Back to Journals » Cancer Management and Research » Volume 13
A CLDN18.2-Targeting Bispecific T Cell Co-Stimulatory Activator for Cancer Immunotherapy
Authors Liang J, Zhang H, Huang Y, Fan L, Li F, Li M, Yan Y, Zhang J, Li Z, Yang X ![]()
Received 27 July 2021
Accepted for publication 27 August 2021
Published 7 September 2021 Volume 2021:13 Pages 6977—6987
DOI https://doi.org/10.2147/CMAR.S330637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Jie Liang,1 Huihui Zhang,1 Yue Huang,1 Lilv Fan,1 Fanlin Li,1 Min Li,1 Yaping Yan,1 Junshi Zhang,1 Zeyu Li,1 Xuanming Yang1,2
1Sheng Yushou Center of Cell Biology and Immunology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, 200240, People’s Republic of China; 2Joint International Research Laboratory of Metabolic & Developmental Sciences, Shanghai Jiao Tong University, Shanghai, 200240, People’s Republic of China
Correspondence: Xuanming Yang
Sheng Yushou Center of Cell Biology and Immunology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, People’s Republic of China
Tel +86-21-34204065
Email [email protected]
Background: Co-stimulatory receptor agonist antibodies have shown promising antitumor efficacy in preclinical models. However, their clinical development lags due to systemic or local adverse effects of non-specific T cell activation. Utilization of a bispecific antibody format to reduce off-tumor immune activation is a focus of co-stimulatory receptor agonist antibody design.
Methods: In this study, a bispecific antibody with anti-CLDN18.2 and anti-CD28 moieties was produced. Its T cell costimulation ability was evaluated in T cell coculture assay in vitro. Its safety and anti-tumor efficacy were explored in mouse tumor models.
Results: Anti-CLDN18.2-anti-CD28 bispecific antibody could co-stimulate T cells and increase the expression of effector cytokines in a CLDN18.2-dependent manner. Treatment of anti-CLDN18.2-anti-CD28 could reduce tumor burden and increase tumor-infiltrated T cells. Immunosuppressive cells including tumor-associated macrophages and myeloid-derived suppressor cells were also reduced without systemic adverse effects.
Conclusion: This work provided proof-of-concept evidence for a new strategy to develop a bispecific co-stimulatory activator for treating CLDN18.2+ tumors.
Keywords: CLDN18.2, CD28, bispecific antibody, cancer immunotherapy
Introduction
Cancer immunotherapies have made great progress in recent decades. Remarkably, various immune checkpoint antibodies and chimeric antigen receptor (CAR) T-cell therapeutics have been clinically approved.1–4 Although CAR T-cell therapy has yielded noteworthy results in treating hematologic malignancies, its efficacy for solid tumors is limited.4,5 Immune checkpoint blockade has shown broad anti-tumor efficacy across different tumor types. However, the overall objective response rate is only approximately 30%, and the majority of patients are resistant to immune checkpoint blockade antibodies.6–8 Immunotherapy against solid tumors remains a challenge owing to the few specific antigen targets, high tumor heterogeneity, hostile tumor microenvironment (TME), etc. Novel therapeutics with tumor-targeted immune activation features are urgently needed for immunotherapies against solid tumors.
Appropriate tumor-specific antigens (TSAs) are the key for immunotherapy to ensure precise immune activation in tumor tissues and to avoid immune damage in normal tissues. The tight junction molecule, claudin-18.2 (CLDN18.2), is an attractive TSA for antibody-based cancer immunotherapy. CLDN18.2 in normal tissues is expressed exclusively in differentiated epithelial cells of the gastric mucosa, with the exception of the gastric stem cell zone. However, it is widely expressed in various tumors, including gastric, pancreatic, esophageal, ovarian, and lung tumors.9–11 All these biological characteristics have paved the way for the development of CLDN18.2-targeted therapies for solid tumors. IMAB362 (zolbetuximab) was the first monoclonal antibody developed to target CLDN18.2; its potent anti-tumor activity was proven using antibody-dependent cytotoxicity, complement-dependent cytotoxicity, induction of apoptosis, and inhibition of cell proliferation in non-clinical pancreatic ductal adenocarcinoma models.12,13 The safety and tolerance of IMAB362 were verified in clinical Phase I and II studies for the treatment of patients with metastatic gastroesophageal adenocarcinomas.14,15 Owing to the tumor specificity of CLDN18.2, other anti-CLDN18.2-based targeted therapeutics such as antibody-drug conjugates and bispecific T cell engagers (BiTEs) have been developed.16,17 They have shown promising outcomes in pancreatic and gastric patient-derived xenograft tumor models, especially the anti-CLDN18.2/CD3 bispecific antibodies.18,19 However, CD3 bispecific antibodies can employ any T cell to act as an effector cell, lacking of selection on tumor antigen-specific T cells. Some CD3 bispecific antibodies have been shown to induce severe cytokine release syndrome (CRS) and other side effects.20
In addition to being activators of the T cell receptor (TCR) signaling pathway, co-stimulation agonist antibodies have shown potent T cell activation ability and anti-tumor efficacy in preclinical models. However, the clinical development of these agonist antibodies has been hampered by undesirable off-tumor adverse effects.21,22 CD28 is expressed constitutively on mouse and human T cells,23 and is a potential target candidate for T cell agonist antibodies. Unfortunately, the CD28 superagonist, TGN1412, was suspended due to severe CRS after infusion.24 It remains unclear whether tumor-targeted CD28 activation can maintain its T cell co-stimulation ability and reduce off-tumor adverse effects.
