Back to Journals » Vascular Health and Risk Management » Volume 12
A clinical approach to obstructive sleep apnea as a risk factor for cardiovascular disease
Authors Maeder M, Schoch O, Rickli H
Received 3 August 2015
Accepted for publication 10 February 2016
Published 21 March 2016 Volume 2016:12 Pages 85—103
DOI https://doi.org/10.2147/VHRM.S74703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Daniel Duprez
Micha T Maeder,1 Otto D Schoch,2 Hans Rickli1
1Department of Cardiology, 2Department of Respiratory Medicine, Kantonsspital St Gallen, St Gallen, Switzerland
Abstract: Obstructive sleep apnea (OSA) is associated with cardiovascular risk factors, cardiovascular diseases, and increased mortality. Epidemiological studies have established these associations, and there are now numerous experimental and clinical studies which have provided information on the possible underlying mechanisms. Mechanistic proof-of-concept studies with surrogate endpoints have been performed to demonstrate that treatment of OSA by continuous positive airway pressure (CPAP) has the potential to reverse or at least to attenuate not only OSA but also the adverse cardiovascular effects associated with OSA. However, no randomized studies have been performed to demonstrate that treatment of OSA by CPAP improves clinical outcomes in patients with cardiovascular risk factors and/or established cardiovascular disease and concomitant OSA. In the present review, we summarize the current knowledge on the role of OSA as a potential cardiovascular risk factor, the impact of OSA on cardiac function, the role of OSA as a modifier of the course of cardiovascular diseases such as coronary artery disease, atrial fibrillation, and heart failure, and the insights from studies evaluating the impact of CPAP therapy on the cardiovascular features associated with OSA.
Keywords: obstructive sleep apnea, cardiovascular, risk, heart failure, atrial fibrillation, hypertension
Introduction
Obstructive sleep apnea (OSA) is a common sleep-related breathing disorder which is caused by recurrent collapse of the upper airway during sleep, leading to intermittent airway obstruction and absence of airflow despite respiratory efforts of the diaphragm against the occluded pharynx, which results in recurrent cycles of hypoxemia and arousals, and thus sleep fragmentation. Patients with OSA, therefore, suffer from poor sleep quality, increased daytime sleepiness, reduced quality of life, depression, reduced vigilance, and patients are at increased risk of motor vehicle accidents.1 Apart from this, there is now strong evidence of an association between the presence and severity of OSA and an increased risk of cardiovascular diseases.2,3 In the present review, we summarize and critically discuss the current knowledge on the role of OSA as a cardiovascular risk factor, the impact of OSA on cardiac function, the role of OSA as a modifier of the course of cardiovascular diseases such as coronary artery disease, atrial fibrillation (AF), and heart failure (HF), and the insights from studies evaluating the impact of continuous positive airway pressure (CPAP) therapy on the cardiovascular features associated with OSA.
This is a narrative review based on a PubMed search using the search term “sleep apn(o)ea” combined with “cardiovascular disease”, “cardiovascular risk”, “hypertension”, “diabetes”, “left ventricular hypertrophy”, “left ventricular diastolic function”, “heart failure”, “coronary artery disease”, and “atrial fibrillation”. Original and review articles were considered, and the bibliography of identified articles was screened for other relevant articles. Only articles published in English until September 2015 were considered.
Definition
OSA is diagnosed based on a sleep study (polysomnography or polygraphy) where the number of apneas and hypopneas is assessed to calculate the apnea–hypopnea index (AHI), that is, the number of apneas and hypopneas per hour of sleep. An apnea is defined as absence of airflow (reduction to less than 10% of baseline for ≥10 seconds), and a hypopnea is defined as a reduction in airflow by ≥30% of baseline for ≥10 seconds and is accompanied by a ≥3% decrease in oxygen saturation or an arousal.4 According to the most recent consensus, the event is obstructive if any of the following criteria are met: 1) there is snoring during the event; b) there is increased inspiratory flattening of the nasal pressure waveform; and/or c) there is associated paradoxical motion of the chest and abdominal respiratory inductance plethysmography excursions.4 The event is central if none of these criteria are met. An AHI of less than five events per hour of sleep is considered to be normal. OSA is mild if AHI is between 5 and 15 h−1, moderate if it is between 15 and 30 h−1, and severe if AHI is >30 h−1. An OSA syndrome is present if OSA is accompanied by symptoms of daytime sleepiness, which is typically defined using the Epworth sleepiness scale (ESS) with a cut-off of a score of 10 points (maximum 24). However, AHI is not the only parameter characterizing OSA, and the definition of OSA is under discussion. Apart from AHI, the arousal frequency and the degree of nocturnal desaturation have been found to be related to relevant pathophysiological aspects of the disease.5 It should also be noted that the definitions of hypopnea have been changed over time, and that depending on these definitions, the prevalence of OSA can vary significantly.6 Some studies have used the respiratory disturbance index to define OSA, a parameter calculated from respiratory polygraphy, describing the number of events per hour of registration (not sleep). Thus, one should also consider that findings from older trials (eg, using the respiratory disturbance index instead of AHI) might not be reproducible under the current definitions.
Predisposition
Predisposing factors for OSA include older age, male sex, smoking, use of muscle relaxants, and endocrine disorders such as hypothyroidism and acromegaly, the most important factor, however, being obesity. In the Wisconsin Sleep Cohort Study (N=690, 56% males, mean age 46 years), a 10% weight gain was associated with a 32% increase in AHI (compared to stable weight) and a sixfold increase in the risk of developing moderate to severe OSA (AHI ≥15 h−1).7 On the other hand, weight loss has been shown to lead to a decrease in the severity of OSA.7,8 From a pathophysiological point of view, this is plausible since with increasing fat accumulation, the upper airway is more likely to be narrowed and to collapse during sleep. Anatomical factors (macroglossia, adenotonsillar hypertrophy, retrognathia, nasal obstruction) also predispose to OSA. Novel concepts regarding predisposition have been proposed, including dysfunction of the pharyngeal dilator muscles and a broad variation in arousal threshold.5
Prevalence
OSA is probably underdiagnosed. The Wisconsin Sleep Cohort Study published in 1993 had revealed that at the age of 30–60 years, 9% of women and 24% of men had any sleep-disordered breathing (AHI ≥5 h−1), and it was estimated that in this age category, 2% of women and 4% of men had OSA syndrome, that is, evidence of sleep-disordered breathing and symptoms of daytime sleepiness.9 A more recent study from the US showed that 10% of men aged 20–49 years and 17% of those aged 50–70 years had at least moderate OSA (AHI ≥15 h−1), whereas the corresponding figures for women in the same age categories were 3% and 9%.10 However, a recent population-based study from Switzerland using contemporary recording technologies revealed a prevalence of 50% of moderate or severe OSA (AHI ≥15 h−1) in men and 23% in women.6 Only a minority had symptoms of sleepiness. Given that this population was considered to be representative of the general population, and that in Switzerland, the average body mass index is lower than in other countries such as the US, the prevalence of OSA worldwide might be markedly higher than previously thought.
Pathophysiology
The following key mechanisms are thought to be of paramount importance for the pathophysiology of the cardiovascular consequences of OSA: intermittent hypoxia, hypercapnia, sleep fragmentation, and intrathoracic pressure swings.2,3 The unique pattern of intermittent hypoxia, that is, short periods of desaturation followed by reoxygenation, seems to be a key pathophysiological driver of adverse cardiovascular effects of OSA. Three mechanisms resulting from intermittent hypoxia/arousals relevant to the pathophysiology of the cardiovascular consequences of OSA have been proposed: sympathetic activation, oxidative stress, and systemic inflammation. A detailed discussion of the literature providing the basis for these concepts is beyond the scope of the present manuscript, but can be found elsewhere.2,3,11 Here, we will focus on the findings in human studies. Mechanical effects resulting from the massive intrathoracic pressure swings also seem to play an important role. Ineffective respiratory efforts against the occluded pharynx generate a negative intrathoracic pressure and, thus, an increased difference between intracardiac and extracardiac pressure, and thereby an increased left ventricular (LV) afterload.12 In addition, the negative intrathoracic pressure results in suction of blood into the right ventricle and, thus, increased right ventricular preload. On the other hand, hypoxia leads to pulmonary vasoconstriction and increased right ventricular afterload. If healthy volunteers perform a Mueller maneuver, that is, voluntary forced inspiration against a fixed resistance, to simulate obstructive apnea, this leads to reduction in LV function either assessed by LV ejection fraction (LVEF) or by strain,13 as well as significant variation in left atrial volume.14 Interestingly, right ventricular strain is also affected by the Mueller maneuver.13 Notably, even sleep restriction alone can induce features of cardiovascular dysfunction, including LV diastolic15 and left atrial dysfunction and endothelial dysfunction16 in healthy subjects, indicating that sleep deprivation per se may also play a role.
OSA and the cardiovascular system
In the following, we will present a summary of studies on the association between OSA and 1) established cardiovascular risk factors, 2) surrogates of cardiovascular risk (measures of autonomic tone, biomarkers), 3) measures of cardiac and vascular structure and function in subjects without overt cardiovascular disease, and 4) important cardiovascular disease states. We will separate studies reporting associations between the presence and severity of OSA and cardiovascular risk factors/diseases and the studies evaluating the impact CPAP on cardiovascular features proposed to be associated with OSA. In Table 1, an overview on these studies is given. Details are discussed in the following paragraphs.
