or sorafenib; HNP containing sorafenib and selumetinib; or both free drugs. Data are expressed as mean ± SD obtained from three independent experiments (n=3). Asterisks represent significant differences in the diameter of the spheroids. Cardinals represent significant differences regarding the activity of caspase 9. Ampersand represent significant differences in caspase 3/7 activity. (***, ### and &&&P< 0.001); (**, ## and &&P< 0.01); (*, # and &P< 0.05) Denotes a statistically significant difference when compared with cells treated with HNP containing sorafenib and selumetinib. |
Regarding the activation of caspases, as illustrated in Figure 10E and F, the observed data showed a robust activation of caspases-3/7 and −9, in both HepG2 and Hep3B spheroids, after treatment with the combination of selumetinib and sorafenib. This activation was particularly evident when the combined strategy was mediated by HNP and was much greater than that obtained with individual drugs. In contract, the activation of caspase-8 was inexistent. These results constituted an evidence of the involvement of the intrinsic apoptotic pathway. Overall, these data showed that the combined strategy mediated by the hybrid nanosystems presented a higher therapeutic efficiency than the individual approaches or the combination of free drugs.
Conclusion
In this work, we have developed a biocompatible and biodegradable hybrid nanosystem with suitable physico-chemical properties, namely, a size lower than 200 nm and a negative zeta potential. This new nanoformulation is specific for HCC cells, due to the presence of the GalNAc ligand, which promoted a strong increase of binding and internalization of the HNP by HCC cells. This formulation allowed the simultaneous and efficient loading of both drugs, selumetinib and sorafenib, substantially and specifically potentiating their antitumor effect in HCC cells. In fact, when these drugs were encapsulated in the hybrid nanosystems, it was observed a significant increase in HCC cell death and a substantial decrease in death of non-tumor cells, comparing to that observed with the combination of the two free drugs. The potentiation of the antitumor effect occurred through the increase of the programmed cell death, since there was a significant enhancement in the number of cells in initial apoptosis, associated to an increased activation of the initiating and effector caspases, and loss of mitochondrial membrane potential. The new hybrid nanosystems were able to potentiate the antitumor effect of selumetinib and sorafenib both in 2D cell cultures and 3D cultures, where it was observed a greater spheroid-mass reduction, due to programmed cell death, than with the combination of free drugs. This new drug delivery formulation has proven not only to be specific for HCC cells, which allows to reduce the side effects of the drugs, but also to potentiate the synergistic antitumor effect of this combination and to induce a reduction of the cell resistance to these drugs. The promising results obtained with our new HNP formulation need to be further verified in vivo, in an animal model of HCC, in order to evaluate its clinical translational potential. These studies should include the analyses of its biodistribution, pharmacokinetics and therapeutic efficacy, involving the evaluation of both the antitumor activity and the possible side effects.
Overall, our data show that this nanoformulation has the potential to overcome the main restrictions associated with conventional chemotherapy and may therefore constitute a promising approach for future clinical application in hepatocellular carcinoma treatment.
Acknowledgments
This work was financed by the European Regional Development Fund (ERDF) through the COMPETE 2020 program (Operational Program for Competitiveness and Internationalization) and Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, under projects: IF/01007/2015, POCI-01-0145-FEDER-30916, and UIDB/04539/2020. Dina Farinha acknowledges FCT for the Grant: SFRH/BD/104894/2014. Selumetinib was kindly provided by AstraZeneca PLC. Sorafenib was kindly provided by Bayer HealthCare Pharmaceutical Inc. Triantennary N‐acetylgalactosamine (GalNAc) cluster was kindly offered by Ionis Pharmaceuticals Inc.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2020.
2. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J Hepatol. 2020;72(2):250–261. doi:10.1016/j.jhep.2019.08.025
3. Wang C, Wang H, Lieftink C, et al. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut. 2020;69(4):727–736. doi:10.1136/gutjnl-2019-318506
4. Leathers JS, Balderramo D, Prieto J, et al. Sorafenib for treatment of hepatocellular carcinoma. J Clin Gastroenterol. 2019;53(6):464–469. doi:10.1097/MCG.0000000000001085
5. Bruix J, Raoul J-L, Sherman M, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a Phase III trial. J Hepatol. 2012;57(4):821–829. doi:10.1016/j.jhep.2012.06.014
6. Keating GM, Santoro A. Sorafenib. A review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69(2):223–240. doi:10.2165/00003495-200969020-00006
7. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24(26):4293–4300. doi:10.1200/JCO.2005.01.3441
8. Zhu Y-J, Zheng B, Wang H-Y, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–622. doi:10.1038/aps.2017.5
9. Personeni N, Rimassa L, Pressiani T, et al. Molecular determinants of outcome in sorafenib-treated patients with hepatocellular carcinoma. J Cancer Res Clin Oncol. 2013;139(7):1179–1187. doi:10.1007/s00432-013-1429-x
10. Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. doi:10.1158/1078-0432.CCR-06-1150
11. Tomkinson H, McBride E, Martin P, et al. Comparison of the pharmacokinetics of the phase II and phase III capsule formulations of selumetinib and the effects of food on exposure: results from two randomized crossover trials in healthy male subjects. Clin Ther. 2017;39(11):2260–2275. e2261. doi:10.1016/j.clinthera.2017.08.022
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi:10.1038/nature00766
13. Riverso M, Montagnani V, Stecca B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene. 2016;36(23):3322–3333. doi:10.1038/onc.2016.481
14. Poustchi F, Amani H, Ahmadian Z, et al. Combination therapy of killing diseases by injectable hydrogels: from concept to medical applications. Adv Healthcare Mater. 2021;10(3):2001571. doi:10.1002/adhm.202001571
15. Liu C, Chen Z, Chen Y, et al. Improving oral bioavailability of sorafenib by optimizing the “spring” and “parachute” based on molecular interaction mechanisms. Mol Pharm. 2016;13(2):599–608. doi:10.1021/acs.molpharmaceut.5b00837
16. Leijen S, Soetekouw PM, Evans TJ, et al. A Phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor Selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1619–1628. doi:10.1007/s00280-011-1732-7
17. Kim BY, Rutka JT, Chan WC. Nanomedicine. New Engl j Med. 2010;363(25):2434–2443. doi:10.1056/NEJMra0912273
18. Zhang L, Zhang L. Lipid-polymer hybrid nanoparticles: synthesis, characterization and applications. Nano Life. 2010;1(01n02):163–173. doi:10.1142/S179398441000016X
19. Grottkau B E, Cai X, Wang J, Yang X, Lin Y. Polymeric nanoparticles for a drug delivery system. Curr Drug Metab. 2013;14(8):840–846. doi:10.2174/138920021131400105
20. Mallick S, Choi JS. Liposomes: versatile and biocompatible nanovesicles for efficient biomolecules delivery. J Nanosci Nanotechnol. 2014;14(1):755–765. doi:10.1166/jnn.2014.9080
21. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A. Préat Vr. PLGA-based nanoparticles: an overview of biomedical applications. J Controlled Release. 2012;161(2):505–522. doi:10.1016/j.jconrel.2012.01.043
22. Huang S-L. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1167–1176. doi:10.1016/j.addr.2008.03.003
23. Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Controlled Release. 2008;126(3):187–204. doi:10.1016/j.jconrel.2007.12.017
24. Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomed Nanotechnol, Biol Med. 2013;9(4):474–491. doi:10.1016/j.nano.2012.11.010
25. Sengupta S, Eavarone D, Capila I, et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436(7050):568–572. doi:10.1038/nature03794
26. Li M, Zhang W, Wang B, Gao Y, Song Z, Zheng QC. Ligand-based targeted therapy: a novel strategy for hepatocellular carcinoma. Int J Nanomedicine. 2016;11:5645. doi:10.2147/IJN.S115727