In this study, we designed an anti-CLDN18.2-anti-CD28 non-Fc antibody with one arm that binds to the TSA, CLDN18.2, and the other arm that targets CD28. It showed tumor-specific T cell co-stimulatory activity in vitro and a good safety profile in vivo. Importantly, this bispecific anti-CLDN18.2-anti-CD28 protein exhibited potential anti-tumor efficacy mainly by increasing the number of cytotoxic CD8+ T cells and reshaping the immune-suppressive TME. Collectively, our data indicate that CLDN18.2-targeted CD28 activation is a potential therapeutic approach for the treatment of solid tumors, with limited adverse effects.
Materials and Methods
Mice
Six- to eight-week-old female C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). OT-I mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All the mice were maintained under pathogen-free conditions and approved by the Animal Care and Use Committee of Shanghai Jiao Tong University. Animal studies were conducted in accordance with the NIH and institutional protocols and guidelines.
Cell Lines and Reagents
The Lenti-X 293 cell line was purchased from Clontech (CA, USA). B16-ovalbumin (OVA) mouse melanoma cells were provided by Hans Schreiber (University of Chicago). The usage of B16-OVA was approved by the Research Ethical Committee of Shanghai Jiao Tong University. B16-OVA-CLDN18.2 and 293-CLDN18.2 cell lines or 293-CD28 cell line were generated by infection with lentiviruses expressing mouse CLDN18.2 or mouse CD28, respectively. All the cells were cultured in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Brazil), 2 mM
Purification of Fusion Protein
The sequence of anti-CLDN18.2 single-chain variable fragment (scFv) was derived from patent WO 2019/242505 A1, which recognizes both human and murine CLDN18.2. The anti-mouse CD28 scFv was synthesized at Genewiz (Suzhou, China) and cloned into the pCDH-EF1-MCS vector. The antibodies, anti-CLDN18.2-anti-CD28 and anti-CLDN18.2, were generated using Lenti-X 293 cells infected with the respective lentivirus. Proteins in the collected supernatant of cells were salted out with ammonium sulfate, dissolved, and dialyzed against PBS. The two proteins were purified using Ni-chelating affinity chromatography.
Animal Studies
On day 0, wild-type C57BL/6 mice at the age of 6–8 weeks were inoculated s.c. in the right flank with 2.5×105 mouse melanoma cells (B16-OVA) overexpressing mouse CLDN18.2. For the B16-OVA-CLDN18.2 animal model, mice were peri-tumor-injected with 20 μg of vehicle buffer (PBS), control protein (anti-CLDN18.2), and anti-CLDN18.2-anti-CD28 on days 8 and 11. The deletion antibody, anti-CD8 (YTS 169.4.2), was injected intraperitoneally along with anti-CLDN18.2-anti-CD28. The tumors were measured along three orthogonal axes (a, b, and c) every 3 days, and the tumor volumes were calculated using the equation, (a × b × c)/2.
Acquisition of Single Cells from Animal Tissues or Tumors
Red blood cells from mouse spleens were lysed using Red Blood Cell Lysis buffer (Sangon Biotech, China). OT-I CD8+ T cells from OT-I transgenic mice were purified using the MojoSortTM Mouse CD3 T cell Isolation kit (BioLegend, USA). Harvested tumors were dissociated with 0.2 mg/mL DNase I (Sigma-Aldrich, USA) and 0.05 mg/mL Liberase TL (Roche, Germany) in RPMI for 25 min at 37°C.
Re-Stimulation of ex vivo Lymphocytes from Spleens and Lymph Nodes
Spleens and lymph nodes of tumor-burdened mice were ground through 70 μm filter strainers and washed with RPMI 1640. Red blood cell lysis was performed according to the protocol described above. Lymphocytes (4 × 105) were incubated in 96-well plates in the absence or presence of OT-I or OT-II peptide at 37 °C and 5% CO2. IFN-γ and TNF-α in the supernatant were measured using a CBA kit (BD Biosciences, USA).
Flow Cytometry and Analysis
The proteins we produced were verified using monoclonal anti-His antibodies (R&D Systems) and then stained with the secondary antibody, anti-mouse Fc-APC (Jackson ImmunoResearch). Single-cell suspensions of tumors were prepared according to the aforementioned procedure. Non-specific labeling was blocked with anti-CD16/32 (anti-FcγRII/III, clone 2.4G2) before specific labeling. Cells were stained with anti-mouse CD45-BV605 (30-F11), anti-mouse CD4-FITC (GK1.5), anti-mouse CD8α-APC700 (53–6.7), anti-mouse CD11c-PE (N418), anti-mouse MHCII-APC (M5/114.15.2), anti-mouse F4/80-FITC (BM8), anti-mouse Ly6C-FITC (HK1.4), and anti-mouse Ly6G-PE (1A8), which were purchased from BioLegend. Anti-mouse CD69-PE (H1-2F3) and anti-mouse CD11b-APC700 (M1/70) were purchased from BD Biosciences and eBioscience, respectively. Anti-CD8 (YTS 169.4.2) was produced in-house. All the samples were analyzed using a Cytoflex Flow Cytometer (Beckman Coulter, Brea, CA, USA), and the data were analyzed using FlowJo software v.10.4 (TreeStar Inc., San Carlos, CA, USA).