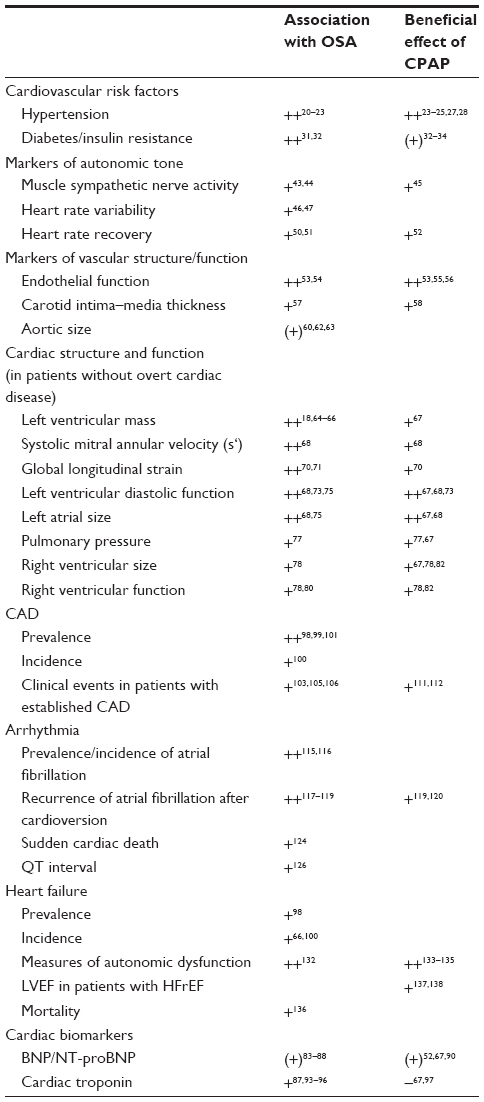 | Table 1 Overview of the association between OSA and cardiovascular risk factors, surrogate markers of cardiovascular dysfunction, and cardiovascular diseases and the impact of treatment with CPAP |
OSA and cardiovascular risk factors
Hypertension
The association between OSA and hypertension is now well established, although the clinical consequences are not clear yet.17 The presence of OSA has been shown to be associated with increased arterial stiffness (as assessed by pulse wave velocity), which is additive to the effect of hypertension alone.18 However, not all patients with OSA are hypertensive. Apart from age and body mass index, low daytime oxygen saturation and more severe nocturnal hypoxia seem to be key factors associated with the presence of hypertension in OSA patients.19 The landmark study by Young et al20 published in 1997 revealed that in a cohort of 1,060 subjects aged between 30 and 60 years, there was a linear rise in blood pressure with rising AHI. Subjects with an AHI of 15 h−1 were 1.8 times more likely to have hypertension that those with an AHI of zero. In a recently published cross-sectional study by Heinzer et al,6 among 2,121 subjects undergoing polysomnography (median AHI 6.9 h–1), the upper AHI quartile (>20.6 h−1) was independently associated with hypertension (odds ratio 1.6). This does not necessarily indicate that OSA causes hypertension, since there are many confounding factors, in particular, obesity. Data on the impact of OSA on the incidence of hypertension are somewhat conflicting. Peppard et al21 followed 709 participants of the Wisconsin Sleep Cohort Study without hypertension at baseline for 4 years. The odds ratios for having hypertension (blood pressure >140/90 mmHg or use of antihypertensive medication) at the 4 years follow-up were 1.42 for an AHI of 0.1–4.9 h−1, 2.03 for an AHI of 5.0–14.9 h−1, and 2.89 for an AHI ≥15 h−1, compared to an AHI of zero. In the study by Cano-Pumarega et al22 in 1,180 subjects without hypertension at baseline followed for 7.5 years, there was also a dose-dependent increase of incident hypertension with increasing OSA severity expressed by the respiratory disturbance index. However, after adjustment for confounders, OSA was not associated with incident hypertension anymore. In contrast, in a prospective observational study of 1,889 participants without hypertension at baseline with a median follow-up of 12.2 years, Marin et al23 found a higher risk of incident hypertension in patients with OSA ineligible for CPAP (adjusted hazard ratio 1.33), in patients with OSA who declined CPAP (hazard ratio 1.96), and in patients with OSA nonadherent to CPAP (hazard ratio 1.78), compared to controls.
The role of CPAP in the prevention and treatment of OSA is also controversial. The observational study by Marin et al23 suggested that the incidence of hypertension was lower in OSA patients treated with CPAP compared to controls (adjusted hazard ratio 0.71). However, this was not a controlled trial, and adherence to CPAP may have been a surrogate for a behavior that is likely to result in a reduction in blood pressure. In a trial among 725 patients with OSA and AHI ≥20 h−1 and an ESS score of <10, that is, nonsleepy patients with significant OSA, randomized to CPAP or no CPAP, there was no significant difference in the combined endpoint of incident hypertension and cardiovascular events between the groups, although the study may not have been adequately powered.24 Interestingly, the study was positive, that is, the incidence of hypertension and cardiovascular events was reduced, in the subset of 230 patients randomized to CPAP and using the device for more than 4 hours per night,24 pointing to the importance of CPAP adherence for such patients.
The antihypertensive effect of CPAP in patients with significant OSA is generally accepted.17 CPAP reduces blood pressure in patients with significant OSA (AHI >15 h−1) and additional cardiovascular risk factors, whereas nocturnal oxygen does not.25 The antihypertensive effect of CPAP in OSA patients is relatively modest, however, and less than that of pharmacological antihypertensive therapy (ie, valsartan).26 In a meta-analysis, a weighted mean decrease in diurnal systolic and diastolic blood pressure of 2.6 and 2.0 mmHg was found.27 Importantly, the antihypertensive effect of CPAP depends on the hours of CPAP use and on several other patient characteristics. Male sex, sleepiness, body mass index, smoking, alcohol use, and baseline blood pressure are predictive of the CPAP-related reduction in 24-hour blood pressure.27 In an open-label, randomized, multicenter trial in 194 patients with resistant hypertension (3.8 antihypertensive drugs) and OSA (mean AHI 40 h−1), the reduction in 24-hour mean blood pressure after 12 weeks was larger in the CPAP group than in the control group (crude difference −3.1 mmHg).28 The extent of blood pressure reduction was related to the average daily CPAP use.28
Diabetes
The basic science literature has established a plausible link between OSA and insulin resistance.29 In healthy volunteers, 5 hours of intermittent hypoxia is sufficient to induce altered insulin sensitivity.30 However, in humans with a cluster of cardiovascular risk factors, it is often hard to delineate whether diabetes is the result of obesity or OSA. Population-based and cross-sectional studies have revealed an association between the presence and severity of OSA and the prevalence and incidence of type 2 diabetes. An analysis of data from 1,387 participants of the Wisconsin Sleep Cohort Study revealed an increasing baseline prevalence of diabetes with increasing OSA severity (adjusted odds ratio of 2.30 for having a diagnosis of diabetes for subjects with AHI ≥15 h−1 compared to those with AHI <5 h−1).31 In the cross-sectional study by Heinzer et al6 among 2,121 subjects undergoing polysomnography (median AHI 6.9 h–1), the upper AHI quartile (>20.6 h−1) was independently associated with diabetes (odds ratio 2.0) and the metabolic syndrome (odds ratio 2.8). In the Wisconsin Sleep Cohort Study, the risk of developing diabetes within 4 years was also related to OSA severity at baseline (adjusted odds ratio 1.62 for subjects with AHI ≥15 h−1 compared to those with AHI <5 h−1).31 In another cohort of 544 patients referred for the evaluation of OSA and who were free of diabetes at baseline, the risk of incident diabetes was shown to be related to OSA severity after adjusting for potential confounders (adjusted hazard ratio per OSA severity quartile 1.43).32
Data on the effect of CPAP on diabetes and markers of the metabolic syndrome are scarce and conflicting, however. In the above-mentioned observational study,32 the 60% of patients with supramedian AHI using CPAP had a lower risk of incident diabetes than the 40% not using CPAP. A small retrospective uncontrolled study in 38 patients with both OSA (mean AHI 53 h−1) and diabetes showed a decrease in glycosylated hemoglobin (from 7.8% to 7.3%) following CPAP therapy (mean duration 134 days).33 In a nonrandomized study among 32 patients with severe OSA, Dorkova et al34 found that patients using CPAP for at least 4 hours per night (n=16) experienced an improvement in insulin sensitivity as measured by the homeostasis model assessment. In a trial by Sharma et al35 in 86 patients with OSA (87% with the metabolic syndrome) using a crossover CPAP versus sham-CPAP design over 3 months, a reduction in glycosylated hemoglobin (by 0.2%) could be shown in the CPAP group. Importantly, in this study, CPAP also reduced body mass index and visceral fat, which was not the case in many other studies. Notably, the study by Sharma et al36 was later retracted. In fact, the majority of OSA patients in other trials experienced a weight gain after initiation of CPAP therapy, as recently shown in a meta-analysis.37 A previous randomized sham-CPAP controlled study in patients with type 2 diabetes mellitus and newly diagnosed OSA had shown no effect of CPAP on glycosylated hemoglobin and insulin resistance after 3 months.38 Thus, at the moment, the role of CPAP in both prevention and treatment of diabetes in patients with OSA has not been clarified.
OSA as a risk marker
In the last 10 years, studies have not only revealed an association between OSA and prevalence/incidence of the major risk factors hypertension and diabetes, but severe OSA per se has been found to be a marker of adverse outcomes in several cohort studies,39–42 which represents an impetus for very intense research in this area. A landmark study by Marin et al39 showed that patients with severe untreated OSA had had a significantly increased risk of fatal (adjusted hazard ratio 2.87) and nonfatal (hazard ratio 3.17) cardiovascular events compared to healthy subjects, after a mean follow-up of 10.1 years. Patients with untreated severe OSA also had a higher risk of fatal and nonfatal events than simple snorers, patients with mild-to-moderate OSA, and patients with OSA treated by CPAP. In a cohort of patients referred to a sleep center (N=1,022, 68% with OSA with a mean AHI of 35 h−1), the risk of the composite endpoint of stroke or death after a median follow-up of 3.4 years was significantly higher in OSA patients compared to controls (adjusted hazard ratio 1.97).42 In the unadjusted analysis, OSA patients also had a higher risk of death (hazard ratio 2.00), which failed to reach statistical significance after adjustment for demographics and cardiovascular risk factors.42 A cohort study among 1,116 women revealed that those with untreated severe OSA (AHI ≥30 h−1) had a severalfold higher risk of cardiovascular mortality (adjusted hazard ratio 3.50) than those without OSA (AHI <10 h−1), while women with CPAP-treated severe OSA and women with mild-to-moderate OSA (AHI 10–29 h−1) had similar outcomes as those without OSA.40 Also, among 939 subjects ≥65 years of age, severe OSA (AHI ≥30 h−1) was shown to be associated with higher cardiovascular mortality (adjusted hazard ratio 2.25) compared with no OSA (AHI <10 h−1), while CPAP-treated patients and control subjects had similar prognosis.41 Notably, in these studies, “no CPAP use” was defined as either insufficient use (<4 hours per night) or no prescription. Thus, the presence of severe OSA clearly indicates an increased cardiovascular risk, and good CPAP adherence (which potentially also is a surrogate of unmeasured confounding factors) seems to be attenuate this risk, but the underlying mechanisms remain to be defined.
Surrogate markers of autonomic imbalance in OSA without overt cardiac disease
The paradigm of sympathetic predominance in OSA is reflected by several studies showing that surrogate markers of sympathetic tone are elevated in OSA patients without obvious cardiovascular disease compared to controls, and that CPAP attenuates these changes. These studies are briefly summarized in the following sections.
Muscle sympathetic nerve activity
Muscle sympathetic nerve activity, as assessed by microneurography, is an established method to quantify sympathetic activity. In their pivotal paper, Somers et al43 showed that patients with OSA (mean AHI 44 h−1) had clearly increased muscle sympathetic nerve activity during wakefulness, compared to age- and body mass index-matched controls (n=5, AHI <10 h−1; 59 versus 34 bursts/minute). Importantly, muscle sympathetic nerve activity in patients with OSA and obesity is higher than in those with obesity alone.44 In a controlled study, muscle sympathetic nerve activity was significantly reduced in patients with OSA using CPAP therapy for 1 year (n=11, mean AHI 27 h−1) compared to those who did not undergo CPAP therapy because they refused it (n=9, mean AHI 23 h−1; from 51 to 41 bursts/minute versus from 45 to 43 bursts/minute).45
Heart rate variability
Similar to patients with HF, OSA patients have reduced heart rate variability with a shift toward sympathetic predominance.46 Narkiewicz et al46 performed spectral analysis of heart rate variability and found an increase in low-frequency RR interval variability (77 versus 58 normalized units), a decrease in high-frequency RR interval variability (17 versus 37 normalized units), and an increase in the ratio of low- to high-frequency RR interval variability in patients with moderate-to-severe OSA (n=11, mean AHI 61 h−1) compared to controls matched for age, body mass index, and blood pressure (n=12). Such findings have initially been obtained in patients with severe OSA. However, even patients with milder forms of OSA seem to have subtle alterations in heart rate variability.47 Although a very interesting field analysis of heart rate variability is complex, a high number of indices have been suggested, and data on the impact of CPAP on heart rate variability are relatively scarce with inconsistent findings in the adult OSA literature.