27. Nair AB, Shah J, Al-Dhubiab BE, et al. Development of asialoglycoprotein receptor-targeted nanoparticles for selective delivery of gemcitabine to hepatocellular carcinoma. Molecules. 2019;24(24):4566. doi:10.3390/molecules24244566
28. Pranatharthiharan S, Patel MD, Malshe VC, et al. Asialoglycoprotein receptor targeted delivery of doxorubicin nanoparticles for hepatocellular carcinoma. Drug Deliv. 2017;24(1):20–29. doi:10.1080/10717544.2016.1225856
29. Steirer LM, Park EI, Townsend RR, Baenziger JU. The asialoglycoprotein receptor regulates levels of plasma glycoproteins terminating with sialic acid α2, 6-galactose. J Biol Chem. 2009;284(6):3777–3783. doi:10.1074/jbc.M808689200
30. Farinha D, de Lima MCP, Faneca H. Specific and efficient gene delivery mediated by an asialofetuin-associated nanosystem. Int J Pharm. 2014;473(1–2):366–374. doi:10.1016/j.ijpharm.2014.07.019
31. Magalhães M, Farinha D, de Lima MCP, Faneca H. Increased gene delivery efficiency and specificity of a lipid-based nanosystem incorporating a glycolipid. Int J Nanomedicine. 2014;9:4979. doi:10.2147/IJN.S69822
32. Prakash TP, Yu J, Migawa MT, et al. Comprehensive structure-activity relationship of triantennary N-acetylgalactosamine conjugated antisense oligonucleotides for targeted delivery to hepatocytes. J Med Chem. 2016;59(6):2718–2733. doi:10.1021/acs.jmedchem.5b01948
33. Kwon H-Y, Lee J-Y, Choi S-W, Jang Y, Kim J-H. Preparation of PLGA nanoparticles containing estrogen by emulsification-diffusion method. Colloids Surf a Physicochem Eng Asp. 2001;182(1–3):123–130. doi:10.1016/S0927-7757(00)00825-6
34. Costa PM, Cardoso AL, Mendonça LS, et al. Tumor-targeted chlorotoxin-coupled nanoparticles for nucleic acid delivery to glioblastoma cells: a promising system for glioblastoma treatment. Mol Ther-Nucleic Acids. 2013;2:e100. doi:10.1038/mtna.2013.30
35. Bhardwaj V, Ankola D, Gupta S, Schneider M, Lehr C-M, Kumar MR. PLGA nanoparticles stabilized with cationic surfactant: safety studies and application in oral delivery of paclitaxel to treat chemical-induced breast cancer in rat. Pharm Res. 2009;26(11):2495–2503. doi:10.1007/s11095-009-9965-4
36. Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17(3):247–289. doi:10.1163/156856206775997322
37. Troutier A-L, Delair T, Pichot C, Ladavière C. Physicochemical and interfacial investigation of lipid/polymer particle assemblies. Langmuir. 2005;21(4):1305–1313. doi:10.1021/la047659t
38. Trere D, Fiume L, De Giorgi LB, Di Stefano G, Migaldi M, Derenzini M. The asialoglycoprotein receptor in human hepatocellular carcinomas: its expression on proliferating cells. Br J Cancer. 1999;81(3):404–408. doi:10.1038/sj.bjc.6690708
39. Yousef S, Alsaab HO, Sau S, Iyer AK. Development of asialoglycoprotein receptor directed nanoparticles for selective delivery of curcumin derivative to hepatocellular carcinoma. Heliyon. 2018;4(12):e01071. doi:10.1016/j.heliyon.2018.e01071
40. Ostergaard ME, Yu J, Kinberger GA, et al. Efficient synthesis and biological evaluation of 5ʹ-GalNAc conjugated antisense oligonucleotides. Bioconjug Chem. 2015;26(8):1451–1455. doi:10.1021/acs.bioconjchem.5b00265
41. Mao X, Liu J, Gong Z, et al. iRGD-conjugated DSPE-PEG2000 nanomicelles for targeted delivery of salinomycin for treatment of both liver cancer cells and cancer stem cells. Nanomedicine. 2015;10(17):2677–2695. doi:10.2217/nnm.15.106
42. Guo Y, Zhang Y, Ma J, et al. Light/magnetic hyperthermia triggered drug released from multi-functional thermo-sensitive magnetoliposomes for precise cancer synergetic theranostics. J Controlled Release. 2018;272:145–158. doi:10.1016/j.jconrel.2017.04.028
43. Li X, Feng Q, Jiang X. Microfluidic Synthesis of Gd-based nanoparticles for fast and ultralong MRI signals in the solid tumor. Adv Healthcare Mater. 8(20):1900672. doi:10.1002/adhm.201900672.