Statistics
Statistical analysis was performed using GraphPad Prism software v.8.0. Data were expressed as means ± standard errors of the means (SEM). The significance of the assays was determined using a two-sided Student’s unpaired t-test. Where indicated, *P < 0.05, **P < 0.01, and ***P < 0.001 were considered statistically significant.
Results
Generation and Characterizations of Anti-CLDN18.2-Anti-CD28 Bispecific T Cell Co-Stimulator
Bispecific antibodies are considered the second generation of antibodies, and are engineered to bind to two different antigens or epitopes. As representative bispecific antibodies, BiTEs are designed to activate TCR signaling in a tumor antigen-dependent manner and have shown great success in both preclinical and clinical settings.25–27 It is unclear whether tumor-specific activation of T cell co-stimulatory signaling can boost anti-tumor activity. Hence, we designed a fusion protein, anti-CLDN18.2-anti-CD28. Two scFvs recognizing murine or human CLDN18.2 and murine CD28 were fused through a flexible (G4S)2 linker, followed by a 6 × His tag for affinity purification (Figure 1A). The purified anti-CLDN18.2-anti-CD28 antibody showed a molecular weight of approximately 58 kDa based on SDS-PAGE analysis (Figure 1B).
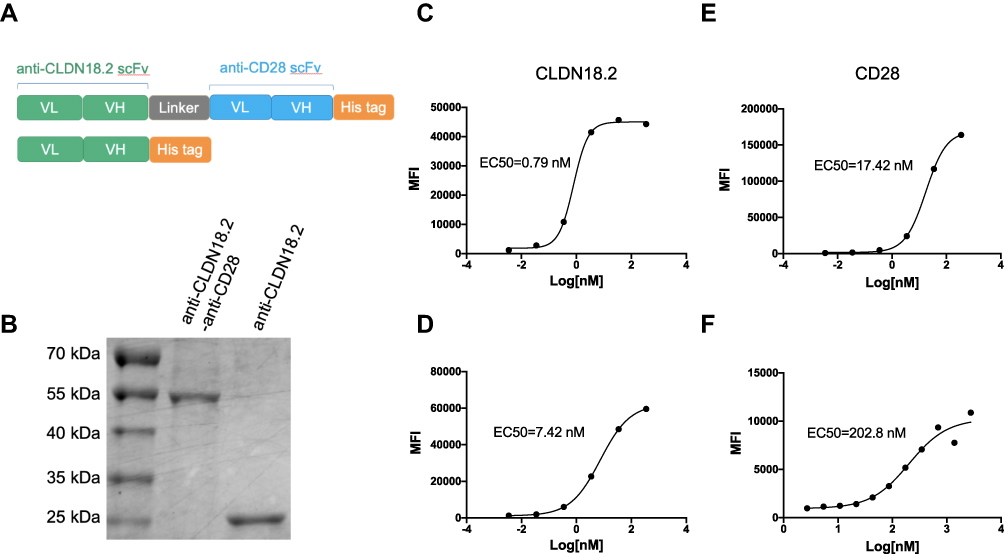 |
Figure 1 Generation and characterization of anti-CLDN18.2-anti-CD28 bispecific antibody. (A) Schematic diagrams of bispecific antibody, anti-CLDN18.2-anti-CD28 (top), and isotype control protein, anti-CLDN18.2 (bottom). (B) SDS-PAGE of purified anti-CLDN18.2-anti-CD28 (58 kDa) and anti-CLDN18.2 (30 kDa) proteins. (C and D) Dose-dependent binding affinity of anti-CLDN18.2 arm of the bispecific protein to Lenti-X 293-CLDN18.2 (C) or B16-OVA-CLDN18.2 cells (D) expressing CLDN18.2. (E and F) Dose-dependent binding affinity of anti-CD28 arm of the bispecific protein to Lenti-X 293-CD28 cells (E) or splenocytes from C57BL/6 mice (F). |
To verify whether the two moieties of anti-CLDN18.2-anti-CD28 were functional, we firstly established Lenti-X 293 and B16-OVA cell lines overexpressing murine CLDN18.2 or murine CD28. The anti-CLDN18.2-anti-CD28 fusion protein showed a strong binding ability to both 293-CLDN18.2 and B16-OVA-CLDN18.2 tumor cells, with an EC50 of 0.79 and 7.42 nM, respectively (Figure 1C and D). Similarly, anti-CLDN18.2-anti-CD28 could specifically bind to 293-CD28 cells with an EC50 of 17.42 nM (Figure 1E). To test whether anti-CLDN18.2-anti-CD28 could bind to endogenously expressed CD28 on T cells, we performed a similar binding assay with mouse splenocytes. The EC50 in splenocytes was 202.8 nM, indicating that anti-CLDN18.2-anti-CD28 had the ability to bind to endogenously expressed CD28 (Figure 1F). Thus, we successfully generated a bispecific antibody, and the functions of the two parts remained intact.