Heart rate recovery
Heart rare recovery is another measure of autonomic tone with established prognostic value in patients with cardiovascular disease.48 Heart rate recovery is a measure derived from an exercise stress test and defined as the reduction in heart rate from peak exercise to the first (HRR-1) or the second (HRR-2) minute of recovery. Its relationship with autonomic tone has been demonstrated in elegant experiments.49 In a cohort of patients with OSA without HF (N=63, mean age 49 years, median AHI 30 h−1), we had found an association between higher AHI and lower HRR-1 (AHI >30 h−1: median HRR-1 20 beats per minute [bpm] versus AHI 5–30 h−1: median HRR-1: 24 bpm).50 In the multivariable analysis, higher AHI remained an independent predictor of lower HRR-1.50 We also found an association between the severity of OSA expressed as higher desaturation index and HRR-2, particularly in younger patients (ie, ≤48 years of age).51
In an uncontrolled study assessing the effect of CPAP (at least 3.5 hours use, mean duration 7.9 months) in 40 patients with OSA (median AHI 37 h−1), we found an improvement in HRR-2 (from 38 to 42 bpm) in the entire cohort and an improvement in HRR-1 (from 20 to 21 bpm) and HRR-2 (from 38 to 42 bpm) in those with AHI >30 h−1 at baseline.52
The cardiovascular system in OSA
In the following sections, studies investigating the effects of OSA per se (ie, in patients without overt cardiovascular disease) on heart and vessels are summarized.
Endothelial function
Endothelial function, a surrogate of vascular function, has been shown to be impaired in OSA patients. Patients with moderate-to-severe OSA (n=28, mean AHI 46 h−1) were shown to have worse forearm flow-mediated vasodilation compared to controls (n=12; mean AHI 2.4/h; 5.3% versus 8.3%).53 This is not only true for severely symptomatic OSA patients, but also for those with few symptoms. Kohler et al54 showed that minimally symptomatic OSA patients (n=64, mean oxygen desaturation index 23.1 h−1, ESS score 8) had worse flow-mediated dilatation (5% versus 7.5%) compared to matched control subjects without OSA (n=15).
A randomized study in patients with moderate-to-severe OSA undergoing CPAP for 4 weeks (n=14) or no CPAP (n=13) revealed a significant improvement in flow-mediated vasodilation in the group allocated to CPAP compared to the group allocated to no CPAP (from 5.1% to 9.6% versus from 5.6% to 4.7%; P<0.001 for between-group difference).53 Interestingly, this is true even for minimally symptomatic patients (who have been shown to respond in a different manner, compared to symptomatic patients with respect to a number of cardiovascular endpoints). In the Multicentre Obstructive Sleep Apnea Interventional Cardiovascular trial, including 208 patients (oxygen desaturation index 13.7 h−1, ESS score 8.3), a subgroup (n=64) had complete data on brachial artery flow-mediated dilatation at baseline and after 6 months.55 Those undergoing CPAP had improved flow-mediated dilatation compared to those not undergoing CPAP (from 3.4% to 4.4% versus from 3.4% to 2.6%; P<0.001 for adjusted treatment effect), and the effect was larger in those using CPAP for more than 4 hours per night compared to those not doing so.55 A recent meta-analysis summarizing four randomized trials on the effect of CPAP on endothelial function (150 patients) revealed an improvement in flow-mediated vasodilation by 3.87% following CPAP.56
Carotid intima–media thickness
Carotid intima–media thickness, an established surrogate of early atherosclerosis, was shown to be higher in patients with OSA (n=36, two thirds with moderate-to-severe OSA) compared to obese controls (n=16; 1,070 versus 710 μm).57 There was also a significant difference in carotid intima–media thickness between subjects with moderate-to-severe (mean AHI 48 h−1) versus mild (mean AHI 11 h−1) OSA (1,160 versus 920 μm). There was a correlation between AHI and carotid intima-media thickness as well as between the degree of nocturnal desaturation and carotid intima-media thickness. A higher percentage of total sleeping time spent with an oxygen saturation less than 90% was the strongest predictor of thicker carotid intima–media in the multivariable analysis.57 A randomized study in 24 patients with severe OSA (AHI >30 h−1, similar baseline carotid intima–media thickness in both groups) assigned to CPAP for 4 months (n=12; mean hours of CPAP use: 6 hours per night) or no treatment (n=12) revealed a significant reduction of carotid intima–media thickness following CPAP (CPAP group: from 707 to 645 μm versus control group: from 732 to 740 μm).58
Large vessels
By large intrathoracic pressure changes and the above-mentioned mechanisms contributing to vascular damage, OSA may also have adverse effects on the aorta. A detailed and excellent summary of available studies and a discussion of possible underlying mechanisms can be found elsewhere.59 Here, we briefly mention some key studies. Marfan’s syndrome was used as a prototype and model of aortic disease where OSA might be a factor contributing to disease progression and aortic events. In a prospective study among 61 patients with Marfan’s syndrome and 26 matched control subjects, Kohler et al60 found a high prevalence of OSA in Marfan patients (33% with AHI >5 h−1, 18% with AHI >15 h−1 versus 12% and 0%, respectively, in controls). In Marfan patients without previous aortic surgery, aortic root diameter was larger in those with AHI >5 h−1 compared to those with AHI <5 h−1 (45 versus 37 mm; correlation between AHI and aortic root diameter: r=0.5). In the same cohort (44 patients with Marfan’s syndrome, 15 with OSA, median follow-up 29 months), the presence of OSA was related to the incidence of aortic events.61 Cross-sectional studies evaluating the association between OSA and aortic size have revealed mixed results. In one of the positive studies in 150 unselected patients referred for evaluation of OSA (mean age 60 years, 73% with AHI >10 h−1), the diameter of the ascending aorta as assessed by computed tomography was larger in patients with OSA than in those without (36.8 versus 31.5 mm), and higher AHI was independently associated with larger aortic size in the multivariable analysis.62 Another positive study was that by Mason et al63 who conducted a retrospective study among 127 patients with abdominal aortic aneurysm who underwent a sleep study (15% with AHI >30 h−1, 10% with oxygen desaturation index >30 h−1), the result of which was correlated to the previously observed aortic aneurysm expansion in a surveillance program (median interval between the first and last scan for aneurysm size assessment: 18 months). Patients with AHI >30 h−1 and oxygen desaturation index >30 h−1 had higher aneurysm expansion rates adjusted for baseline diameter and cardiovascular risk factors, compared to subjects with AHI and oxygen desaturation index 0–5 and 6–15 h−1, respectively.63 Prospective randomized studies on the effect on CPAP on aortic size are not yet available.
LV mass
Figure 1 is a schematic representation of the OSA-related changes to the heart which are discussed in the following sections. Cross-sectional studies reported an association between OSA and OSA severity and LV mass (typically expressed as LV mass index, where LV mass is indexed to body surface area, height, or height2.7), but not in all studies the association remained significant after adjustment for important confounders such as obesity and hypertension.18,64,65 Drager et al18 demonstrated an additive effect of severe OSA and hypertension on LV mass. They found that interventricular septal thickness, posterior wall thickness, and LV mass index were lowest in control subjects without OSA or hypertension, intermediate in patients with hypertension or severe OSA, and highest in those with both severe OSA and hypertension. These authors also demonstrated a link between OSA and increased arterial stiffness (as assessed by pulse wave velocity) on the one hand, and between increased arterial stiffness and increased LV mass index on the other.18 Niroumand et al65 reported higher LV mass indexed to height in men with OSA (n=224, AHI 34 h−1) compared to those without (n=108, AHI 1.2 h-1; 98.9 versus 92.3 g/m), but this association was explained by the confounding effects of body mass index, age, and hypertension. There was no significant association between LV mass index and AHI. Contrasting findings were obtained in a much larger cohort of 2,058 participants of the Sleep Heart Healthy Study, where even after extensive adjustment, Chami et al64 found a higher LV mass index in patients with severe OSA (AHI 30 h−1; n=84) compared to that in those without OSA (AHI <5 h−1, n=957; 44.1 versus 41.3 g/m2.7). There was an independent association between LV mass index and presence and severity of OSA expressed as AHI or the percentage of total sleeping time spent with an oxygen saturation <90% in men, while in women, only the association between LV mass index and the percentage of total sleeping time spent with an oxygen saturation <90% was statistically significant.64 Seemingly opposing findings were reported very recently by Roca et al66 who found an independent association between OSA severity expressed by AHI and LV mass index in women, but not in men. However, in this study, subjects from the Atherosclerosis Risk In Communities study and the Sleep Heart Health Study (371 men and 535 women) underwent an echocardiogram approximately 13 years after the baseline assessment including polysomnography. These findings were interpreted as evidence of a more unfavorable cardiac remodeling response to OSA in women than in men.66
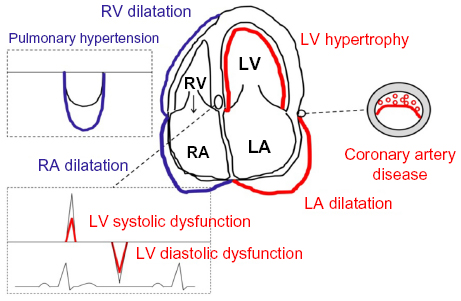 | Figure 1 Schematic representation of the OSA-related effects on cardiac structure and function. |
An uncontrolled study in symptomatic patients with moderate-to-severe OSA (N=47, mean AHI 63 h−1, mean ESS score 14) using cardiac magnetic resonance imaging showed a significant reduction in LV mass index (from 159 to 137 g/m2) following 12 months of CPAP therapy, although there was no change in blood pressure.67 Another uncontrolled study found a reduction in LV septal and posterior wall thickness in 37 patients with OSA treated with CPAP (mean duration 26 weeks), whereas LV mass index was not reduced.68 The latter was due to the fact that there was an increase in LV end-diastolic dimensions (and ejection fraction) following CPAP. Interestingly, no change in LV mass index, as assessed by cardiac magnetic resonance imaging, was found following 6 months of CPAP therapy in only minimally symptomatic patients with OSA (n=68, mean oxygen desaturation index 13.5 h−1, mean ESS score 8), even though there was a reduction in oxygen desaturation index and a symptomatic improvement with a significant improvement of the ESS score in patients randomized to CPAP.69
LV systolic function
LVEF is the most commonly used measure of LV systolic function. However, LVEF is a very crude measure of LV systolic function, and LV systolic function can be impaired before LVEF is reduced. There is no good evidence that OSA per se is associated with significantly impaired LVEF in patients without overt cardiovascular disease. In contrast, several studies reported associations between OSA and OSA severity and measures of LV myocardial velocity (tissue Doppler)68 and myocardial deformation (strain),70,71 that is, measures of LV systolic function which are more sensitive than LVEF. In an interesting study by Butt et al,68 patients with OSA and borderline blood pressure (AHI >15 h−1; n=40, mean systolic blood pressure 142 mmHg) showed a similar impairment of the systolic mitral annular velocity as age-matched patients with hypertension and no OSA (n=40, mean systolic blood pressure 152 mmHg; 6 versus 6 cm/s), but lower systolic mitral annular velocity when compared to age-matched controls (6 versus 7 cm/s). In a study by Haruki et al,70 a reduction in global longitudinal LV strain could be demonstrated in OSA patients (n=32, mean AHI 37 h−1) immediately after sleep as compared to before sleep (before sleep: −20.7 versus after sleep: −19.1; no control group).