44. Tripathi P, Dwivedi P, Khatik R, et al. Development of 4-sulfated N-acetyl galactosamine anchored chitosan nanoparticles: a dual strategy for effective management of Leishmaniasis. Colloids Surf B Biointerfaces. 2015;136:150–159. doi:10.1016/j.colsurfb.2015.08.037
45. Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Controlled Release. 2001;70(1–2):1–20. doi:10.1016/S0168-3659(00)00339-4
46. Drozdek S, Bazylinska U. Biocompatible oil core nanocapsules as potential co-carriers of paclitaxel and fluorescent markers: preparation, characterization, and bioimaging. Colloid Polym Sci. 2016;294(1):225–237. doi:10.1007/s00396-015-3767-5
47. Zou P, Stern ST, Sun D. PLGA/liposome hybrid nanoparticles for short-chain ceramide delivery. Pharm Res. 2013;31(3):684–693. doi:10.1007/s11095-013-1190-5
48. Gao D-Y, Lin -T-T, Sung Y-C, et al. CXCR4-targeted lipid-coated PLGA nanoparticles deliver sorafenib and overcome acquired drug resistance in liver cancer. Biomaterials. 2015;67:194–203. doi:10.1016/j.biomaterials.2015.07.035
49. Hu H, You Y, He L, Chen T. The rational design of NAMI-A-loaded mesoporous silica nanoparticles as antiangiogenic nanosystems. J Mater Chem B. 2015;3(30):6338–6346. doi:10.1039/C5TB00612K
50. Neves AR, Queiroz JF, Lima SAC, Reis S. Apo E-functionalization of solid lipid nanoparticles enhances brain drug delivery: uptake mechanism and transport pathways. Bioconjug Chem. 2017;28(4):995–1004. doi:10.1021/acs.bioconjchem.6b00705
51. Huang W-C, Hsieh Y-L, Hung C-M, et al. BCRP/ABCG2 inhibition sensitizes hepatocellular carcinoma cells to sorafenib. PLoS One. 2013;8(12). doi:10.1371/journal.pone.0083627.
52. Tummala S, Gowthamarajan K, Satish kumar M, Wadhwani A. Oxaliplatin immuno hybrid nanoparticles for active targeting: an approach for enhanced apoptotic activity and drug delivery to colorectal tumors. Drug Deliv. 2016;23(5):1773–1787. doi:10.3109/10717544.2015.1084400
53. Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb Protoc. 2016;2016(11):
54. Kato A, Miyazaki M, Ambiru S, et al. Multidrug resistance gene (MDR-1) expression as a useful prognostic factor in patients with human hepatocellular carcinoma after surgical resection. J Surg Oncol. 2001;78(2):110–115. doi:10.1002/jso.1129
55. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
56. Brentnall M, Rodriguez-Menocal L, De Guevara RL, Cepero E, Boise LH. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14(1):32. doi:10.1186/1471-2121-14-32
57. Ferraro E, Fuoco C, Strappazzon F, Cecconi F Apoptosome structure and regulation. Apoptosome: Springer; 2010:27–39.
58. Cossarizza A, Baccaranicontri M, Kalashnikova G, Franceschi C. A new method for the cytofluorometric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5, 5ʹ, 6, 6ʹ-tetrachloro-1, 1ʹ, 3, 3ʹ-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun. 1993;197(1):40–45. doi:10.1006/bbrc.1993.2438
59. Smiley ST, Reers M, Mottola-Hartshorn C, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Nat Acad Sci. 1991;88(9):3671–3675. doi:10.1073/pnas.88.9.3671
60. Feng G, Qin W, Hu Q, Tang BZ, Liu B. Cellular and mitochondrial dual-targeted organic dots with aggregation-induced emission characteristics for image-guided photodynamic therapy. Adv Healthcare Mater. 4(17):2667–2676. doi:10.1002/adhm.201500431.
61. Kerstjens M, Driessen EM, Willekes M, et al. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget. 2016;8(9):14835. doi:10.18632/oncotarget.11730
62. Lake D, Corrêa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73(23):4397–4413. doi:10.1007/s00018-016-2297-8
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.