Anti-CLDN18.2-Anti-CD28 Co-Stimulated T Cells in an Antigen-Dependent Manner
CD28 is constitutively expressed on T cells, and provides critical co-stimulatory signals upon engaging its natural ligands, CD80 or CD86.28 We tested whether anti-CLDN18.2-anti-CD28 could co-stimulate T cells in a CLDN18.2-dependent manner. To this end, we established CLDN18.2-negative B16-OVA and CLDN18.2-overexpressing B16-OVA-CLDN18.2 cell lines. In both cell lines, OVA was stably expressed, mimicking a TSA. The dominant peptide, OT-I, from OVA was recognized by CD8+ T cells from OT-I transgenic mice.29 We then co-cultured OT-I T cells, and B16-OVA and B16-OVA-CLDN18.2 cells, and tested the activation marker of T cells (CD69). When co-cultured with B16-OVA-CLDN18.2 cells, the expression of murine CD69 on OT-I T cells was increased by anti-CLDN18.2-anti-CD28 in a dose-dependent manner (Figure 2A). However, when co-cultured with B16-OVA, there was no difference in the expression of CD69 in the presence of different concentrations of anti-CLDN18.2-anti-CD28, suggesting an antigen-dependent activation mechanism. TNF-α and IFN-γ are critical effector cytokines in activated CD8+ T cells. We also observed similar dose- and CLDN18.2-dependent enhancement of TNF-α and IFN-γ production by anti-CLDN18.2-anti-CD28 (Figure 2B and C). These data collectively suggested that anti-CLDN18.2-anti-CD28 could co-stimulate T cells in a CLDN18.2-dependent manner.
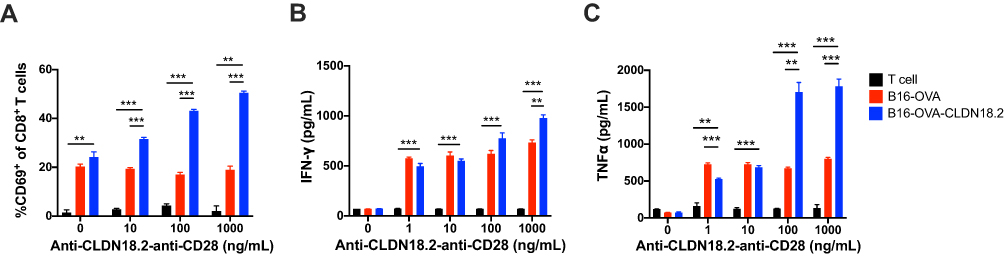 |
Figure 2 Co-stimulatory activation of anti-CLDN18.2-anti-CD28 is dependent on the recognition of CLDN18.2. 2×105 OT-I CD8+ T cells were incubated with 1×104 B16-OVA or B16-OVA-CLDN18.2 cells in the absence or presence of different concentrations of anti-CLDN18.2-anti-CD28 in vitro. (A–C) Percentage of CD69+CD8+ T cells after 72 h of incubation (A) and release of IFN-γ (B) and TNF-α (C) in the supernatant, measured after 24 h of incubation; n = 3, data are shown as means ± SEM; **P < 0.01, ***P < 0.001. |
Safety Profile of Anti-CLDN18.2-Anti-CD28 Fusion Protein in Naïve Mice
Co-stimulatory receptor agonist antibodies have been reported to induce mild to severe immune-related adverse effects in both preclinical models and clinical settings.24,28,29 To test whether our tumor antigen-specific activation of co-stimulatory receptor strategy could reduce unwanted immune activation in non-tumor tissues, non-tumor-bearing C57BL/6 mice were injected intraperitoneally with 100 μg of anti-CLDN18.2-anti-CD28. Peripheral blood was collected for hematological analysis, and various tissues were collected to analyze immune cell infiltration as the evidence of immune activation. There were no obvious differences in body weight, hematological features, and infiltration of immune cells in the heart, liver, spleen, lung, kidney, and stomach (Figure 3A–E). Although gastric epithelial cells express CLDN18.2, anti-CLDN18.2-anti-CD28 treatment did not induce inflammatory immune cell infiltration in the stomach. Overall, our findings suggested that anti-CLDN18.2-anti-CD28 was well tolerated in mice, without obvious side effects.