In the above-mentioned study by Butt et al,68 CPAP (mean duration 26 weeks) improved the systolic mitral annular velocity in OSA patients (from 6 to 7 cm/s; no control group). In the study by Haruki et al,70 the overnight reduction in global longitudinal LV strain was shown to be abolished after 3 months of CPAP in 14 patients with moderate-or-severe OSA
Importantly, subtle changes in LV systolic function have also been found in patients with obesity (no assessment of OSA was performed),72 and again, it is difficult to separate the effects attributable to obesity or OSA.
LV diastolic function
Studies concur that OSA is associated with LV diastolic dysfunction.73–75 In an early study by Arias et al73 using Doppler echocardiography but no tissue Doppler, OSA patients (mean AHI 44 h−1) were found to be more likely to have a pattern of impaired LV relaxation than controls with similar age and body mass index (15/27 versus 3/15). In studies including tissue Doppler measurements, other investigators consistently found lower early mitral annular velocity (e’)75 and/or higher ratio of the early peak mitral inflow velocity (E) to e’ (E/e’,68,75 a higher value indicating worse diastolic function) in patients with OSA compared to controls. Notably, e’ is currently regarded as the most valid measure of LV diastolic function.76 Oliveira et al75 compared 56 patients with OSA (mean AHI 30 h−1) and 50 age-matched control subjects with similar body mass index and blood pressure (mean AHI 2.6 h−1) and found lower e’ (averaged measure from septal and lateral annulus: 7.0 versus 7.9 cm/s) and higher E/e’ (10.6 versus 9.4) in OSA patients. In the study by Butt et al,68 E/e’ in patients with hypertension was higher than in healthy controls, and in OSA patients, it was similar to those with hypertension, but the difference between OSA patients and controls failed to reach statistical significance.
Arias et al73 were able to show in a sham-CPAP-controlled crossover study in 25 OSA patients (mean AHI ~44 h−1) that CPAP over 12 weeks increased the ratio of the early to atrial transmitral inflow velocity (E/A ratio; from 1.07 to 1.24), a measure of LV relaxation, and reduced the deceleration time (from 244 to 223 ms), that is, CPAP changed the diastolic function from an impaired relaxation pattern toward a normal pattern. Butt et al68 reported improved measures of LV diastolic function including an increase in the E/A ratio (from 1.0 to 1.4) and a reduction in the isovolemic relaxation time (from 0.09 to 0.07 seconds) and E/e’ (from 9.0 to 8.0) in 37 OSA patients following CPAP therapy (mean duration 26 weeks). Colish et al67 found a dramatic increase in e’, both measured at the septal (from 5.0 to 9.2 cm/s) and lateral (from 5.5 to 9.8 cm/s) annulus, and a reduction in E/e’ (from 18 to 8) in an uncontrolled study in 47 patients with severe OSA (mean AHI 63 h−1) after 1 year of CPAP therapy (more than 4.5 hours use per night in all patients).
Left atrial size and function
The size of the left atrium is regarded as a mirror of LV diastolic function. Several authors have reported worse diastolic function in conjunction with larger left atrial volume in OSA patients compared to controls.68,75 Oliveira et al75 reported larger left atrial volume index (24.9 versus 18.5 mL/m2) in 56 patients with OSA (mean AHI 30 h−1) than in 50 age-matched control subjects (mean AHI 2.6 h−1), and there was a rise in left atrial volume index across AHI categories. Similarly, Butt et al68 found a larger left atrial volume index, as assessed by echocardiography, in OSA patients compared to controls, either assessed using 2D (28.1 versus 18.6 mL/m2) or 3D (26.3 versus 18.1 mL/m2) echocardiography, and the left atrial volume index was similar in the OSA patients (having borderline hypertension) and in patients with hypertension but no OSA.
In the study by Butt et al,68 a reduction in left atrial volume index (2D: from 28 to 23 mL/m2, 3D: from 26 to 22 mL/m2) was reported in 37 patients following CPAP therapy. A significant reduction in left atrial size volume index (from 45 to 31 mL/m2) along with an improvement in LV diastolic function following 12 months of CPAP was also reported by Colish et al67 in an uncontrolled study.
Pulmonary pressure and right heart
It is generally assumed that sleep-disordered breathing including OSA can lead to pulmonary hypertension, the underlying mechanism being chronic or intermittent hypoxia (class III pulmonary hypertension, which is a form of pulmonary arterial hypertension). However, detailed invasive studies are not available. A carefully conducted noninvasive study had shown that patients with OSA (n=23, mean AHI 44 h−1) and without other obvious cause of pulmonary hypertension had higher estimated systolic pulmonary artery pressure (sPAP) than age-matched controls (n=10).77 However, sPAP was low in both groups (30 versus 23 mmHg). Oliveira et al78 found slightly higher noninvasively estimated pulmonary vascular resistance (2.1 versus 1.8 Wood units), larger right ventricular end-diastolic (52.2 versus 49.9 mL/m2) and end-systolic (18.7 versus 15.4 mL/m2) volume index, and lower right ventricular ejection fraction (64.3% versus 68.4%) in OSA patients (n=56, mean AHI 30 h−1) compared to control subjects (n=50, mean AHI 2.6 h−1). In contrast to this study using 3D echocardiography, an earlier study using conventional 2D echocardiography had revealed a greater right ventricular wall thickness (after multivariable adjustment: 0.8 versus 0.7), but no differences in right ventricular and right atrial volumes and right ventricular function between patients with significant OSA (n=90, mean respiratory disturbance index 42 h−1) compared to those without/mild OSA (n=90, mean respiratory disturbance index 5 h−1).79 In a more recent study, patients with severe OSA (mean AHI 58 h−1) had worse right ventricular function, as assessed by tricuspid annular systolic excursion (19 versus 25 mm) and strain (−21 versus −34%), compared to subjects without OSA, while pulmonary pressures were similar.80
In the above-mentioned study by Arias et al,77 sham-controlled CPAP therapy for 12 weeks resulted in a significant reduction of sPAP (from 29 to 24 mmHg) among the OSA patients of this population. Given the lack of invasive hemodynamics in this study, the mechanisms underlying the results remain unknown, but there was also a difference in deceleration time (from 246 to 224 ms) after CPAP, which may suggest that the effects on sPAP were driven by improved LV diastolic function. Indeed, it is currently suspected that pulmonary venous hypertension due to LV systolic and diastolic dysfunction is the most common form of pulmonary hypertension in OSA patients.81 An uncontrolled study also found a reduction in echocardiographically assessed sPAP (from 54 to 39 mmHg) in 47 patients with severe OSA (AHI 63 h−1) after 12 months of effective CPAP, which went in parallel to an improvement in LV diastolic function and was also accompanied by a reduction in the size of the right atrium (right atrial volume index; from 48 to 31 mL/m2) and ventricle (as assessed by echocardiography: right ventricular end-diastolic diameter, from 41 to 30 mm, and cardiac magnetic resonance imaging: right ventricular end-diastolic volume index, from 62 to 46 mL/m2).67 Somewhat contradictory to this, a randomized study by Oliveira et al78 revealed a reduction in noninvasively assessed pulmonary vascular resistance (from 2.2 to 1.8 Wood units), which suggests an effect on the pulmonary vasculature, accompanied by a reduction in right ventricular end-systolic volume index (from 20.3 to 16.0 mL/m2) and an improvement in right ventricular ejection fraction (from 63% to 71%) in patients with significant OSA (AHI >20 h−1) treated with CPAP during 24 weeks (n=15), whereas this was not the case in patients not treated with CPAP. Similar findings on the right ventricular size and function were obtained in a very recent uncontrolled study among 44 patients with moderate or severe OSA following 12 months of CPAP.82
Cardiac biomarkers in OSA
Natriuretic peptide system
Natriuretic peptides were investigated in OSA patients with the idea that B-type natriuretic peptide (BNP) and/or N-terminal-proBNP (NT-proBNP) might reflect myocardial stress in the context of OSA. Findings are conflicting in that most studies did not find significant differences in natriuretic peptide concentrations between OSA patients without overt cardiac disease and controls, and that there was no association between OSA severity and natriuretic peptide plasma concentration.83–87 However, in one study in women, an association between higher BNP and presence and severity of OSA was found.88 In this study, BNP was measured directly after sleep.88 We recently found a larger relative overnight reduction in BNP, but not in NT-proBNP and no difference in the absolute overnight change in BNP between patients with moderate/severe OSA compared to those with mild OSA/without OSA.89
Studies have evaluated the effect of CPAP on BNP and NT-proBNP in patients with OSA without overt cardiac disease,52,90 but an effect was seen in only one study.90 BNP and NT-proBNP are influenced by a number of factors other than LV wall stress, including age, sex, renal function, and body mass index.91 Patients with higher body mass index have lower natriuretic peptide plasma concentrations.92 The exact mechanism underlying this association is unknown, but it might play an important role in patients with OSA and it may dilute the effect of LV wall stress.
Cardiac troponin
Plasma concentrations of cardiac troponins are not specific for myocardial ischemia, but can be elevated following all types of myocyte damage/stress. Studies have, therefore, been undertaken to assess the relationship between the presence and severity of OSA and cardiac troponin. Some studies using traditional and newer troponin assays have found no significant relationship between the presence and severity of OSA and cardiac troponin plasma concentrations.93,94 In one study, the severity of OSA was related to cardiac troponin, but the association was no longer significant after adjustment for potential confounders.95 However, in two population-based studies, high-sensitivity cardiac troponin was associated with the severity of OSA, and this remained significant after multivariable adjustment.87,96 In 1,645 subjects from the Atherosclerosis Risk In Communities study and the Sleep Heart Health Study, the authors found an independent association between high-sensitivity cardiac troponin T and presence and severity categories of OSA.87 In addition, high-sensitivity cardiac troponin T was a predictor of death or incident HF across all OSA categories.87 In another study based on this population, the authors recently showed that an independent association between high-sensitivity cardiac troponin T and presence of OSA was present in women only.66 Einvik et al96 performed a cross-sectional study (N=514) targeting subjects at high risk of OSA (45% with AHI >5 h−1). They nicely demonstrated in several multivariable models that high-sensitivity cardiac troponin I was not only related to AHI, but also to the extent of nocturnal desaturations expressed as mean nocturnal oxygen saturation, nadir of nocturnal desaturation, and percent total sleeping time spent with an oxygen saturation <90%. Thus, these studies were in line with the hypothesis that OSA and its severity are related to subclinical myocardial damage. However, the mechanisms underlying the association between OSA and raised cardiac troponin have not been explored in detail.