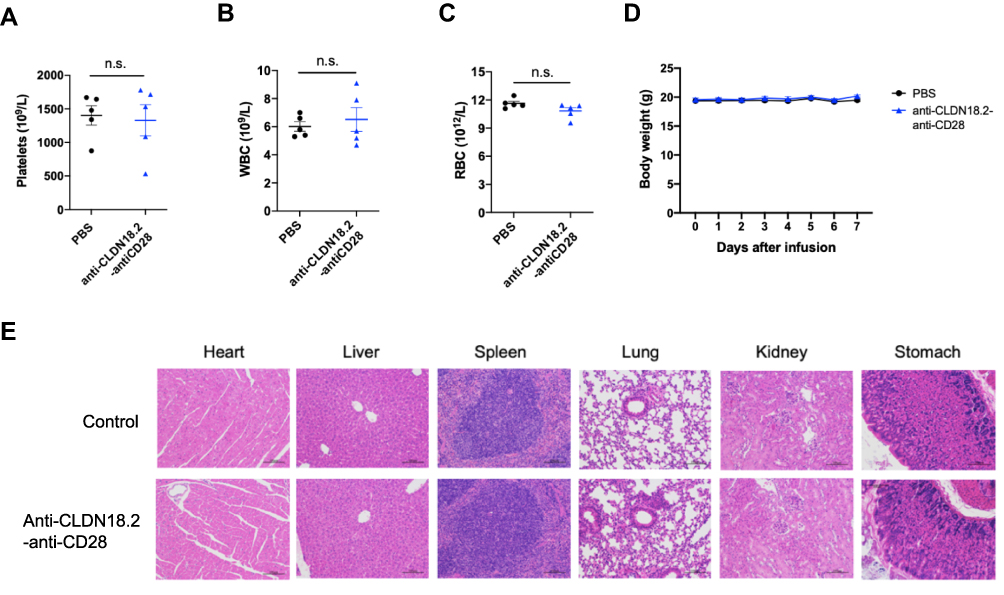 |
Figure 3 Safety profile of anti-CLDN18.2-anti-CD28 in naïve C57BL/6 mice. Mice were injected i.p. with vehicle buffer (PBS) or 100 μg anti-CLDN18.2-anti-CD28 on day 0. (A–C) Platelet (A), WBC (B), and RBC (C) counts in the blood were measured on day 7. (D) The weights of the mice were measured every day after treatment with vehicle buffer (PBS) or anti-CLDN18.2-anti-CD28. Data are shown as means ± SEM; n = 5, n.s., P > 0.05. (E) H&E staining of major organs (heart, liver, spleen, lung, kidney, and stomach) from treated mice on day 7. |
Anti-CLDN18.2-Anti-CD28 Mainly Reduced Tumor Burden by Promoting T Cell Responses
Owing to the enhanced ability of anti-CLDN18.2-anti-CD28 to co-stimulate T cells in vitro, we hypothesized that it could re-activate T cells in the TME by providing co-stimulation signals. To test this hypothesis, B16-OVA-CLDN18.2 tumor-bearing mice were treated with PBS, anti-CLDN18.2, or anti-CLDN18.2-anti-CD28. The percentage of CD4+ and CD8+ T cells and the activation marker (CD69) in tumor tissues were analyzed by flow cytometry 2 days after the last treatment. Although both CD4+ and CD8+ T cells express CD28, we only observed an increase in the percentage of CD8+ T cells and higher expression of CD69 on CD8+ T cells after treatment with anti-CLDN18.2-anti-CD28 (Figure 4A and B). We further tested whether anti-CLDN18.2-anti-CD28 could activate tumor-specific T cell responses. Splenocytes from treated mice were re-stimulated with OT-I or OT-II peptide, which mimics the TSA. The splenocytes produced more IFN-γ and TNF-α in the anti-CLDN18.2-anti-CD28-treated group, indicating elevated tumor-specific CD4+ and CD8+ T cell responses (Figure 4C and D). Consistent with the increase in the number of T cells and the activation status, anti-CLDN18.2-anti-CD28 reduced tumor burden in vivo compared with that in the PBS- and anti-CLDN18.2-treated groups (Figure 4E). There was no difference in body weight among the groups (Figure 4F). Moreover, we used the deletion antibody to verify the necessity of CD8+ T cells for anti-CLDN18.2-anti-CD28 antibody efficacy in B16-OVA-CLDN18.2 mouse models. As expected, the presence of CD8+ T cells affected the function of anti-CLDN18.2-anti-CD28 (Figure 4G and H). Therefore, the anti-CLDN18.2 -anti-CD28 bispecific antibody could provide co-stimulation signals in the TME to promote anti-tumor T cell responses, which subsequently reduced the tumor burden.
 |
Figure 4 Anti-CLDN18.2-anti-CD28 mainly reduced the tumor burden by activating long-term tumor-specific T cells in TME. (A–D) C57BL/6 mice were inoculated subcutaneously with 2.5×105 B16-OVA-CLDN18.2 cells on day 0. Vehicle buffer (PBS), anti-CLDN18.2, and bispecific antibody (anti-CLDN18.2-anti-CD28) were peri-tumor-injected on days 8 and 11. Mice were euthanized on day 13. Percentage of CD8+ or CD4+ T cells (A) in tumor-infiltrating myeloid populations and expression of CD69 on CD8+ or CD4+ T cells (B) in tumor-infiltrating lymphocytes were analyzed on day 13. (C and D) Splenic and lymph node cells from mice were separated and incubated in the absence or presence of OT-I or OT-II peptide. IFN-γ and TNF-α in the supernatants of splenocyte (C) and lymph node cell (D) cultures were measured after 48 h; n = 5. (E and F) C57BL/6 mice were inoculated subcutaneously with 2.5×105 B16-OVA-CLDN18.2 tumor cells. Tumor size (E) and weight (F) of mice. Black arrows indicate the time points of antibody injection; n = 5–6. (G and H) Mice inoculated subcutaneously with B16-OVA-CLDN18.2 were treated with vehicle buffer and anti-CLDN18.2-anti-CD28 with or without deletion antibody (anti-CD8) on days 8 and 11. The tumor size (G) and survival curves (H) were measured and recorded, respectively. Black arrows indicate the time points of antibody injection; n = 6. Statistical significance of survival experiments was determined using the Mantel-Cox test. All the data are shown as means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001. |
Anti-CLDN18.2-Anti-CD28 Reduced the Immunosuppressive Components in Tumor Microenvironment
Cancer responding to immunotherapy does not directly depend only on tumor infiltration by cytotoxic CD8+ T cells; other tumor-associated immune cells, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), also play a protective or opposite role in the process of tumor killing. To test whether anti-CLDN18.2-anti-CD28 reshaped the immunosuppressive TME, we analyzed other types of immune cells in TME on days 4 and 10 after treatment with anti-CLDN18.2-anti-CD28. Decreased TAMs of intra-tumoral infiltration were found in mice treated with anti-CLDN18.2-anti-CD28 (Figure 5A). TAMs in tumors could release cytokines and chemokines that suppress anti-tumor immunity, indicating infusion of anti-CLDN18.2-anti-CD28 might affect the suppressive microenvironment.30,31 The number of MDSCs, another important immunosuppressive myeloid-derived population, including Ly6C+CD11b+ (mMDSCs) and Ly6G+CD11b+ (gMDSCs) of mice treated with anti-CLDN18.2-anti-CD28 showed a trend of reduction (Figure 5B and C) on day 4 after treatment. Previous studies have reported that early infiltration of MDSCs within primary tumors was low and it would gradually increase in the late stage.32,33 Hence, we investigated the MDSCs again after several days. Treatment of anti-CLDN18.2-anti-CD28 reduced both types of MDSCs in TME (Figure 5D and E). Taken together, anti-CLDN18.2-anti-CD28 could also influence the growth of tumor by improving the suppressive TME, suggesting the validity of antigen CLDN18.2 and potent activation capability of CD28 in tumor killing.
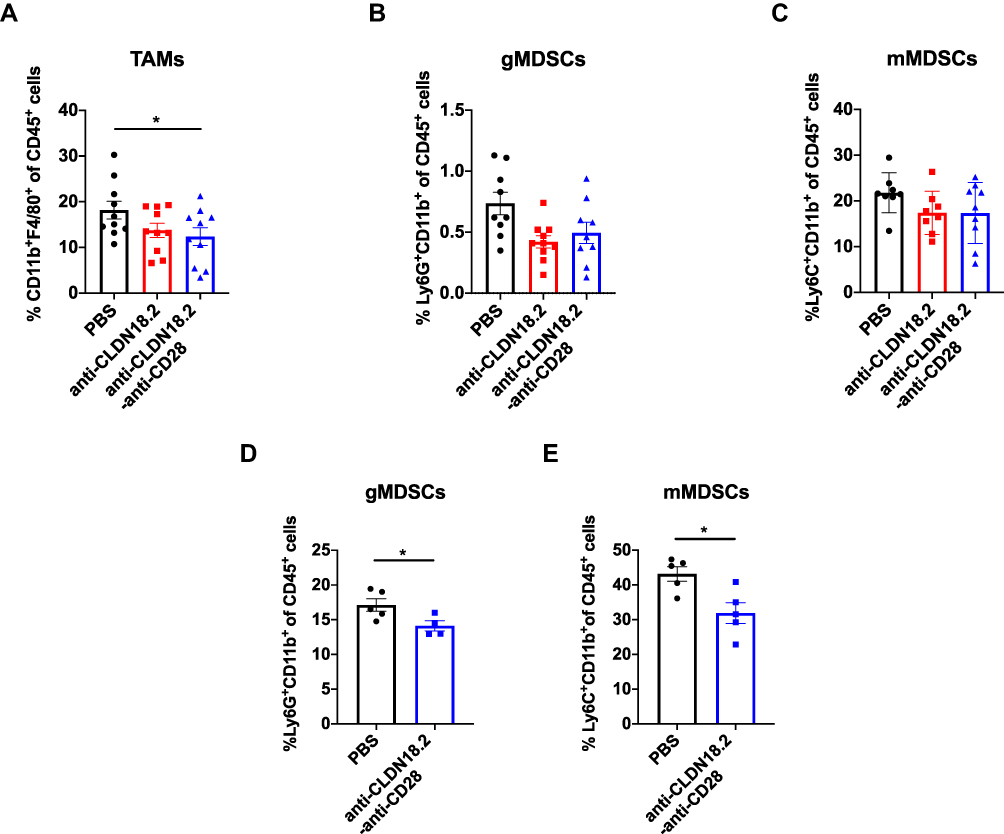 |
Figure 5 Anti-CLDN18.2-anti-CD28 reduced the immunosuppressive components in tumor microenvironment. Tumor-infiltrating immune cells were acquired from B16-OVA-CLDN18.2 mouse models. (A) Proportion of intra-tumoral macrophage subpopulation from C57BL/6 mice. (B and C) Proportion of intra-tumoral CD11b+Ly6G+ MDSCs (B) and CD11b+Ly6C+ MDSCs (C) from C57BL/6 mice. In (A–C), tumors were resected and analyzed on day 12 after B16-OVA-CLDN18.2 inoculation; n = 8–10, data were shown as the mean ± SEM; *P < 0.05. (D and E) Proportion of intra-tumoral CD11b+Ly6G+ MDSCs (D) and CD11b+Ly6C+ MDSCs (E) from C57BL/6 mice. Tumors were resected and analyzed on day 18 after B16-OVA-CLDN18.2 tumor cells inoculation; n = 5, data were shown as means ± SEM; *P < 0.05. |
Discussion
In this study, we designed a CLDN18.2-targeted bispecific antibody by connecting two scFvs against CLDN18.2 and CD28. It co-stimulated T cells in a TSA-specific manner in vitro, increased T cell infiltration and activation in vivo, and showed a promising anti-tumor effect in mouse tumor models.