Studies on the effect of CPAP on cardiac troponin have been relatively small and uncontrolled. One uncontrolled study in OSA patients found no effect of CPAP, while another study even found an increase in cardiac troponin following CPAP.97
Cardiac diseases and OSA
Coronary artery disease
An association between OSA and atherosclerotic diseases, in particular, coronary artery disease, is plausible from the suspected underlying pathophysiology and highly suggestive from a number of observational and epidemiological studies. In a cross-sectional study of 6,424 subjects undergoing polysomnography (median AHI 4.4 h−1), the adjusted odds ratio for self-reported coronary artery disease for the highest compared to the lowest AHI quartile was 1.27 (borderline statistical significance).98 In a case–control study (62 patients admitted to the intensive care unit because of a coronary event, 62 matched control subjects), the prevalence of OSA (respiratory disturbance index ≥10 h−1) was higher in patients than in controls (19 versus 8), and in the multivariable analysis, OSA remained independently associated with coronary artery disease (adjusted odds ratio 3.1).99 In the Sleep Heart Health Study (1,927 men with median AHI of 6.2 h−1, 2,495 women with median AHI of 2.7 h−1, age ≥40 years, no known coronary disease or HF), the incidence of a clinically manifest coronary artery disease (ie, a coronary event including myocardial infarction, revascularization, or death due to coronary artery disease) was higher in men <70 years with OSA (adjusted hazard ratio: 1.10 per 10 h−1 increase in AHI; adjusted hazard ratio 1.68 for AHI ≥30 h−1 compared to AHI <5 h−1).100 A significant association between OSA and incident coronary artery disease was not found in older men or women, however.100
Whereas these studies looked at clinically manifest coronary artery disease, a recent analysis from the Multi-Ethnic Study of Atherosclerosis selected a surrogate for silent coronary artery disease as the endpoint.101 In this study, 1,465 participants free of clinical cardiovascular disease (15% with AHI ≥30 h−1) underwent polysomnography and coronary computed tomography. The study revealed that subjects with severe OSA were more likely to have any coronary artery diseases (as defined by a coronary calcium score >0 Agatston units) than those without OSA, after adjusting for smoking, body mass index and other traditional cardiovascular risk factors. A higher arousal index, but not a higher AHI was associated with a higher likelihood of a high coronary disease burden (coronary calcium score >400 Agatston units).101 In 2,603 participants of the Multi-Ethnic Study of Atherosclerosis, a higher disease progression rate (based on two coronary artery computed tomography scans 8 years apart) could be demonstrated for subjects with a diagnosis of OSA at baseline compared to those without OSA or habitual snoring (mean increase in Agatston units: 204 versus 136), but this association failed to reach statistical significance (P=0.06) after adjustment for traditional cardiovascular risk factors.102
In patients with established coronary artery disease, OSA is common and seems to have an adverse effect on the course of the disease, as suggested by a small study on 59 patients admitted because of a coronary event which showed higher mortality during a follow-up of 5 years in patients with OSA (respiratory disturbance index ≥10 h−1; death in 6/16 patients compared to 4/43 in those without OSA).103 In patients with OSA, the likelihood of a nocturnal acute myocardial infarction is higher than in patients without OSA,104 possibly pointing to OSA as a trigger. A higher rate of in-stent restenosis after percutaneous coronary intervention105 and a higher incidence of events during the long-term follow-up after coronary bypass grafting106 have been shown for patients with OSA compared to those without OSA. Interestingly, nonobese patients with coronary artery disease and OSA have higher inflammatory activity than nonobese patients with coronary artery disease but no OSA.107 This may play a role in the progression of the disease in terms of healing after interventions and new events. Among patients with acute myocardial infarction, those with OSA have worse endothelial function,108 potentially explaining the higher risk of complications. In addition, patients with coronary artery disease and OSA have been shown to have more advanced LV diastolic dysfunction, compared to those without OSA.109 On the other hand, patients with chronic total occlusion of coronary arteries and OSA have better collaterals than those without OSA,110 which may be protective.
One observational study (allocation to CPAP or upper airway surgery was not randomized, but was accepted or declined by patients) in patients with both coronary artery disease and OSA (AHI ≥15 h−1) revealed better outcomes (composite of cardiovascular death, acute coronary syndrome, coronary revascularization, hospitalization for HF) in those treated for OSA (6/25 with endpoint) compared to those not treated (17/29 with endpoint), after a median follow-up of 87 months (hazard ratio 0.24 for treatment versus no treatment).111 Similarly, a retrospective cohort study revealed a lower risk of cardiac death (3% versus 10%) after percutaneous coronary intervention in OSA patients (AHI ≥15 h–1) treated with CPAP (n=175) compared to those without CPAP (n=196).112 Prospective data on the impact of CPAP therapy on outcomes in patients with coronary artery disease and OSA are awaited.
Arrhythmia
Patients with OSA have an underlying substrate for AF, that is, LV hypertrophy, LV diastolic dysfunction, and left atrial dilatation. In addition, OSA patients were shown to have structural and electrical atrial remodeling.113 Recently, a study also showed that simulation of OSA with the Müller maneuver leads to an increasing number of atrial premature beats which may precipitate AF.114 A higher prevalence of OSA has been found in patients with AF than in patients without AF referred to a cardiology practice with similar age, sex, and cardiovascular risk profile including body mass (49% versus 32%).115 The incidence of AF is also higher in patients with OSA as shown in retrospective cohort study in 3,542 patients without past or current AF undergoing polysomnography (mean AHI 26.7 h−1) and followed for a mean duration of 4.7 years. OSA (AHI ≥5 h−1; hazard ratio 2.18) and measures of OSA severity including the lowest nocturnal oxygen saturation (hazard ratio 3.08 per 1% decrease) were related to incident AF in the univariable analysis.116 In the multivariable analysis, the lowest nocturnal oxygen saturation (hazard ratio 3.29 per 1% decrease), but not AHI was an independent predictor of incident AF in patients younger than 65 years, whereas in older patients, there was no independent association between OSA and incident AF.116 However, obesity is also strongly associated with the incidence of AF, and thus, the association between OSA and AF is likely to be at least, in part, explained by the high burden of coexisting risk factors. Patients with OSA also have a higher risk of recurrence of AF following electrical cardioversion117 or catheter ablation.118,119
There is good evidence from epidemiological studies and cohort studies that CPAP may reduce the risk of AF recurrence after cardioversion and ablation.119,120 In an interesting study, Fein et al120 identified 62 patients with confirmed OSA in a population of 426 patients undergoing pulmonary vein isolation. The 32 patients using CPAP had a significantly higher AF-free survival compared to the 30 patients not using CPAP (72% versus 37%), and the AF-free survival rate of CPAP users was similar to that of patients without OSA. However, no randomized study has been performed in this field yet. Although sleep-disordered breathing is very common in patients with AF, most of them are not sleepy,121 and thus, the long-term adherence to CPAP may be problematic since the latter depends on sleepiness before initiation of therapy.122 Not surprisingly, there is not only an association between OSA and AF, but also between OSA and stroke,42,123 and interestingly, CPAP seems to attenuate this risk.123 However, again, this has not been studied prospectively.
In addition, OSA and its severity are predictors of sudden cardiac death,124 and sudden cardiac death in OSA patients typically occurs during the night.125 CPAP withdrawal, a model to study to effects of OSA, results in prolongation of the QT interval.126 The clinical consequences of these findings regarding OSA and ventricular arrhythmia are not known yet. A detailed summary of the available preliminary evidence on OSA and arrhythmia other than AF can be found elsewhere.127
HF with reduced ejection fraction
OSA has been found to be a factor associated with the development of HF, which may be explained by the fact that OSA represents a cluster of risk factors which can lead to cardiac disease and eventually HF, and also, the effects of OSA on LV structure and function per se. In a cross-sectional study of 6,424 subjects undergoing polysomnography (median AHI 4.4 h−1), the adjusted odds ratio for self-reported HF for the highest compared to the lowest AHI quartile was 2.38.98 In the Sleep Heart Health Study (1,927 men with median AHI of 6.2 h−1, 2,495 women with median AHI of 2.7 h−1, age ≥40 years, no known coronary disease or HF), the risk of incident HF was higher in men with OSA than in those without (adjusted hazard ratio 1.13 per 10 AHI units increase, adjusted hazard ratio 1.58 for AHI ≥30 h−1 versus AHI <5 h−1).100 In contrast, no significant association between OSA and incident HF was found in women.100 In a recent analysis, however, based on 752 men and 893 women free of cardiovascular disease at baseline from the Atherosclerosis Risk In Communities study and the Sleep Heart Health Study, a significant association between OSA and incident HF or death after a mean follow-up of 13.6 years was found only in women.66
In patients with established HF, sleep-disordered breathing is very common.128 In patients with HF and reduced LVEF (HFrEF), two forms of sleep-disordered breathing exist: OSA and central sleep apnea. Central sleep apnea is not the topic of the present study, but it is important to realize that a recent multicenter trial has shown that treatment of central sleep apnea by adaptive servo-ventilation did not improve outcomes.129 In contrast, higher mortality was reported in patients with HFrEF undergoing adaptive servo-ventilation.129
The role of OSA in HFrEF is different. Many HFrEF patients have concomitant OSA,128 which may be coexisting due to risk factors such as obesity or which may be triggered by nocturnal rostral fluid shift, that is, edema of the neck following redistribution of fluid from the legs.130,131 HFrEF patients with concomitant OSA have more pronounced features of sympathetic overactivity compared to those without, and CPAP has been shown to attenuate this. Spaak et al132 have shown that in 60 patients with HFrEF (mean LVEF 22%), those with sleep-disordered breathing (predominantly OSA, AHI ≥15 h−1, n=43) had higher muscle sympathetic nerve activity than those without OSA (AHI <15 h−1, n=17; 58 versus 50 bursts/minute). In 17 patients with HFrEF and OSA (AHI >20 h−1), CPAP for 1 month (n=9) versus no CPAP (n=8) led to a significant reduction in muscle sympathetic nerve activity (from 58 to 48 bursts/minute versus from 63 to 63 bursts/minute).133 Similar data have been obtained for heart rate variability134 and baroreflex sensitivity,135 that is, attenuation by CPAP therapy.
An observational study in 164 patients with HFrEF (LVEF <45%) revealed that patients with untreated moderate-to-severe OSA (mean AHI 33 h−1; n=37) had higher mortality (24% versus 12%) than those without/mild OSA (mean AHI 7 h−1, n=113), and that there was a signal for better outcome in patients with moderate-to-severe OSA treated with CPAP (n=14) compared to those with untreated moderate-to-severe OSA (P=0.07).136 Two trials evaluated the impact of CPAP therapy on LVEF in HFrEF patients.137,138 In a small randomized study among 24 HFrEF patients (mean LVEF ~27%), those undergoing CPAP therapy (n=12) for 1 month improved their LVEF significantly (from 25.0% to 30.8%; P<0.001), whereas no effect was observed in those not treated by CPAP (from 28.5% to 30%; P<0.001 for difference between changes).138 Mansfield et al137 studied the effect of 3 months of CPAP therapy versus no CPAP on LVEF in 55 HFrEF patients (data from 40 patients were available for final analysis). They found an improvement in LVEF in patients treated with CPAP (from 37.6% to 42.6%) compared to those not treated with CPAP (from 33.6% to 35.1%; P=0.04 for difference between changes).137 In this study, the authors also found a significant reduction in overnight urinary norepinephrine excretion in patients treated with CPAP compared to those not treated with CPAP.137 These two studies have been criticized for not using a placebo-controlled design, that is, sham-CPAP in the control group. Thus, overall data regarding the effects of CPAP therapy in HFrEF and OSA are promising, but no randomized trial has so far proven that CPAP therapy for OSA improves clinical outcomes in HFrEF patients. The clinical utility of CPAP for OSA will have to be demonstrated on the background of optimal medical therapy before it will receive a role in the management of HFrEF patients in clinical practice, with the exception of patients with symptomatic OSA, where CPAP is indicated as a symptomatic treatment, irrespective of the cardiac disease status.
HF with preserved ejection fraction
In contrast to HFrEF, there is no established therapy for patients with HF and preserved LVEF (HFpEF).139 Sleep-disordered breathing, and predominantly OSA, is common in HFpEF also.140 There has been much debate over the pathophysiology of HFpEF during the last years, and more recently, it has been proposed that HFpEF results from coronary endothelial dysfunction under the influence of comorbidities including OSA.141 It has also been proposed that there are different HFpEF phenotypes which require different treatments, and that OSA may play an important role for a subset of patients, that is, overweight, hypertensive patients with concomitant severe OSA.142 A recent study among patients with AF and mainly preserved LVEF undergoing cardiac magnetic resonance imaging prior to catheter ablation revealed that sleep apnea (probably OSA in most cases) was an independent predictor of death and hospitalization for HF.143 However, prospective mechanistic studies evaluating the impact of OSA and disease severity in HFpEF, the impact of CPAP on surrogate markers, and the clinical effects of treating OSA in HFpEF are lacking.
Valvular heart disease
Data on the role of sleep-disordered breathing are relatively scarce. A recent study among 140 elderly patients with severe aortic stenosis and significant comorbidities scheduled for transcatheter aortic valve implantation revealed a high prevalence (71%) of sleep-disordered breathing, the predominant type being central sleep apnea, particularly in those with severe sleep apnea.144 However, 25% of patients also had OSA. In a subgroup, it was shown that after transcatheter aortic valve implantation, AHI improved, driven by a strong effect in the central sleep apnea group.145 However, the presence and type of sleep-disordered breathing was not associated with clinical outcomes.145 Obviously, the relatively mild form of OSA had no prognostic impact in an old and highly comorbid population. It remains unknown, however, whether in younger patients with valve disease, severe OSA is of prognostic impact.
Summary and outlook
There is strong evidence of an association between OSA and cardiovascular risk factors, cardiac dysfunction, and cardiac diseases including coronary artery disease, AF, and HF. Figure 2 illustrates the complex and incompletely understood interplay between OSA, diabetes, hypertension, and cardiac diseases. OSA has impact on the major risk factors hypertension and diabetes, but may also directly influence the development and/or progression of coronary artery disease, cardiac structure and function, and AF, which in turn eventually can lead to HFrEF or HFpEF, probably depending on the predominant mechanism, that is, HFrEF in a coronary artery disease-dominant phenotype and HFpEF in a hypertensive heart disease/AF-dominant phenotype. Given that we assume the effects of OSA on hypertension and diabetes on the one hand, and its direct effects on coronary artery disease, AF, and LV function on the other, and we also know about the impact of hypertension and diabetes on coronary artery disease and HF and the interaction between cardiac function and AF in HF, the effects attributable to OSA per se are very hard to differentiate. Therefore, only controlled intervention studies with clinically important endpoints will show the clinical relevance of OSA and its treatment. Until now, observational studies on the effect of CPAP in patients with cardiovascular risk factors and/or established cardiac disease and OSA with clinical endpoints and small prospective studies with surrogate endpoints have revealed promising results. In Table 2, the key findings of previous studies in different areas are summarized. We also suggest important clinical questions which should be addressed in future studies in this table. For this purpose, prospective and randomized studies with clearly defined study populations and clinical endpoints will be required to define to role of OSA, not only as a risk marker but also as a risk factor, disease modifier, and therapeutic target. In the meantime, a clear CPAP indication is restricted to symptomatic OSA patients, where improvement of symptoms can be expected.
 | Figure 2 Illustration of the possible interplay between classical cardiovascular risk factors and cardiac diseases, and the role of OSA. |
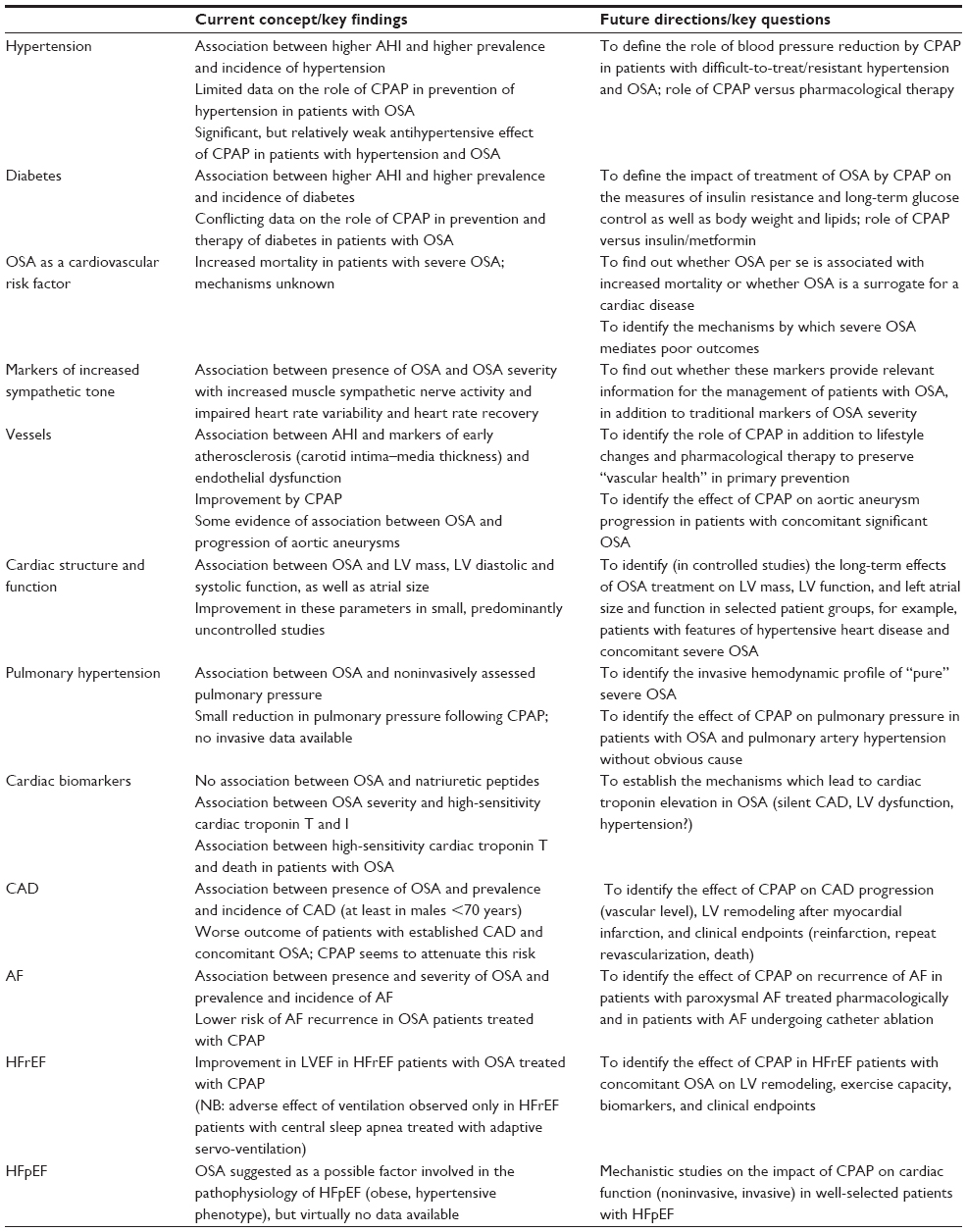 | Table 2 Key findings and future directions regarding the impact of OSA on different aspects of cardiovascular diseases |
Disclosure
The authors report no conflicts of interest in this work.
References
Jordan AS, McSharry DG, Malhotra A. Adult obstructive sleep apnoea. Lancet. 2014;383(9918):736–747. | |
Levy P, Ryan S, Oldenburg O, Parati G. Sleep apnoea and the heart. Eur Respir Rev. 2013;22(129):333–352. | |
Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82–93. | |
Berry RB, Budhiraja R, Gottlieb DJ, et al; American Academy of Sleep Medicine. Rules for scoring respiratory events in sleep: Update of the 2007 aasm manual for the scoring of sleep and associated events. Deliberations of the sleep apnea definitions task force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597–619. | |
Malhotra A, Orr JE, Owens RL. On the cutting edge of obstructive sleep apnoea: Where next? Lancet Respir Med. 2015;3(5):397–403. | |
Heinzer R, Vat S, Marques-Vidal P, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310–318. | |
Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA. 2000;284(23):3015–3021. | |
Tuomilehto HP, Seppa JM, Partinen MM, et al. Kuopio Sleep Apnea Group. Lifestyle intervention with weight reduction: First-line treatment in mild obstructive sleep apnea. Am J Respir Crit Care Med. 2009; 179(4):320–327. | |
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–1235. | |
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–1014. | |
Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010;7(12):677–685. | |
Tkacova R, Rankin F, Fitzgerald FS, Floras JS, Bradley TD. Effects of continuous positive airway pressure on obstructive sleep apnea and left ventricular afterload in patients with heart failure. Circulation. 1998;98(21):2269–2275. | |
Koshino Y, Villarraga HR, Orban M, et al. Changes in left and right ventricular mechanics during the Mueller maneuver in healthy adults: A possible mechanism for abnormal cardiac function in patients with obstructive sleep apnea. Circ Cardiovasc Imaging. 2010;3(3):282–289. | |
Orban M, Bruce CJ, Pressman GS, et al. Dynamic changes of left ventricular performance and left atrial volume induced by the Mueller maneuver in healthy young adults and implications for obstructive sleep apnea, atrial fibrillation, and heart failure. Am J Cardiol. 2008; 102(11):1557–1561. | |
Cincin A, Sari I, Sunbul M, et al. Effect of acute sleep deprivation on left atrial mechanics assessed by three-dimensional echocardiography. Sleep Breath. Epub 2015 Jun 16. | |
Calvin AD, Covassin N, Kremers WK, et al. Experimental sleep restriction causes endothelial dysfunction in healthy humans. J Am Heart Assoc. 2014;3(6):e001143. | |
Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: The task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2013;34(28):2159–2219. | |
Drager LF, Bortolotto LA, Figueiredo AC, Silva BC, Krieger EM, Lorenzi-Filho G. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest. 2007; 131(5):1379–1386. | |
Natsios G, Pastaka C, Vavougios G, et al. Age, body mass index, and daytime and nocturnal hypoxia as predictors of hypertension in patients with obstructive sleep apnea. J Clin Hypertens (Greenwich). Epub 2015 August 7. | |
Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157(15):1746–1752. | |
Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–1384. | |
Cano-Pumarega I, Duran-Cantolla J, Aizpuru F, et al. Obstructive sleep apnea and systemic hypertension: Longitudinal study in the general population: the Vitoria Sleep Cohort. Am J Respir Crit Care Med. 2011;184(11):1299–1304. | |
Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA. 2012;307(20):2169–2176. | |
Barbe F, Duran-Cantolla J, Sanchez-de-la-Torre M, et al. Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: A randomized controlled trial. JAMA. 2012;307(20):2161–2168. | |
Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med. 2014;370(24):2276–2285. | |
Pepin JL, Tamisier R, Barone-Rochette G, Launois SH, Levy P, Baguet JP. Comparison of continuous positive airway pressure and valsartan in hypertensive patients with sleep apnea. Am J Respir Crit Care Med. 2010;182(7):954–960. | |
Montesi SB, Edwards BA, Malhotra A, Bakker JP. The effect of continuous positive airway pressure treatment on blood pressure: A systematic review and meta-analysis of randomized controlled trials. J Clin Sleep Med. 2012;8(5):587–596. | |
Martinez-Garcia MA, Capote F, Campos-Rodriguez F, et al. Spanish Sleep N. Effect of CPAP on blood pressure in patients with obstructive sleep apnea and resistant hypertension: the HIPARCO randomized clinical trial. JAMA. 2013;310(22):2407–2415. | |
Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: A cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7):569–576. | |
Louis M, Punjabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol. 2009;106(5):1538–1544. | |
Reichmuth KJ, Austin D, Skatrud JB, Young T. Association of sleep apnea and type II diabetes: A population-based study. Am J Respir Crit Care Med. 2005;172(12):1590–1595. | |
Botros N, Concato J, Mohsenin V, Selim B, Doctor K, Yaggi HK. Obstructive sleep apnea as a risk factor for type 2 diabetes. Am J Med. 2009;122(12):1122–1127. | |
Hassaballa HA, Tulaimat A, Herdegen JJ, Mokhlesi B. The effect of continuous positive airway pressure on glucose control in diabetic patients with severe obstructive sleep apnea. Sleep Breath. 2005;9(4):176–180. | |
Dorkova Z, Petrasova D, Molcanyiova A, Popovnakova M, Tkacova R. Effects of continuous positive airway pressure on cardiovascular risk profile in patients with severe obstructive sleep apnea and metabolic syndrome. Chest. 2008;134(4):686–692. | |