The initial idea for a bispecific T cell activation antibody was proposed by linking a TSA with CD3 to one molecule; this antibody showed potent anti-tumor efficacy.18 However, this strategy transiently activates all T cells without antigen specificity. The risk of excessive stimulation in a cytokine storm cannot be ignored.24 CD28, due to its co-stimulatory nature, is activated upon TCR signal activation, showing a tumor antigen-specific T cell activation. A recent study described a type of bispecific antibody that combined another TSA and CD28, which promoted long-term anti-tumor immunity, suggesting the potential of CD28 in the development of bispecific antibodies.16
CD28 is a co-stimulatory receptor of T cells that augments and sustains T cell activation and growth. Since CD28 is constitutively expressed on T cells, we believe that CD28, which targets T cell activation, has a broader application potential than other transiently expressed co-stimulatory receptors. We observed enhanced activity of most intra-tumoral CD8+ T cells of mice treated with anti-CLDN18.2-anti-CD28 in B16-CLDN18.2 mouse models, in accordance with the in vitro experimental results. Besides, the immunosuppressive cells including TAMs and MDSCs also showed a trend of reduction. It is possible that the treatment of anti-CLDN18.2-anti-CD28 reduced the tumor burden through enhanced anti-tumor T cell response, which indirectly suppressed MDSCs proliferation by limiting tumor-derived MDSC-promoting factors.34 Since CD28-agonistic antibodies induce severe side effects in clinical settings, we also evaluated the safety profile after anti-CLDN18.2-anti-CD28 infusion. We did not observe obvious side effects after anti-CLDN18.2-anti-CD28 treatment, even in CLDN18.2-expressing gastric tissues. These data collectively suggested that anti-CLDN18.2-anti-CD28 was both effective and safe in mouse tumor models.
The data on the curve of tumor growth in vivo indicated that the control protein, anti-CLDN18.2, also exhibited anti-tumor efficacy to some extent; however, its specific mechanism was unclear. To test whether the anti-CLDN18.2 scFv could directly inhibit tumor growth in vitro, we incubated B16-OVA or B16-OVA-CLDN18.2 cells with different concentrations of the scFv and found that it had no effect on the growth of both tumor cells (Figure S1). It is possible that anti-CLDN18.2 blocks the interaction of CLDN18.2 with other components from the TME, thereby indirectly affecting tumor cells or the tumor-promoting TME.
Although checkpoint inhibition with monoclonal antibodies such as anti-PD-1/PD-L1 has been widely used to treat solid tumors in clinical settings, the objective response rate is approximately 20–30%.35 Low tumor mutation burdens, less tumor infiltration, and low expression of PD-1/PD-L1 may contribute to the unresponsiveness to PD-1/PD-L1 antibodies. Instead of blocking the inhibitory mechanism, our antibody focused on activating T cells, independent of the inhibitory mechanism; this is a potential strategy to treat patients unresponsive to PD-1/PD-L1 antibodies. A recent study described a type of antibody that merged TSAs and anti-CD28 into one molecule; it synergized with anti-PD-1 and enhanced the overall efficacy of the immunotherapy.36 Therefore, the combination of anti-CLDN18.2-anti-CD28 and anti-PD-1 can be considered for further exploration.
The limitations of this study include the following. Owing to the lack of an Fc domain, our anti-CLDN18.2-anti-CD28 fusion protein has limited durability and stability in vivo. We performed peri-tumor injection to overcome these limitations in proof-of-concept experiments. Due to the lack of a CLDN18.2-expressing mouse tumor cell line, we used ectopic CLDN18.2-expressing melanoma B16-OVA cells and pancreatic tumor Pan02 cells in our tumor models. However, anti-CLDN18.2-anti-CD28 showed only modest anti-tumor efficacy in the Pan02-CLDN18.2 mouse model (data not shown). The endogenous expression of CLDN18.2 probably varies in different tumor cells, and it is important to investigate the influence of different CLDN18.2 densities on the anti-CLDN18.2-anti-CD28-induced T cell activation capability.
In summary, bispecific antibodies are useful for generating site-specific immune responses. Our CLDN18.2-targeted CD28 activation bispecific antibody shows tumor-specific T cell co-stimulatory activity in vitro and potent anti-tumor efficacy in mouse models, with limited adverse effects, providing support for extensive exploration of CD28 for the treatment of solid tumors in clinical settings.
Institutional Review Board Statement
All the animal studies were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University (ethics code: A2017043, approved on July 18, 2017).
Acknowledgments
We thank Dr. Hans Schreiber (University of Chicago) for providing B16-OVA mouse melanoma cells.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
Xuanming Yang was supported by the National Natural Science Foundation of China (81971467) and Shanghai Jiao Tong University Scientific and Technological Innovation Funds (2019QYA11).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
1. Maury S, Chevret S, Thomas X, et al. Addition of rituximab improves the outcome of adult patients with CD20-positive, Ph-negative, B-cell precursor acute lymphoblastic leukemia (BCP-ALL): results of the randomized Graall-R 2005 study. Blood. 2015;126(23):1. doi:10.1182/blood.V126.23.1.1
2. Jabbour E, O’Brien S, Ravandi F, Kantarjian H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood. 2015;125(26):4010–4016. doi:10.1182/blood-2014-08-596403
3. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi:10.1056/NEJMoa1411087
4. Kosti P, Maher J, Arnold JN. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front Immunol. 2018;9:1104. doi:10.3389/fimmu.2018.01104
5. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–152. doi:10.1146/annurev-med-062315-120245
6. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, Phase 3 trial. Lancet Oncol. 2015;16(4):375–384. doi:10.1016/S1470-2045(15)70076-8
7. Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. doi:10.1200/JCO.2014.56.2736
8. Fuchs CS, Ozguroglu M, Bang YJ, et al. Pembrolizumab versus paclitaxel for previously treated patients with PD-L1-positive advanced gastric or gastroesophageal junction cancer (GC): update from the phase III KEYNOTE-061 trial. J Clin Oncol. 2020;38(15):4503.