Sharma SK, Agrawal S, Damodaran D, et al. CPAP for the metabolic syndrome in patients with obstructive sleep apnea. N Engl J Med. 2011;365(24):2277–2286. | |
Sharma SK, Agrawal S, Damodaran D, et al. Retraction: CPAP for the metabolic syndrome in patients with obstructive sleep apnea. N Engl J Med. 2013;369(18):1770. | |
Drager LF, Brunoni AR, Jenner R, Lorenzi-Filho G, Bensenor IM, Lotufo PA. Effects of CPAP on body weight in patients with obstructive sleep apnoea: a meta-analysis of randomised trials. Thorax. 2015;70(3):258–264. | |
West SD, Nicoll DJ, Wallace TM, Matthews DR, Stradling JR. Effect of cpap on insulin resistance and hba1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax. 2007;62(11):969–974. | |
Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365(9464):1046–1053. | |
Campos-Rodriguez F, Martinez-Garcia MA, de la Cruz-Moron I, Almeida-Gonzalez C, Catalan-Serra P, Montserrat JM. Cardiovascular mortality in women with obstructive sleep apnea with or without continuous positive airway pressure treatment: A cohort study. Ann Intern Med. 2012;156(2):115–122. | |
Martinez-Garcia MA, Campos-Rodriguez F, Catalan-Serra P, et al. Cardiovascular mortality in obstructive sleep apnea in the elderly: role of long-term continuous positive airway pressure treatment: a prospective observational study. Am J Respir Crit Care Med. 2012;186(9):909–916. | |
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353(19):2034–2041. | |
Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96(4):1897–1904. | |
Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation.1998;98(8):772–776. | |
Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK. Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation. 1999;100(23):2332–2335. | |
Narkiewicz K, Montano N, Cogliati C, van de Borne PJ, Dyken ME, Somers VK. Altered cardiovascular variability in obstructive sleep apnea. Circulation. 1998;98(11):1071–1077. | |
Trimer R, Mendes RG, Costa FS, et al. Is there a chronic sleep stage-dependent linear and nonlinear cardiac autonomic impairment in obstructive sleep apnea? Sleep Breath. 2014;18(2):403–409. | |
Cole CR, Blackstone EH, Pashkow FJ, Snader CE, Lauer MS. Heart-rate recovery immediately after exercise as a predictor of mortality. N Engl J Med. 1999;341(18):1351–1357. | |
Imai K, Sato H, Hori M, et al. Vagally mediated heart rate recovery after exercise is accelerated in athletes but blunted in patients with chronic heart failure. J Am Coll Cardiol. 1994;24(6):1529–1535. | |
Maeder MT, Munzer T, Rickli H, et al. Association between heart rate recovery and severity of obstructive sleep apnea syndrome. Sleep Med. 2008;9(7):753–761. | |
Maeder MT, Ammann P, Schoch OD, et al. Determinants of postexercise heart rate recovery in patients with the obstructive sleep apnea syndrome. Chest. 2010;137:310–317. | |
Maeder MT, Ammann P, Munzer T, et al. Continuous positive airway pressure improves exercise capacity and heart rate recovery in obstructive sleep apnea. Int J Cardiol. 2009;132(1):75–83. | |
Ip MS, Tse HF, Lam B, Tsang KW, Lam WK. Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med. 2004;169(3):348–353. | |
Kohler M, Craig S, Nicoll D, Leeson P, Davies RJ, Stradling JR. Endothelial function and arterial stiffness in minimally symptomatic obstructive sleep apnea. Am J Respir Crit Care Med. 2008;178(9):984–988. | |
Kohler M, Craig S, Pepperell JC, et al. CPAP improves endothelial function in patients with minimally symptomatic OSA: Results from a subset study of the mosaic trial. Chest. 2013;144(3):896–902. | |
Schwarz EI, Puhan MA, Schlatzer C, Stradling JR, Kohler M. Effect of CPAP therapy on endothelial function in obstructive sleep apnoea: A systematic review and meta-analysis. Respirology. 2015;20(6):889–895. | |
Minoguchi K, Yokoe T, Tazaki T, et al. Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172(5):625–630. | |
Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF. Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2007;176(7):706–712. | |
Gaisl T, Bratton DJ, Kohler M. The impact of obstructive sleep apnoea on the aorta. Eur Respir J. 2015;46(2):532–544. | |
Kohler M, Blair E, Risby P, et al. The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan’s syndrome. Thorax. 2009;64(2):162–166. | |
Kohler M, Pitcher A, Blair E, et al. The impact of obstructive sleep apnea on aortic disease in Marfan’s syndrome. Respiration. 2013;86(1):39–44. | |
Serizawa N, Yumino D, Takagi A, et al. Obstructive sleep apnea is associated with greater thoracic aortic size. J Am Coll Cardiol. 2008;52(10):885–886. | |
Mason RH, Ruegg G, Perkins J, et al. Obstructive sleep apnea in patients with abdominal aortic aneurysms: Highly prevalent and associated with aneurysm expansion. Am J Resp Crit Care Med. 2011;183(5):668–674. | |
Chami HA, Devereux RB, Gottdiener JS, et al. Left ventricular morphology and systolic function in sleep-disordered breathing: The sleep heart health study. Circulation. 2008;117(20):2599–2607. | |
Niroumand M, Kuperstein R, Sasson Z, Hanly PJ. Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J Respir Crit Care Med. 2001;163(7):1632–1636. | |
Roca GQ, Redline S, Claggett B, et al. Sex-specific association of sleep apnea severity with subclinical myocardial injury, ventricular hypertrophy, and heart failure risk in a community-dwelling cohort: The atherosclerosis risk in communities-sleep heart health study. Circulation. 2015;132(14):1329–1337. | |
Colish J, Walker JR, Elmayergi N, et al. Obstructive sleep apnea: Effects of continuous positive airway pressure on cardiac remodeling as assessed by cardiac biomarkers, echocardiography, and cardiac MRI. Chest. 2012;141(3):674–681. | |
Butt M, Dwivedi G, Shantsila A, Khair OA, Lip GY. Left ventricular systolic and diastolic function in obstructive sleep apnea: Impact of continuous positive airway pressure therapy. Circ Heart Fail. 2012;5(2):226–233. | |
Craig S, Kylintireas I, Kohler M, et al. Effect of CPAP on cardiac function in minimally symptomatic patients with OSA: Results from a subset of the mosaic randomized trial. J Clin Sleep Med. 2015;11(9):967–973. | |
Haruki N, Takeuchi M, Kanazawa Y, et al. Continuous positive airway pressure ameliorates sleep-induced subclinical left ventricular systolic dysfunction: Demonstration by two-dimensional speckle-tracking echocardiography. Eur J Echocardiogr. 2010;11(4):352–358. | |
Wang D, Ma GS, Wang XY, Lu QQ, Wang Y, Liu NF. Left ventricular subclinical dysfunction associated with myocardial deformation changes in obstructive sleep apnea patients estimated by real-time 3d speckle-tracking echocardiography. Sleep Breath. Epub 2015 May 24. | |
Wong CY, O’Moore-Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation. 2004;110(19):3081–3087. | |
Arias MA, Garcia-Rio F, Alonso-Fernandez A, Mediano O, Martinez I, Villamor J. Obstructive sleep apnea syndrome affects left ventricular diastolic function: Effects of nasal continuous positive airway pressure in men. Circulation. 2005;112(3):375–383. | |
Baguet JP, Barone-Rochette G, Levy P, et al. Left ventricular diastolic dysfunction is linked to severity of obstructive sleep apnoea. Eur Respir J. 2010;36(6):1323–1329. | |
Oliveira W, Campos O, Bezerra Lira-Filho E, et al. Left atrial volume and function in patients with obstructive sleep apnea assessed by real-time three-dimensional echocardiography. J Am Soc Echocardiogr. 2008;21(12):1355–1361. | |
Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr. 2009;22(2):107–133. | |
Arias MA, Garcia-Rio F, Alonso-Fernandez A, Martinez I, Villamor J. Pulmonary hypertension in obstructive sleep apnoea: Effects of continuous positive airway pressure: a randomized, controlled cross-over study. Eur Heart J. 2006;27(9):1106–1113. | |
Oliveira W, Poyares D, Cintra F, et al. Impact of continuous positive airway pressure treatment on right ventricle performance in patients with obstructive sleep apnoea, assessed by three-dimensional echocardiography. Sleep Med. 2012;13(5):510–516. | |
Guidry UC, Mendes LA, Evans JC, et al. Echocardiographic features of the right heart in sleep-disordered breathing: The Framingham Heart Study. Am J Respir Crit Care Med. 2001;164(6):933–938. | |
Altekin RE, Karakas MS, Yanikoglu A, et al. Determination of right ventricular dysfunction using the speckle tracking echocardiography method in patients with obstructive sleep apnea. Cardiol J. 2012;19(2):130–139. | |
Kholdani C, Fares WH, Mohsenin V. Pulmonary hypertension in obstructive sleep apnea: Is it clinically significant? A critical analysis of the association and pathophysiology. Pulm Circ. 2015;5(2):220–227. | |
Karamanzanis G, Panou F, Lazaros G, et al. Impact of continuous positive airway pressure treatment on myocardial performance in patients with obstructive sleep apnea. A conventional and tissue Doppler echocardiographic study. Sleep Breath. 2015;19(1):343–350. | |
Hubner RH, El Mokhtari NE, Freitag S, et al. NT-proBNP is not elevated in patients with obstructive sleep apnoea. Respir Med. 2008;102(1):134–142. | |
Maeder MT, Ammann P, Rickli H, et al. N-terminal pro-B-type natriuretic peptide and functional capacity in patients with obstructive sleep apnea. Sleep Breath. 2008;12(1):7–16. | |
Patwardhan AA, Larson MG, Levy D, et al. Obstructive sleep apnea and plasma natriuretic peptide levels in a community-based sample. Sleep. 2006;29(10):1301–1306. | |
Vartany E, Imevbore M, O’Malley M, et al. N-terminal pro-brain natriuretic peptide for detection of cardiovascular stress in patients with obstructive sleep apnea syndrome. J Sleep Res. 2006;15(4):424–429. | |
Querejeta Roca G, Redline S, Punjabi N, et al. Sleep apnea is associated with subclinical myocardial injury in the community. The ARIC-SHHS study. Am J Respir Crit Care Med. 2013;188(12):1460–1465. | |
Ljunggren M, Lindahl B, Theorell-Haglow J, Lindberg E. Association between obstructive sleep apnea and elevated levels of type b natriuretic peptide in a community-based sample of women. Sleep. 2012;35(11):1521–1527. | |