9. Sahin U, Koslowski M, Dhaene K, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res. 2008;14:7624–7634. doi:10.1158/1078-0432.CCR-08-1547
10. Tanaka M, Shibahara J, Fukushima N, et al. Claudin-18 is an early-stage marker of pancreatic carcinogenesis. J Histochem Cytochem. 2011;59(10):942–952. doi:10.1369/0022155411420569
11. Soini Y, Takasawa A, Eskelinen M, et al. Expression of claudins 7 and 18 in pancreatic ductal adenocarcinoma: association with features of differentiation. J Clin Pathol. 2012;65(5):431–436. doi:10.1136/jclinpath-2011-200400
12. Woll S, Schlitter AM, Dhaene K, et al. Claudin 18.2 is a target for IMAB362 antibody in pancreatic neoplasms. Int J Cancer. 2014;134(3):731–739. doi:10.1002/ijc.28400
13. Tureci O, Mitnacht-Kraus R, Woll S, Yamada T, Sahin U. Characterization of zolbetuximab in pancreatic cancer models. Oncoimmunology. 2019;8(1):e1523096. doi:10.1080/2162402x.2018.1523096
14. Schuler MH, Zvirbule Z, Lordick F, et al. Safety, tolerability, and efficacy of the first-in-class antibody IMAB362 targeting claudin 18.2 in patients with metastatic gastroesophageal adenocarcinomas. J Clin Oncol. 2013;31(15):4080. doi:10.1200/jco.2013.31.15_suppl.4080
15. Sahin U, Schuler M, Richly H, et al. A phase I dose-escalation study of IMAB362 (Zolbetuximab) in patients with advanced gastric and gastro-oesophageal junction cancer. Eur J Cancer. 2018;100:17–26. doi:10.1016/j.ejca.2018.05.007
16. Zhu G, Foletti D, Liu X, et al. Targeting CLDN18.2 by CD3 bispecific and ADC modalities for the treatments of gastric and pancreatic cancer. Sci Rep. 2019;9(1):8420. doi:10.1038/s41598-019-44874-0
17. Chasov V, Zaripov M, Mirgayazova R, et al. Promising new tools for targeting p53 mutant cancers: humoral and cell-based immunotherapies. Front Immunol. 2021;12:3223. doi:10.3389/fimmu.2021.707734
18. Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316(6026):354–356. doi:10.1038/316354a0
19. Hoffman L, Gore L. Blinatumomab, a bi-specific anti-CD19/CD3 BiTE® antibody for the treatment of acute lymphoblastic leukemia: perspectives and current pediatric applications. Front Oncol. 2014;4:63. doi:10.3389/fonc.2014.00063
20. Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–175. doi:10.1016/j.pharmthera.2017.08.005
21. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–1043. doi:10.1158/1078-0432.CCR-12-2064
22. Qi X, Li F, Wu Y, et al. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat Commun. 2019;10(1):2141. doi:10.1038/s41467-019-10088-1
23. Correnti CE, Laszlo GS, de van der Schueren WJ, et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia. 2018;32(5):1239–1243. doi:10.1038/s41375-018-0014-3
24. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a Phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi:10.1056/NEJMoa063842
25. Jabbour EJ, Gokbuget N, Kantarjian HM, et al. Transplantation in adults with relapsed/refractory acute lymphoblastic leukemia who are treated with blinatumomab from a phase 3 study. Cancer. 2019;125(23):4181–4192. doi:10.1002/cncr.32335
26. Dombret H, Topp MS, Schuh AC, et al. Blinatumomab versus chemotherapy in first salvage or in later salvage for B-cell precursor acute lymphoblastic leukemia. Leuk Lymphoma. 2019;60(9):2214–2222. doi:10.1080/10428194.2019.1576872
27. Loffler A, Kufer P, Lutterbuse R, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95(6):2098–2103. doi:10.1182/blood.V95.6.2098
28. Vonderheide RH, Flaherty KT, Khalil M, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–883. doi:10.1200/Jco.2006.08.3311
29. Dubrot J, Milheiro F, Alfaro C, et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol Immun. 2010;59(8):1223–1233. doi:10.1007/s00262-010-0846-9
30. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–78. doi:10.1038/nrc1256
31. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. doi:10.1038/nri3789
32. Jordan KR, Amaria RN, Ramirez O, et al. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother. 2013;62(11):1711–1722. doi:10.1007/s00262-013-1475-x
33. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. doi:10.1016/j.it.2016.01.004
34. Groth C, Hu X, Weber R, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25. doi:10.1038/s41416-018-0333-1
35. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
36. Waite JC, Wang B, Haber L, et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci Transl Med. 2020;12(549):eaba2325. doi:10.1126/scitranslmed.aba2325
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.