Maeder MT, Strobel W, Christ M, et al. Comprehensive biomarker profiling in patients with obstructive sleep apnea. Clin Biochem. 2015;48(4–5):340–346. | |
Tasci S, Manka R, Scholtyssek S, et al. NT-pro-BNP in obstructive sleep apnea syndrome is decreased by nasal continuous positive airway pressure. Clin Res Cardiol. 2006;95(1):23–30. | |
Maeder MT, Mueller C, Pfisterer ME, Buser PT, Brunner-La Rocca HP. Use of B-type natriuretic peptide outside of the emergency department. Int J Cardiol. 2008;127(1):5–16. | |
Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation. 2004;109(5):594–600. | |
Hall TS, Herrscher T, Jarolim P, et al. Obstructive sleep apnea: no independent association to troponins. Sleep Breath. 2014;18(2):351–358. | |
Gami AS, Svatikova A, Wolk R, et al. Cardiac troponin T in obstructive sleep apnea. Chest. 2004;125(6):2097–2100. | |
Randby A, Namtvedt SK, Einvik G, et al. Obstructive sleep apnea is associated with increased high-sensitivity cardiac troponin t levels. Chest. 2012;142(3):639–646. | |
Einvik G, Rosjo H, Randby A, et al. Severity of obstructive sleep apnea is associated with cardiac troponin I concentrations in a community-based sample: data from the Akershus Sleep Apnea Project. Sleep. 2014;37(6):1111–1116. | |
Barcelo A, Esquinas C, Bauca JM, et al. Effect of CPAP treatment on plasma high sensitivity troponin levels in patients with obstructive sleep apnea. Respir Med. 2014;108(7):1060–1063. | |
Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: Cross-sectional results of the sleep heart health study. Am J Respir Crit Care Med. 2001;163(1):19–25. | |
Peker Y, Kraiczi H, Hedner J, Loth S, Johansson A, Bende M. An independent association between obstructive sleep apnoea and coronary artery disease. Eur Respir J. 1999;14(1):179–184. | |
Gottlieb DJ, Yenokyan G, Newman AB, et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation. 2010;122(4):352–360. | |
Lutsey PL, McClelland RL, Duprez D, et al. Objectively measured sleep characteristics and prevalence of coronary artery calcification: the multi-ethnic study of atherosclerosis sleep study. Thorax. 2015; 70(9):880–887. | |
Kwon Y, Duprez DA, Jacobs DR, et al. Obstructive sleep apnea and progression of coronary artery calcium: the multi-ethnic study of atherosclerosis study. J Am Heart Assoc. 2014;3(5):e001241. | |
Peker Y, Hedner J, Kraiczi H, Loth S. Respiratory disturbance index: An independent predictor of mortality in coronary artery disease. Am J Respir Crit Care Med. 2000;162(1):81–86. | |
Kuniyoshi FH, Garcia-Touchard A, Gami AS, et al. Day-night variation of acute myocardial infarction in obstructive sleep apnea. J Am Coll Cardiol. 2008;52(5):343–346. | |
Yumino D, Tsurumi Y, Takagi A, Suzuki K, Kasanuki H. Impact of obstructive sleep apnea on clinical and angiographic outcomes following percutaneous coronary intervention in patients with acute coronary syndrome. Am J Cardiol. 2007;99(1):26–30. | |
Uchoa CH, Danzi-Soares Nde J, Nunes FS, et al. Impact of OSA on cardiovascular events after coronary artery bypass surgery. Chest. 2015;147(5):1352–1360. | |
Thunstrom E, Glantz H, Fu M, et al. Increased inflammatory activity in nonobese patients with coronary artery disease and obstructive sleep apnea. Sleep. 2015;38(3):463–471. | |
Sert Kuniyoshi FH, Singh P, Gami AS, et al. Patients with obstructive sleep apnea exhibit impaired endothelial function after myocardial infarction. Chest. 2011;140(1):62–67. | |
Glantz H, Thunstrom E, Johansson MC, et al. Obstructive sleep apnea is independently associated with worse diastolic function in coronary artery disease. Sleep Med. 2015;16(1):160–167. | |
Steiner S, Schueller PO, Schulze V, Strauer BE. Occurrence of coronary collateral vessels in patients with sleep apnea and total coronary occlusion. Chest. 2010;137(3):516–520. | |
Milleron O, Pilliere R, Foucher A, et al. Benefits of obstructive sleep apnoea treatment in coronary artery disease: A long-term follow-up study. Eur Heart J. 2004;25(9):728–734. | |
Cassar A, Morgenthaler TI, Lennon RJ, Rihal CS, Lerman A. Treatment of obstructive sleep apnea is associated with decreased cardiac death after percutaneous coronary intervention. J Am Coll Cardiol. 2007;50(14):1310–1314. | |
Dimitri H, Ng M, Brooks AG, et al. Atrial remodeling in obstructive sleep apnea: Implications for atrial fibrillation. Heart rhythm. 2012;9(3):321–327. | |
Schlatzer C, Schwarz EI, Sievi NA, et al. Intrathoracic pressure swings induced by simulated obstructive sleep apnoea promote arrhythmias in paroxysmal atrial fibrillation. Europace. 2015;18(1):64–70. | |
Gami AS, Pressman G, Caples SM, et al. Association of atrial fibrillation and obstructive sleep apnea. Circulation. 2004;110(4):364–367. | |
Gami AS, Hodge DO, Herges RM, et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49(5):565–571. | |
Kanagala R, Murali NS, Friedman PA, et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation. 2003; 107(20):2589–2594. | |
Matiello M, Nadal M, Tamborero D, et al. Low efficacy of atrial fibrillation ablation in severe obstructive sleep apnoea patients. Europace. 2010;12(8):1084–1089. | |
Neilan TG, Farhad H, Dodson JA, et al. Effect of sleep apnea and continuous positive airway pressure on cardiac structure and recurrence of atrial fibrillation. J Am Heart Assoc. 2013;2(6):e000421. | |
Fein AS, Shvilkin A, Shah D, et al. Treatment of obstructive sleep apnea reduces the risk of atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. 2013;62(4):300–305. | |
Altmann DR, Ullmer E, Rickli H, et al. Clinical impact of screening for sleep related breathing disorders in atrial fibrillation. Int J Cardiol. 2012;154(3):256–258. | |
Schoch OD, Baty F, Niedermann J, Rudiger JJ, Brutsche MH. Baseline predictors of adherence to positive airway pressure therapy for sleep apnea: a 10-year single-center observational cohort study. Respiration. 2014;87(2):121–128. | |
Campos-Rodriguez F, Martinez-Garcia MA, Reyes-Nunez N, Caballero-Martinez I, Catalan-Serra P, Almeida-Gonzalez CV. Role of sleep apnea and continuous positive airway pressure therapy in the incidence of stroke or coronary heart disease in women. Am J Respir Crit Care Med. 2014;189(12):1544–1550. | |
Gami AS, Olson EJ, Shen WK, et al. Obstructive sleep apnea and the risk of sudden cardiac death: A longitudinal study of 10,701 adults. J Am Coll Cardiol. 2013;62(7):610–616. | |
Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med. 2005; 352(12):1206–1214. | |
Rossi VA, Stoewhas AC, Camen G, et al. The effects of continuous positive airway pressure therapy withdrawal on cardiac repolarization: data from a randomized controlled trial. Eur Heart J. 2012;33(17):2206–2212. | |
Rossi VA, Stradling JR, Kohler M. Effects of obstructive sleep apnoea on heart rhythm. Eur Respir J. 2013;41(6):1439–1451. | |
Oldenburg O, Lamp B, Faber L, Teschler H, Horstkotte D, Topfer V. Sleep-disordered breathing in patients with symptomatic heart failure: A contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail. 2007;9(8):251–257. | |
Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373(12):1095–1105. | |
Redolfi S, Yumino D, Ruttanaumpawan P, et al. Relationship between overnight rostral fluid shift and obstructive sleep apnea in nonobese men. Am J Respir Crit Care Med. 2009;179(3):241–246. | |
Yumino D, Redolfi S, Ruttanaumpawan P, et al. Nocturnal rostral fluid shift: a unifying concept for the pathogenesis of obstructive and central sleep apnea in men with heart failure. Circulation. 2010;121(14):1598–1605. | |
Spaak J, Egri ZJ, Kubo T, et al. Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension. 2005;46(6):1327–1332. | |
Usui K, Bradley TD, Spaak J, et al. Inhibition of awake sympathetic nerve activity of heart failure patients with obstructive sleep apnea by nocturnal continuous positive airway pressure. J Am Coll Cardiol. 2005;45(12):2008–2011. | |
Gilman MP, Floras JS, Usui K, Kaneko Y, Leung RS, Bradley TD. Continuous positive airway pressure increases heart rate variability in heart failure patients with obstructive sleep apnoea. Clin Sci(Lond). 2008;114(3):243–249. | |
Ruttanaumpawan P, Gilman MP, Usui K, Floras JS, Bradley TD. Sustained effect of continuous positive airway pressure on baroreflex sensitivity in congestive heart failure patients with obstructive sleep apnea. J Hypertens. 2008;26(6):1163–1168. | |
Wang H, Parker JD, Newton GE, et al. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol. 2007;49(15):1625–1631. | |
Mansfield DR, Gollogly NC, Kaye DM, Richardson M, Bergin P, Naughton MT. Controlled trial of continuous positive airway pressure in obstructive sleep apnea and heart failure. Am J Respir Crit Care Med. 2004;169:361–366. | |
Kaneko Y, Floras JS, Usui K, et al. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med. 2003;348(13):1233–1241. | |
Maeder MT, Kaye DM. Heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol. 2009;53(11):905–918. | |
Herrscher TE, Akre H, Overland B, Sandvik L, Westheim AS. High prevalence of sleep apnea in heart failure outpatients: Even in patients with preserved systolic function. J Card Fail. 2011;17(5):420–425. | |
Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–271. | |
Senni M, Paulus WJ, Gavazzi A, et al. New strategies for heart failure with preserved ejection fraction: The importance of targeted therapies for heart failure phenotypes. Eur Heart J. 2014;35(40):2797–2815. | |
Shah RV, Abbasi SA, Heydari B, et al. Obesity and sleep apnea are independently associated with adverse left ventricular remodeling and clinical outcome in patients with atrial fibrillation and preserved ventricular function. Am Heart J. 2014;167(4):620–626. | |
Linhart M, Sinning JM, Ghanem A, et al. Prevalence and impact of sleep disordered breathing in patients with severe aortic stenosis. PLoS One. 2015;10(7):e0133176. | |
Linhart M, Pabst S, Fistera R, et al. Transcatheter valve implantation improves central sleep apnoea in severe aortic stenosis. EuroIntervention. 2013;9(8):923–928. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.